 Review
Review

Affiliation:
1Department of Pharmacology and Toxicology, Temerty Faculty of Medicine, University of Toronto, Toronto, ON M5G 2C8, Canada
ORCID: https://orcid.org/0000-0001-7371-7262
Affiliation:
1Department of Pharmacology and Toxicology, Temerty Faculty of Medicine, University of Toronto, Toronto, ON M5G 2C8, Canada
2Department of Medicine, Temerty Faculty of Medicine, University of Toronto, Toronto, ON M5G 2C8, Canada
Email: s.george@utoronto.ca
Explor Neuroprot Ther. 2025;5:100498 DOI: https://doi.org/10.37349/ent.2025.100498
Received: August 30, 2024 Accepted: February 09, 2025 Published: March 09, 2025
Academic Editor: Dasiel Oscar Borroto-Escuela, Karolinska Institutet, Sweden, University of Malaga, Spain; Rafael Franco, Universidad de Barcelona, Spain
The article belongs to the special issue GPCR Heteroreceptor Complexes as Key Players in Neuroprotection
Neurodegenerative diseases are a complex ensemble of ailments characterized by progressive neuronal deterioration and ultimate loss, resulting in drastic impairments of memory, cognition and other brain functions. These incapacitating conditions are challenging for the public health system worldwide, with unfortunately no real cure and lack of efficient drugs capable of delaying or reversing these diseases. In this context, the endocannabinoid system and exogenous cannabinoids represent an interesting field of research due to numerous studies highlighting the neuroprotective effect of cannabinoids from different sources, i.e., endogenous, phytocannabinoids, and synthetic cannabinoids. This review highlights the multilayered effects of cannabinoids and the endocannabinoid system to block the progression of neurodegeneration and minimize the deleterious effects of insults that affect the brain. We illustrate examples showing that the main effects of cannabinoids modulate different components of the brain response to these insults at the level of three major mechanisms involved in neurodegeneration: neuroinflammation, excitotoxicity, and oxidative stress.
Neurodegeneration is a complex and multifaceted phenomenon, which could be defined as a progressive deterioration and loss of neuronal functions often leading to significant impairments in cognitive, motor, and sensory abilities [1, 2]. The triggers of neuronal deterioration and ultimate loss can be attributed to a myriad of factors, including genetic predisposition, environmental exposures, aging, and various pathological processes [3, 4]. The most known and studied neurodegenerative diseases are age-related such as Alzheimer’s disease (AD) and Parkinson’s disease (PD). Other important neurodegenerative diseases include Huntington’s disease, amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), spinocerebellar ataxia, and the neuronal loss following ischemia resulting from stroke or brain trauma and injuries [4–8]. Whether the characteristics of these diseases include protein aggregates such as β-amyloid or α-synuclein in AD and PD, respectively, or oxidized lipid molecules and reactive oxygen species (ROS) in some neurological disorders, including AD, PD, and stroke, all these neurodegenerative diseases are marked by chronic neuroinflammation [5, 8–10]. Indeed, studies have shown that the protein aggregates and/or oxidized lipid molecules cause microglial activation, a key feature in neurodegenerative diseases. The activated microglia release pro-inflammatory molecules, which in turn activate astrocytes, and both these activated glial cells increase the levels of inflammatory cytokines and the production of ROS. A feed-forward loop that supports chronic oxidative stress coupled to persistent neuroinflammation is thus established [5, 9–12]. This is exacerbated by the higher susceptibility of neurons to oxidative stress due to their high metabolic rate [11], and the fact that multiple neuroprotective mechanisms are declining during aging [5, 9–12]. The global burden of neurodegenerative diseases is staggering, with the World Health Organization (WHO) estimating that over 50 million people are currently affected by these debilitating conditions, a number that is expected to rise greatly in the coming decades [13–16].
Contemporary medications for neurodegenerative and neurological disorders are absent and mostly focus on treating the symptoms rather than having a substantial effect on stopping or delaying disease progression [17–19]. Novel therapeutic interventions and preventive strategies are under investigation, among them targeting of the endocannabinoid (eCB) system (ECS), and cannabinoids in general, have garnered significant interest in recent years.
Indeed, since the demonstration in earlier studies that extracted phytocannabinoids had some potential in neuroprotection, and following the discovery of an endogenous cannabinoid system that may play a similar role, the use of cannabinoids as alternative medicines has gathered a growing research interest. A large amount of dedicated research has demonstrated the neuroprotective properties of various cannabinoids, whether endogenous or exogenous, under different conditions, and both in vitro and in vivo models. In most cases, the use of cannabinoids and the modulation of the ECS were shown to limit the effects of different insults that could lead to neuronal damage and neuronal loss [5, 18, 20–23].
The present mini-review will describe briefly the main elements of the ECS, its involvement in neuroprotection, the main mechanisms responsible for neurodegeneration, and how the cannabinoids may act at different levels to limit the impact of these critical mechanisms.
Cannabis sativa has been used for centuries by humans [24, 25], but its extracts have been studied more intensively in the last six to seven decades, after the identification of the two main constituents, the psychoactive Δ9-trans-tetrahydrocannabinol (THC) [26], and the non-psychoactive cannabidiol (CBD) ([27]; reviewed in [28, 29]). Currently, the number of natural compounds from Cannabis sativa is close to 565, among which, 120 cannabinoids have been isolated and classified [24, 25, 29]. The discovery of THC and CBD was followed by the identification of the receptors that mediate their actions: the cannabinoid receptors, and then followed by the identification of endogenous ligands for these receptors, called eCBs, within the physiological ECS [29–35].
The ECS comprises the cannabinoid receptors, their ligands eCBs, the enzymes responsible for eCBs synthesis and degradation, as well as a not well-known reuptake system. The present description is relatively succinct and for further detailed reviews please see the following [5, 21, 23, 29, 33, 35, 36].
Two major receptors, the cannabinoid CB1 and CB2 receptors (CB1Rs, CB2Rs), were identified, cloned, and extensively studied. Both receptors, CB1R and CB2R belong to the G protein-coupled receptor (GPCR) superfamily and couple to Gi/o to inhibit adenylyl cyclase and protein kinase A (PKA) (reviewed in [21, 28, 29, 32, 35]). They also modulate the mitogen-activated protein kinase (MAPK) pathway by regulating different kinases such as extracellular signal-regulated kinase (ERK), p38 kinase, and c-Jun N-terminal kinases [21, 28]. CB1R is highly expressed in the brain and represents the main target of the eCBs. CB1R is expressed at presynaptic terminals and their activation leads to the inhibition of neurotransmitter release resulting in the inhibition of excitatory and inhibitory neurotransmission [21, 28, 37, 38]. There is also evidence for CB1R expression at postsynaptic locations where its activation leads to the modulation of ion channels, notably the N-methyl-D-aspartate (NMDA) receptors [21, 39]. The CB2R is expressed mostly by immune cells and seems to be involved in the modulation of the immune system principally. In the brain, CB2R expression increases during neuroinflammation but is very low under normal physiological conditions ([40]; reviewed in [21, 41]). Both CB1R and CB2R are expressed by glial cells, astrocytes and microglia [42]. The CB1R expression has also been shown in oligodendrocytes and human vascular endothelial cells of the blood-brain barrier (BBB) [21, 43, 44]. Although CB2R expression is generally very low leading to some controversy around their existence in certain types of cells such as astrocytes, it has been shown that this receptor is increased in astrocytes and microglia after induced neurotoxicity and during neuroinflammation (reviewed in [42]).
Outside the CB1Rs and CB2Rs, multiple other receptors were shown to be involved in the transduction of cannabinoid signaling (reviewed in [28, 29, 35, 38, 45]). That is the case of nuclear peroxisome proliferator-activated receptors (PPARs) [14, 46], and some categories of transient potential receptors (TRP) such as the vanilloid type1 channel (TRPV1) [47, 48]. Also reported as potential receptors for cannabinoids are serotonin 1A receptor (5HT1A) [49], opioid receptors mu and delta (µ-OR and δ-OR) [28, 38], as well as other receptors including some orphan receptors such as GPR55, GPR119 and GPR18 [38, 50–52], and multiple receptor complexes (heteromers) formed between CB1Rs or CB2Rs and other GPCRs or non-GPCRs receptors [28, 29, 33, 46, 50, 53–56].
Eicosanoids N-arachidonoyl ethanolamide (anandamide, AEA) and 2-arachidonoyl glycerol (2-AG) represent the primary eCBs and are the most studied and characterized [34, 35, 38, 57–59]. Both AEA and 2-AG act as endogenous ligands for CB1Rs and CB2Rs [28, 34, 35]. There is however a long list of endogenous congeners capable of modulating CB1Rs or CB2Rs or related receptors, among them 2-AG ether [59], virodhamine, with antagonistic activity at the CB1R [60], and oleamide [61]. 2-AG is a full agonist for both cannabinoid receptors, whereas AEA acts as a partial agonist, and both eCBs show higher affinity for the CB1R in comparison to the CB2R [28, 54, 62].
It is well known that these lipidic molecules are synthesized on demand by the actions of specific lipases in response to increased intracellular Ca2+ levels and are immediately released. Both AEA and 2-AG are produced by post-synaptic neurons by the action of different enzymes and act via retrograde transsynaptic action to activate CB1R. AEA is produced by the action of two main enzymes, Ca2+-dependent N-acyltransferase and N-acyl-phosphatidylethanolamine-hydrolyzing phospholipase D (NAPE-PLD) from membrane glycerophospholipids and its levels are regulated by its hydrolysis by fatty acid amide hydrolase (FAAH) (reviewed in [5, 21, 29, 45]). 2-AG is also produced in post-synaptic neurons on demand from diacylglycerol (DAG) in a two-step process involving the action of phospholipase C, and diacylglycerol lipase (DAGL) ([58]; reviewed in [35]). Other studies show that 2-AG is also synthesized through two additional major pathways: from 2-acyl lysophosphatidic acid (LPA) by 2-LPA phosphatase, and from 2-acyl lyso-phosphatidylinositol (LPI) by lyso-phospholipase C (reviewed in [5, 35, 45]). An important enzyme, the monoacylglycerol lipase (MAGL), controls 2-AG levels by assuming its degradation. The degradation of 2-AG occurs in the pre-synaptic neurons after release from post-synaptic neurons, whereas the degradation of AEA occurs in the post-synaptic neurons, where it is synthesized [5, 21]. In addition, other enzymes, such as cyclooxygenase-2 (COX2) and lipoxygenases can metabolize AEA or 2-AG into other bioactive derivative compounds, some of which may also behave as endocannabinoid-like molecules [21, 29, 63, 64]. Some of these derivatives may signal either through the CB1Rs or CB2Rs or through non-cannabinoid targets [21, 29, 63, 64]. Also important in the mechanism of eCB action is their possible removal from the synaptic space by a reuptake mechanism followed by hydrolysis [65]. There is a need for more work in this regard to identify these potential specific endocannabinoid transporters [65].
It is well accepted now that the eCBs synthesized in postsynaptic neurons are released and activate CB1Rs on pre-synaptic neurons in a retrograde manner (reviewed in [5, 21, 29, 45]). In some cases, eCBs can also activate CB1Rs expressed on the membranes of postsynaptic neurons [37, 38]. The generally acknowledged mechanism is that the activation of CB1Rs by eCBs decreases presynaptic neurotransmitter release, which may occur via several mechanisms, including inhibition of calcium influx and activation of potassium channels [37, 38, 58, 66]. Reuptake and then degradation terminate the eCBs (AEA and 2-AG) actions per se, but signal transduction is likely to continue by the actions of the derivatives and bioactive compounds resulting from their degradation. It has been established that the eCBs using this mechanism of action would inhibit pre-synaptic neurotransmitter release at both GABA and glutamate terminals, thus modulating several neurotransmitter systems [5, 45, 67, 68].
The literature abounds with evidence for the neuroprotective role of cannabinoids and the amelioration of outcomes in different conditions and models depicting various neurodegenerative diseases. That is the case for PD models [6, 69–80], where different eCBs and exogenous cannabinoids showed neuroprotective effects and amelioration of affected motor and/or cognitive functions. That is also the case in models for AD [81–85], in models for ischemic stroke or brain trauma injuries [22, 49, 86–97], as well as in disorders such as ALS/MS [98–101], Huntington disease [69, 102–105], glaucoma and retinal neurodegenerative diseases [82, 106–112], and other neurological diseases such as epilepsy, autism and dementia [113–118].
These are only a few examples from a long list of research articles and systematic reviews that underscore a clear neuroprotective role played by multiple cannabinoids including eCBs, synthetic or phytocannabinoids. This suggests that the ECS and related receptors and enzymes play a key role in neuroprotection and consequently, this large body of evidence should render the system a major target for the development of novel pharmacotherapies aiming to relieve and diminish the impact of age-related and other neurodegenerative diseases.
Interestingly, the role of cannabinoids as neuroprotectants was illustrated in all the different mechanisms disrupted during the development of neurodegenerative diseases. As will be detailed, cannabinoids block the release of neurotransmitters, notably glutamate, by their retrograde action, minimizing thus the potential excitotoxicity of high glutamate concentrations. They also inhibit the release of pro-inflammatory molecules, including cytokines and chemokines, by astrocytes and microglia. In addition, an important role of cannabinoids in inhibiting oxidative stress was shown by many studies.
Due to the immense amount of data published in this regard, only a few examples will be detailed. The majority of studies depict a positive role for cannabinoids in neuroprotection and amelioration of outcomes in the progression of neurodegenerative diseases. However, some studies showed the converse, wherein under certain conditions, the cannabinoids had a neurotoxic role and may contribute to development of neurodegeneration [119–123], which is not in the scope of this discussion.
There are multiple mechanisms by which activation of the cannabinoid system by either exogenous cannabinoids or mobilization of eCBs exerts neuroprotective effects. One key mechanism is by reducing neuroinflammation, a major contributor to neuronal damage and cell loss [35, 124]. Secondly, the cannabinoid system has been shown to modulate oxidative stress, another key factor in the pathogenesis of various degenerative neurological disorders. Further, the regulation of excitotoxic and apoptotic pathways, which play crucial roles in neurodegenerative diseases, is a third mechanism by which the cannabinoid system exerts its neuroprotective effects, as will be developed in the next paragraphs.
As mentioned, the connection between neuroinflammation, excitotoxicity, oxidative stress, and neurodegeneration is rather multifaceted and interconnected (Figure 1). To simplify, neurodegeneration could be considered as resulting from the overlapping effects of these three mechanisms (see for more detailed reviews [5, 10, 11, 17, 84, 125–130]). Neuroinflammation is regarded as the brain’s response to injuries and disease processes, which may involve the activation of glial cells, microglia and astrocytes, compromising the integrity of the BBB and its functions, increasing its permeability, with infiltration of immune cells. While the first response is protective, its persistence can lead to the release of pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), which proliferate the inflammation and other molecules such as chemokines (e.g., COX2) that recruit additional immune cells, that prove to be harmful to and destroy the neurons. Elevated glutamate can lead to excitotoxicity, with excessive activation of glutamate receptors notably of extra-synaptic NMDA receptors [10, 17, 131] causing excessive calcium influx into neurons. This may trigger a cascade of events including increased generation of ROS, which are unstable, free radical, highly reactive molecules, generated within mitochondria, leading to mitochondrial dysfunction, contributing to oxidative stress and neuronal damage. An imbalance between the production of ROS and the ability to enzymatically detoxify these molecules in the brain characterizes oxidative stress [10, 17]. Neuroinflammation can exacerbate oxidative stress by increasing the production of ROS, which further damages cellular components including DNA, RNA, proteins, and lipids. In summary, the combination and interconnectedness of these factors create a vicious cycle where each one exacerbates the others, resulting in progressive neuronal damage and cell death that ultimately leads to neurodegeneration (Figure 1). As will be detailed bellow, several studies have demonstrated the ability of cannabinoids to mitigate neuronal damage and death in various pathological conditions by modulating these main mechanisms.
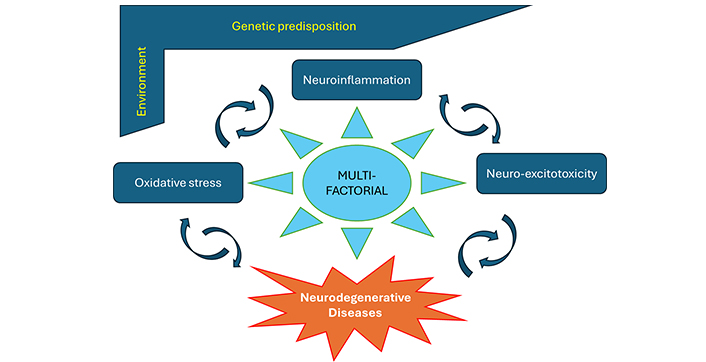
Neurodegenerative diseases are multi-factorial. Neurodegenerative diseases involve a progressive neuronal loss due to a number of factors, including genetic predisposition, environmental exposures, aging, and share multiple pathological processes notably neuroinflammation, excitotoxicity, and oxidative stress. These factors are interconnected. To simplify, neuroinflammation is considered as the brain response to injuries and disease, involving activation of glial cells, microglia and astrocytes, weakening of the blood-brain barrier, and infiltration of immune cells. While the first response is protective, its persistence can be harmful to the neurons. Excitotoxicity is due to elevated glutamate and excessive activation of NMDA receptors causing excessive calcium influx into neurons, increased ROS production and mitochondrial dysfunction, contributing thus to oxidative stress. The combination and interconnection of these factors create a vicious cycle where each one exacerbates the others, resulting in progressive neuronal damage that ultimately leads to neurodegeneration
One major source of neurotoxicity in the brain is sustained glutamate signaling. The glutamate-mediated neurotoxicity is due essentially to an overstimulation of ionotropic glutamate receptors, primarily NMDA receptors (NMDAR), leading to a massive Ca2+ influx into the postsynaptic terminal [5, 21]. For a long time, there was an enduring controversy whether NMDAR activation promoted neuronal protection or neuronal death. It was later resolved that NMDAR-induced responses depended on the receptor location, with synaptic NMDAR activation leading to a neuroprotective effect, whereas stimulation of extra-synaptic NMDAR leads to cell death [131], though it was shown that NMDA receptor-mediated excitotoxicity depends on the coactivation of synaptic and extra-synaptic NMDARs [132]. Perturbations in the balance between synaptic and extra-synaptic NMDAR activity seem to impact neuronal function and appear to be common in some neurodegenerative diseases [131]. The elevated glutamate levels result in the dysfunction of mitochondria and Ca2+-mediated activation of a myriad of enzymes and proteases, which contribute to the degradation of neuronal structure and consequently lead to neuronal death [5, 21, 131]. This type of neurotoxicity was shown to be involved in the neuropathogenesis of different conditions such as AD, PD, ALS, and Huntington’s disease.
The eCBs are neuronal activity-induced and it was shown that activation of glutamate receptors and the resulting Ca2+ influx are influential in the induction of the main eCBs 2-AG and AEA, and their related congeners [133, 134]. Glutamate may originate from neurons but also from glial cells, i.e., astrocytes and microglia. The activation of CB1Rs and CB2Rs in glial cells was shown to lead to the release of gliotransmitters including glutamate [67, 68]. Studies have shown that abnormal glutamate release also triggers an interaction between dendritic spines and activated microglia, which induces process extension from microglia toward neurons, with some studies suggesting that the released eCBs at the sites of synaptic activity (or injury) may play a chemo-attractant role to recruit microglia in a CB2R-dependent manner, notably toward neuroinflammatory lesion sites [5].
Indeed, studies have shown that glutamate elevations and release of eCBs would lead to an activation of microglia [135, 136]. Microglial transcriptomes indicate that microglia can perform three important functions: (i) sense their natural environment, (ii) oversee CNS physiological maintenance, and (iii) defend against damaging agents [137]. In contrast to the prevalent belief, there are no resting microglia, but they are rather functionally engaged continuously, and any dysregulation in their functions could be detrimental to neurons and lead to neurodegeneration [136, 137]. During infection or injury, there is a release of factors called pathogen-associated and danger-associated molecular patterns (PAMPs and DAMPs), respectively, which are recognized by pattern recognition receptors such as the toll-like receptors (TLRs) which are broadly expressed by microglia and astrocytes (reviewed in detail in [136–138]). Microglia and astrocytes both can be activated by TLR-mediated mechanisms, and thence release cytokines and chemokines, which can either promote neuronal survival or induce neuroinflammation and heighten neuronal damage, in extreme conditions such as after ischemia or spinal cord injury [127, 137–139]. The acute activation of these glial types of cells is usually accompanied by the release of glutamate, eCBs, but also proinflammatory cytokines such as IL-1β, TNF-α, IL-2, and IL-6 [136]. These generated proinflammatory cytokines and other excitotoxic molecules increase the production of free radical and lipid peroxidation molecules, leading to mitochondrial dysfunction and further intensification of the detrimental effects of excitotoxicity (Figure 2). However, microglia can also have a pro-survival profile leading to activation of repair mechanisms [140, 141] and regeneration by releasing trophic molecules such as brain-derived neurotrophic factor (BDNF) and cytokines with dual (pro- and anti-inflammatory) potential, like TGF-β and IL-10. Functional coupling of microglia, astrocytes, and neurons during normal synaptic activity but also during excitotoxic injury involves, among other mechanisms, active participation of the ECS. For example, 2-AG from organotypic hippocampal slice cultures has been shown to mediate neuroprotection against NMDA-mediated excitotoxicity through a mechanism involving abnormal-CBD (abn-CBD)-receptor, believed to be GPR18 exclusively expressed on microglia [142]. Besides the important role of microglia, there is a highly significant role played by astrocytes in the neuroprotection mediated by the ECS. Firstly, astrocytes express CB1Rs through which eCBs can elicit the release of gliotransmitters including glutamate, as was shown with AEA in the nucleus accumbens core of rats [143]. Secondly, there is evidence that astrocytes, notably in culture, once activated or in response to CB1R activation can release eCBs including 2-AG, AEA, homo-γ-linolenylethanolamide (HEA) and docosatetraenylethanolamide (DEA) [144–146]. In contrast, there is evidence that astrocytes express MAGL, and consequently are actively involved in the degradation of 2-AG [2, 42, 147–149]. It is important to note that there is a robust interdependence between microglia and astrocytes [127, 136]. Neuroinflammation has been shown to induce two types of reactive astrocytes, A1 and A2, capable of releasing pro- and anti-inflammatory mediators, as well as neurotrophic factors (reviewed in [42]). The eCBs can block the activation of astrocytes as was shown with 2-AG and palmitoylethanolamide (PEA) [150–154] and neutralize reactive astrogliosis in different models, resulting in a better prognosis of neuronal repair and survival.
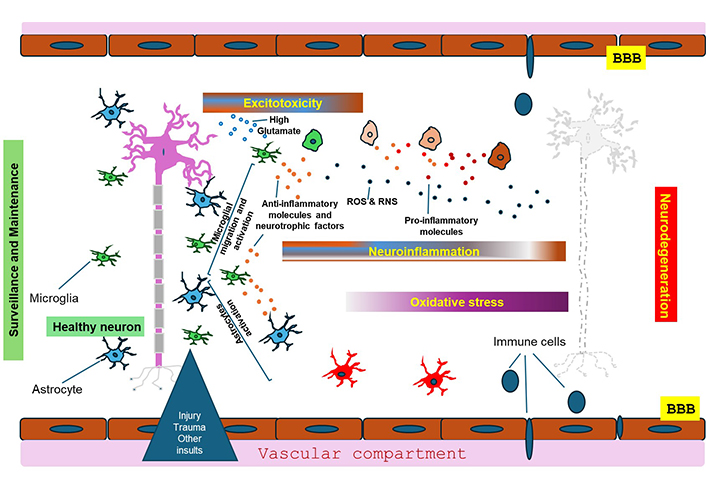
Mechanisms leading to neurodegeneration. Microglia and astrocytes are vital for keeping neurons healthy and preserving normal homeostasis and neurotransmission. Injuries, infections and other insults lead to the activation of microglia and astrocytes and release of glutamate, the main source of excitotoxicity, and also chemokines and cytokines, some of which are involved in anti-inflammation and others in pro-inflammation. These molecular mixtures can lead to either neuronal survival or severe neuro-inflammation, in extreme conditions. The generated pro-inflammatory cytokines and other neurotoxic molecules increase the production of free reactive oxygen species (ROS) and lipid peroxidation, leading to mitochondrial dysfunction and further intensification of the damaging effects of neurotoxicity. The persistence of neuroinflammation and the failing of the blood-brain barrier (BBB) leads to infiltration of immune cells that exacerbate neuroinflammation and ultimately lead to neuronal death. See the text for more details
Cytokines such as IL-6, IL-1β, and TNF-α [5, 42, 94, 127, 136] are released by astrocyte, microglia and endothelial cells in response to brain injuries or during the development of neurodegenerative diseases and their sustained release is a key factor in the modification and vulnerability of the BBB [155]. This vulnerability leads to the infiltration of different peripheral immune cells and chronic neuroinflammation, loss of neurons, and a build-up of the process of neurodegeneration in response to the persistence of insults and the protracted release of proinflammatory cytokines and generation of ROS [17, 127]. This multifactorial mechanism is very important in the context of chronic neurodegenerative diseases such as AD, PD, and MS [127]. Many studies have shown that the ECS modulates neuroinflammation through regulation of cytokine production. Several inhibitors for the enzymes responsible for eCB degradation, including the main enzymes FAAH, MAGL, COX2, and α,β-hydrolase domain-6 (ABHD6) and α,β-hydrolase domain-12 (ABHD12) have been synthesized and characterized [156]. Elevation in eCBs by blocking their degradation was shown to reduce the production of cytokines. That is the case with the inhibitors of FAAH, URB597 or PF3845, which inhibited TNF-α and IL-1β levels in the hippocampi of aged mice [157], the MAGL inhibitor CPD-4645 that reduced IL-1β and IL-6 brain levels after induced inflammation [158], or a selective inhibitor of ABHD6 [148]. These inhibitors among others have been shown to increase the levels of different eCBs and their derivatives, and participate in reducing cytokines and chemokines in different neuroinflammatory conditions, including in MS patients [159], in models of PD [160, 161], and in experimental models of spinal cord and brain injury ([162–164]; reviewed in [5, 21]).
Another strategy that was explored to reduce the levels of cytokines and chemokines was the use of exogenous cannabinoid agonists or antagonists. Some agonists for CB2R were efficient in reducing the levels of pro-inflammatory cytokines in different conditions including PD [165], intracerebral hemorrhage [166], and lipopolysaccharide (LPS)-induced neuroinflammation [167]. Some studies using antagonists for CB1Rs or CB2Rs were not as conclusive. For example, while the CB1R inverse agonist SR141716A (rimonabant) and the CB2R antagonist SR144528 significantly reduced LPS-induced IL-1β production in the brain [168], another study showed that SR141716A in contrast was able to increase pro-inflammatory cytokines in an experimental different model [169].
When it comes to protection of the BBB by the ECS, studies have shown that both 2-AG and AEA decrease BBB permeability while decreasing pro-inflammatory cytokines in different models (reviewed in [5]). They probably also alter immune cell movement at the BBB [5, 170], and may thus provide protection to the BBB during conditions such as ischemic stroke and brain injury [171]. As mentioned, it is no surprise that multiple studies used different inhibitors targeting the degradative enzymes of the eCBs, to directly increase the endogenous levels of the eCBs and their derivatives as possible treatment alternatives to lower the vulnerability of BBB disruption and at the same time decrease neuroinflammation (reviewed in [5]). Indeed, many FAAH and MAGL inhibitors are in clinical trials, at Phase I or II (reviewed in [155]). Many of these trials may not continue, however, due to severe adverse effects encountered during the study of an inhibitor of FAAH called BIA 10-2474 (reviewed in [5, 155]). Many exogenous ligands for CB1R, CB2R, or other associated receptors were also tested [74, 92, 96, 99, 155, 172, 173]. A great interest was notably dedicated to the effects of CBD and derivatives, given the knowledge that this non-psychoactive agent is devoid of severe side effects and showed promise in different models including for AD, PD, ALS, brain trauma, and ischemia [16, 119, 156, 174–177]. An equivalent interest was also dedicated to the use of CB2R agonists to show their efficacy in preventing or reducing neuroinflammation and BBB leakage in several experimental models [5, 89, 177, 178]. Many studies showed that the effects mediated by CB2R may be very effective in neuroprotection in different models [95, 100, 165, 167, 178–182]. It is known that CB2R expression increases during neuroinflammation. Indeed, after earlier being considered as an exclusively peripheral receptor, CB2R was shown to be present in low amounts in the brains of several animal species, including humans [183, 184]. It was repeatedly shown to increase during neuroinflammation, and its activation and increased expression have been shown in various neurodegenerative diseases [184–187]. These receptors have been intensively studied as possible pharmacological targets against neuroinflammation and neuroinflammation-related neurodegeneration. For example, a recent study that used a highly selective agonist, HU-308, as well as its enantiomer, HU-433, which is a putative selective agonist to target CB2R, showed that stimulation with either drug effectively reduced the accumulation of cytokines and related signaling in vitro and in a model of retinopathy, in support of CB2R as a valuable target for drugs targeting inflammation and cellular death [140, 188].
Allosteric modulators of cannabinoid receptors represent another interesting category of drug to modulate the ECS. These allosteric modulators do not possess intrinsic efficacy, but instead enhance or diminish the cannabinoid receptor’s response to orthosteric ligands and may have many advantages over the orthosteric ligands such as higher selectivity, biased signaling, and potentially increased therapeutic benefits with lower side effect profiles (reviewed in [188–190]).
While describing the utility of cannabinoids and the ECS in oxidative stress, the strong interactions among neuroinflammation, excitotoxicity, and oxidative stress are paramount and have been reported in neurodegenerative diseases such as AD, PD, and Huntington’s disease [137, 180, 191–195]. ROS are regular products of the mitochondrial respiratory chain, and their regulation is important for cell survival. However, both excitotoxicity and the activation of microglia lead to an increase in ROS and reactive nitrogen species (RNS) [9, 11, 21, 127, 128, 196]. The accumulation of ROS and RNS contributes highly to oxidative stress, which has many detrimental consequences such as degradation of DNA, carbohydrates and lipid peroxidation, and mitochondrial functions [196–201]. Though some of these molecules do not escape the cell membrane, others that are more stable (e.g., H2O2) can traverse the cell membrane and spread to adjacent tissues. Multiple studies suggest that oxidative stress plays a critical role in the progression of different diseases including CNS disorders and neurodegenerative diseases [196–201], although the use of antioxidants showed limited or, in some cases, negative results [202, 203].
Interestingly, activated oxidative stress pathways were shown to impair ECS-mediated signaling, whereas activation of CB1R as well as the upregulation of brain CB2R reduce oxidative stress in the brain. This results in reduced neuroinflammation and consequently, attenuated neuronal and tissue damage. Multiple studies have shown that activation of the ECS has neuroprotective effects at different levels. For instance, the inhibition of FAAH by URB597, and the consequent increase in the endogenous levels of AEA, which prevented excitotoxic damage, attenuated motor and biochemical (lipid peroxidation and protein carbonylation) alterations in rats, while preserving the structural integrity of the striatum and inhibiting the neuronal loss [204]. This study among many others is in favor of the idea that pharmacological manipulation of the ECS plays a neuroprotective role against excitotoxic insults and the resulting oxidative stress as shown by the biochemical results in the central nervous system [204]. In another study, the cannabinoid trans-caryophyllene has been shown to increase neuronal viability through inhibition of mitochondrial depolarization and oxidative stress, and by increasing the expression of BDNF in rat neuronal-glial cultures [205]. Many studies showed that CB2R expression is increased in microglia and other immune cells and may participate in the reduction of oxidative stress [184]. This is only a limited window into a large volume of research showing that the ECS and cannabinoids are involved in the neuroprotective effects against oxidative stress, but are not antioxidants by themselves.
We provide an overview of the main mechanisms actively participating in neurodegeneration and the active role of cannabinoids and the ECS in inhibiting these mechanisms at different levels. Cannabinoids and the ECS participate in inhibiting the excitotoxicity, reducing the release of cytokines, chemokines, and other molecules with deleterious effects, and inhibiting the overactivation of astrocytes and microglia, as well as the development of ROS, participating thus in the inhibition of intensification of neuroinflammation (Figure 3). They also act to minimize the BBB disruption vulnerability, and some indications suggest a role in neurogenesis under certain conditions [171]. The role of cannabinoids and the ECS seems to involve their simultaneous regulation of multiple pathways that converge to execute the goal of saving or restoring neuronal homeostasis and facilitating neuronal survival without blocking the essential functions in brain of removing cellular debris and apoptotic cells. Their multilayered role in neuroprotection and minimizing cell death has been emphasized by many (see for example [136, 156, 172, 206, 207]).
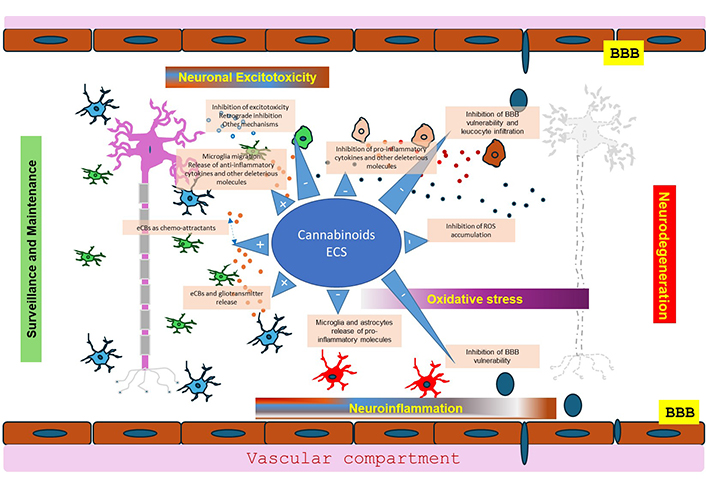
Neuroprotection of endocannabinoids (eCBs) and the ECS (endocannabinoid system). The neuroprotective role of endocannabinoids, exogenous cannabinoids and the ECS is multifactorial helping to maintain or restore neuronal homeostasis and facilitating neuronal survival without preventing the clearing of debris and apoptotic cells. This multilayered role in neuroprotection is due to the ability of cannabinoids to mitigate various pathological processes, notably inflammation, oxidative stress, and excitotoxicity. Cannabinoids and the ECS participate in inhibiting excitotoxicity, reducing the release of cytokines, chemokines, and other deleterious molecules, and inhibiting overactivation of astrocytes and microglia, as well as the development of reactive oxygen species (ROS), and minimize the vulnerability of the blood-brain barrier (BBB) and the influx of immune cells participating thus in the inhibition of neuroinflammation. There are also some indications suggesting a role in neurogenesis not depicted in the figure [174]
In conclusion, the various components of the ECS represent a promising therapeutic target for neuroprotection as numerous studies have demonstrated the ability of cannabinoids and endocannabinoids to mitigate various pathological processes, such as inflammation, oxidative stress, and excitotoxicity, which contribute to neuronal damage and death in a wide range of neurological disorders. It is however understood that rarely, cannabinoids and the ECS may promote neurodegeneration [119, 121, 122, 208, 209], with the underlying mechanisms remaining not as well understood. There are multiple strategies to target the ECS under investigation, ranging from the use of different synthetic and the less studied phytocannabinoids to the development of novel modulators for cannabinoid receptors and other related receptors. Inhibitors for the enzymes involved in the metabolism of eCBs and derivatives represent viable options. There is high interest in CBD or its derivatives in different combinations [78–80]. This is due in part to the fact that CBD is one of the main pharmacologically active phytocannabinoids, devoid of any psychoactive or addictive activity, and has been shown to exert a number of beneficial pharmacological effects, including anti-inflammatory and antioxidant properties [126]. The CB2R ligands are a focus of multiple investigations due to the beneficial role of this receptor, its expression in glial and immune cells, and its increase after injuries or insults [167, 178, 179, 182, 187, 205]. Another theme that was not explored in this mini-review although it is of notable importance, is the involvement of receptor complexes containing cannabinoid receptors called receptor heteromers that were shown to exert a role in cannabinoid signaling (see for example [49, 53, 70, 210, 211]). These are multiple heteromers under investigation in vitro and in vivo regarding their role in neuropsychiatric and neurodegenerative diseases. Investigation of this multiplicity of targets is necessary due to the very large burden of neurodegenerative diseases and the absence of any drugs currently that can stop, let alone reverse, the progression of these diseases, and hopefully ameliorate the quality of patients’ daily lives. We anticipate that combining some of these strategies may also present viable options.
2-AG: 2-arachidonoyl glycerol
ABHD6: α,β-hydrolase domain-6
AD: Alzheimer’s disease
AEA: anandamide
ALS: amyotrophic lateral sclerosis
BBB: blood-brain barrier
BDNF: brain-derived neurotrophic factor
CB1R: CB1 receptor
CB2R: CB2 receptor
CBD: cannabidiol
COX2: cyclooxygenase-2
eCB: endocannabinoid
ECS: endocannabinoid system
FAAH: fatty acid amide hydrolase
GPCR: G protein-coupled receptor
IL-1β: interleukin-1β
LPS: lipopolysaccharide
MAGL: monoacylglycerol lipase
MS: multiple sclerosis
NMDA: N-methyl-D-aspartate
PD: Parkinson’s disease
RNS: reactive nitrogen species
ROS: reactive oxygen species
THC: Δ9-trans-tetrahydrocannabinol
TLRs: toll-like receptors
TNF-α: tumor necrosis factor-α
AH: Writing—original draft. SRG: Writing—review & editing.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was funded by a Canadian Institute of Health Research (CIHR) grant [PJT-189976]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Diego Guidolin ... Luigi F. Agnati
Adèle Vilette ... Peter Vanhoutte
Neelakanta Sarvashiva Kiran, Senthilkumar Rajagopal
