 Review
Review

Affiliation:
1Cancer Sciences, Faculty of Medicine, University of Southampton, SO16 6YD Southampton, UK
ORCID: https://orcid.org/0000-0002-3454-8723
Affiliation:
1Cancer Sciences, Faculty of Medicine, University of Southampton, SO16 6YD Southampton, UK
ORCID: https://orcid.org/0000-0001-8308-9781
Affiliation:
1Cancer Sciences, Faculty of Medicine, University of Southampton, SO16 6YD Southampton, UK
2Department of Pathology, University Hospital Southampton NHS Foundation Trust, SO16 6YD Southampton, UK
ORCID: https://orcid.org/0000-0002-7451-363X
Affiliation:
1Cancer Sciences, Faculty of Medicine, University of Southampton, SO16 6YD Southampton, UK
ORCID: https://orcid.org/0009-0001-4516-8945
Affiliation:
3Department of Molecular Pathology, University Hospitals Dorset, SO16 6YD Bournemouth, UK
Affiliation:
4Institute of Applied Biosciences, Centre for Research and Technology Hellas, 57001 Thessaloniki, Greece
ORCID: https://orcid.org/0000-0001-8529-640X
Affiliation:
1Cancer Sciences, Faculty of Medicine, University of Southampton, SO16 6YD Southampton, UK
ORCID: https://orcid.org/0000-0003-3163-608X
Affiliation:
3Department of Molecular Pathology, University Hospitals Dorset, SO16 6YD Bournemouth, UK
Affiliation:
1Cancer Sciences, Faculty of Medicine, University of Southampton, SO16 6YD Southampton, UK
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0002-0973-8285
Affiliation:
1Cancer Sciences, Faculty of Medicine, University of Southampton, SO16 6YD Southampton, UK
†These authors contributed equally to this work.
Email: JCS@soton.ac.uk
ORCID: https://orcid.org/0000-0002-0972-2881
Explor Target Antitumor Ther. 2024;5:877–901 DOI: https://doi.org/10.37349/etat.2024.00253
Received: February 27, 2024 Accepted: May 08, 2024 Published: July 23, 2024
Academic Editor: Pier Paolo Piccaluga, University of Bologna, Italy
Splenic marginal zone lymphoma (SMZL) is a rare, predominantly indolent B-cell lymphoma constituting fewer than 2% of lymphoid neoplasms. However, around 30% of patients have a shorter survival despite currently available treatments and the prognosis is especially poor for the 5–15% of cases that transform to a large cell lymphoma. Mounting evidence suggests that the molecular pathogenesis of SMZL is critically shaped by microenvironmental triggering and cell-intrinsic aberrations. Immunogenetic investigations have revealed biases in the immunoglobulin gene repertoire, indicating a role of antigen selection. Furthermore, cytogenetic studies have identified recurrent chromosomal abnormalities such as deletion of the long arm of chromosome 7, though specific disease-associated genes remain elusive. Our knowledge of SMZL’s mutational landscape, based on a limited number of cases, has identified recurring mutations in KLF2, NOTCH2, and TP53, as well as genes clustering within vital B-cell differentiation pathways. These mutations can be clustered within patient subgroups with different patterns of chromosomal lesions, immunogenetic features, transcriptional signatures, immune microenvironments, and clinical outcomes. Regarding SMZL epigenetics, initial DNA methylation profiling has unveiled epigenetically distinct patient subgroups, including one characterized by elevated expression of Polycomb repressor complex 2 (PRC2) components. Furthermore, it has also demonstrated that patients with evidence of high historical cell division, inferred from methylation data, exhibit inferior treatment-free survival. This review provides an overview of our current understanding of SMZL’s molecular basis and its implications for patient outcomes. Additionally, it addresses existing knowledge gaps, proposes future research directions, and discusses how a comprehensive molecular understanding of the disease will lead to improved management and treatment choices for patients.
The term “splenic marginal zone lymphoma” (SMZL) was introduced by Schmid in 1992 to describe the splenic histology and immunophenotype in four patients who presented with splenomegaly, anaemia, and weight loss [1]. All patients showed expansion of the splenic marginal zone by B-cells with morphological and immunohistochemical features that were distinct from follicular lymphomas (FLs) or mantle cell lymphomas (MCLs). Like other marginal zone lymphomas (MZLs), including nodal MZL (NMZL) and extranodal MZL (EMZL), SMZL is associated with a history of prior chronic infections, auto-immune conditions, and nuclear factor kappa B (NF-κΒ) pathway dysfunction. There remains uncertainty as to whether all MZLs share a common cell of origin.
Recently, two separate classifications of haematolymphoid tumours have been published. The International Consensus Classification of Mature Lymphoid Neoplasms retains the definition of SMZL, like that in the 4th edition of the World Health Organization (WHO) classification [2], as a mature B-cell neoplasm primarily involving the spleen alongside hairy cell leukaemia (HCL), HCL variant (HCL-V), splenic diffuse red pulp lymphoma (SDRPL), and splenic B-cell lymphoma/leukaemia, unclassifiable. In contrast, the 5th edition of the WHO classification lists SMZL as one of four splenic leukaemia and lymphomas together with HCL, SDRPL, and a new category: splenic B-cell lymphoma/leukaemia with prominent nucleoli (SBLPN) [3]. The latter encompasses cases previously diagnosed as HCL-V, some cases of B-prolymphocytic leukaemia and a small subset of cases previously diagnosed as SMZL. It seems likely that further biological studies in this subset will determine whether SBLPN represents a distinct condition or variants of previously recognised disorders [4].
SMZL comprises about 2% of lymphoid malignancies and approximately 0.6% of non-Hodgkin lymphoma (NHL) cases, with an incidence rate of 0.13 per 100,000 individuals per year [5, 6]. The median age range of diagnosis is 65–70 years and epidemiological studies have shown no consistent male or female preponderance [7–9]. Diagnosis may occur following the incidental discovery of lymphocytosis or the detection of splenomegaly during examination or imaging. However, the typical presentation often involves abdominal fullness or pain due to significant splenomegaly, and/or symptoms associated with cytopenia [10, 11]. SMZL has been associated with autoimmune conditions [12], and there is a significant correlation between SMZL and hepatitis C virus infection in southern European populations and China [13, 14]. Lymphadenopathy, when present, is usually intra-abdominal (especially splenic hilar or intra-thoracic) and rarely involves peripheral nodes. Systemic symptoms such as fever and night sweats should raise the possibility of a high-grade transformation.
Whilst splenic histology provides a definitive diagnosis, splenectomy is now rarely performed for either diagnostic or therapeutic reasons and diagnosis is usually based on a combination of clinical findings, lymphocyte morphology, bone marrow histology, immunohistochemical and immunophenotypic features [15]. The typical immunophenotype includes surface expression of IgM, CD20, CD27, CD49d, and variable expression of IgD, CD5, CD11c, and CD25 [16]. Lack of expression of CD103, CD123, annexin A1, and cyclin D1 serve to distinguish SMZL from other splenic lymphomas (Table 1). Additionally, the CD200/CD180 median fluorescence (MFI) ratio may help to distinguish SDRPL diagnosis over HCL, SMZL, and SBLPN, where a ratio of 0.5 or less is in favour of SDRPL [3]. Cases presenting with isolated lymphocytosis, morphological and immunophenotypic features suggestive of SMZL, but of which lacks splenomegaly (as well as lymphadenopathy) on imaging fulfil the diagnostic features of clonal B-cell lymphocytosis of marginal zone origin (CBL-MZ). CBL-MZ has been included in the new WHO classification, as a non-chronic lymphocytic leukaemia (CLL)/small lymphocytic lymphoma (SLL)-type monoclonal B-cell lymphocytosis (MBL) entity, that frequently harbour features consistent with a MZ origin [3]. On follow-up, a minority of CBL-MZ cases progress to SMZL or another MZL [17].
Similarity and differences in features of SMZL, HCL, and SDRPL
| Features | Type of feature | SMZL | HCL | SDRPL |
|---|---|---|---|---|
| Demographics | Incidence | 0.13/100,000 | 0.4/100,000 | ? |
| Median age | 65–69 | 55–63 | 70 | |
| M:F ratio | 1:1.1 to 1:1.85 | 3:1 to 4:1 | 1.6:1 to 2.4:1 | |
| Immunophenotype | CD25 | + | ++ | - |
| CD27 | ++ | -- | - | |
| CD5 | + | - | - | |
| CD200 | - | ++ | - | |
| Outcome | Need for treatment | 70% | Yes | 50% |
| Transformation | 10–20% into DLBCL | 5–6% | Rarely |
+: indicates positive; ++: indicates strong positive; -: indicates negative; --: indicates strong negative; ?: unknown. SMZL: splenic marginal zone lymphoma; HCL: hairy cell leukaemia; DLBCL: diffuse large B-cell lymphoma; SDRPL: splenic diffuse red pulp lymphoma; M:F: male: female
SMZL frequently pursues an indolent course with a median survival of 10–15 years and only 40–50% of deaths are disease or treatment-related. Despite this generally favourable outcome, the prognosis of SMZL can be heterogenous, wherein 30% of cases have a shorter survival despite currently available treatments [18] and the prognosis is especially poor for the 5–15% of cases that undergo transformation to a large cell lymphoma [19–21]. Importantly, in comparison to other MZLs, SMZL patients have the greatest risk of transformation, which results in significantly decreased survival [22, 23]. The rarity and indolence of the disease have hindered the development of specific treatment options for SMZL, with most current treatment regimens only being supported by retrospective studies or based on retrospective series or trials involving broader indolent B-cell lymphoma subtypes [7].
Indication to begin treatment should be based on evidence of symptomatic or progressive disease, as outlined by the latest guidance for the diagnosis and treatment of MZLs [18, 24] (Figure 1). An overview of the consensus guidance includes the following: (1) the minority of cases with hepatitis C infection should receive up-front anti-viral therapy; (2) for the majority of cases, rituximab monotherapy is the preferred initial therapy with high response rates and durable responses; (3) combination immune-chemotherapy such as rituximab + bendamustine is also highly effective, and can result in prolonged responses especially in cases who achieve minimal residual disease (MRD) negativity after 3 cycles of treatment [25] but in view of the high incidence of adverse events, is only an option for younger, fitter patients; (4) although long term outcome data for splenectomy is comparable to rituximab in many studies, especially when performed in experienced centres, the advantage of obtaining a precise diagnosis is offset by the risk of post-operative complications and requirement for infection prophylaxis. However, if splenectomy is undertaken, access to splenic tissue provides a unique opportunity to study tumour-microenvironmental interactions, let alone establish an unequivocal diagnosis; (5) patients who relapse after 2 years from initial rituximab treatment are candidates for re-treatment with the same regimen; (6) for those with early relapse or with refractory disease, Bruton’s tyrosine kinase (BTK) inhibitors, especially second-generation inhibitors zanubrutinib and acalabrutinib, have shown benefit in clinical trials. Given the activation of phosphoinositide 3-kinase (PI3K) signaling in a broad spectrum of B-cell tumours [26, 27], it is unsurprising that these inhibitors are also active in relapsed/refractory MZL, though they are not licensed for use due to their significant toxicity [24]. Lastly, as in other B-cell tumours, there is increasing interest in the use of immune therapies including immune checkpoint inhibitors, bispecific antibodies, and chimeric antigen receptor T-cell (CAR-T) therapy.
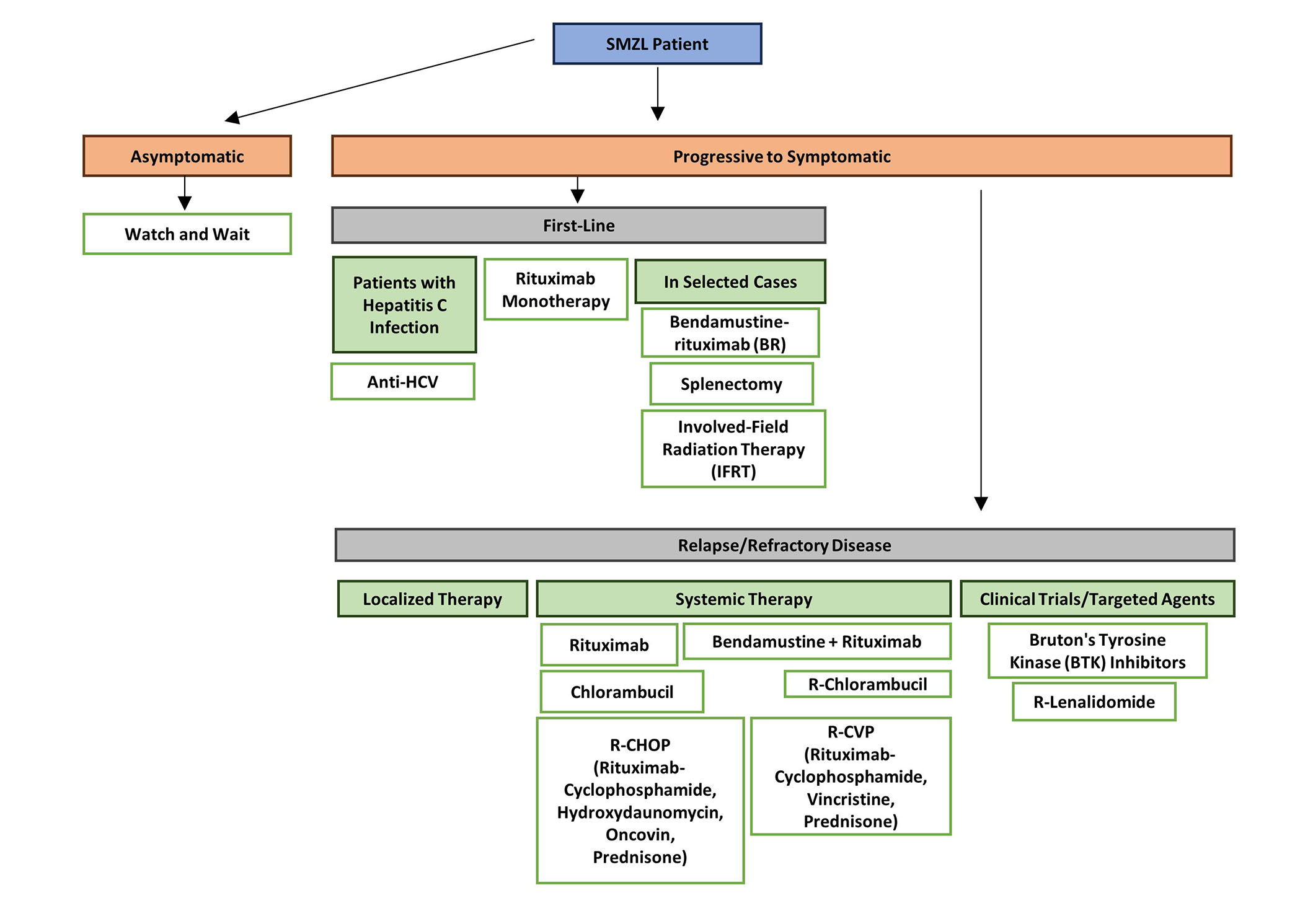
Flowchart of treatment guidelines for SMZL [24]. SMZL: splenic marginal zone lymphoma; HCV: hepatitis C virus
SMZL is associated with a number of molecular characteristics, including recurring genomic abnormalities which include del(7q) (approximately 40% cases), KLF2 (20–30%), NOTCH2 (10–25%), TP53 mutations (10–15%), biased IGHV (immunoglobulin heavy variable) gene repertoire with usage of IGHV1-2*04 in 30% of cases, as well as key transcriptomic and epigenetic features of the tumour cells. As of now, there are yet to be any actionable and predictive biomarkers to stratify SMZL patients with a risk of undergoing a more aggressive clinical course. The biological and clinical implications of such genetic and molecular characteristics are discussed in detail in this review.
SMZL development is shaped significantly by antigen selection and B-cell receptor (BCR) stimulation, demonstrated by the preferential usage of specific IGHV genes, particularly IGHV1-2 (30%), IGHV4-34 (11%), and IGHV3-23 (9%) [20, 21]. The IGHV1-2*04 allele is notably enriched in SMZL cases, representing 30% of all cases and 90% of IGHV1-2 users [28–30]. The usage of this allele is remarkable as it is much less frequent in other types of B-cell lymphomas [31] and is significantly associated with del(7q), KLF2, and NOTCH2 mutations [28, 29, 32].
SMZL displays somatic hypermutation (SHM) in the great majority of cases [33]. Of note, most (approximately 75%) IGHV1-2*04 cases exhibit borderline mutated status (97–99% germline identity), displaying a precise pattern often targeting framework regions (FRs) [30]. Intra-clonal diversification with targeted ongoing SHM is also observed [34]. A minority of cases harbour 100% germline IGHV identity and exhibit reduced time to treatment [29].
The IGHV1-2*04 allele is particularly distinctive as it carries a tryptophan instead of an arginine at position R-75 of its FR3. This alteration is theorized to lead to structural changes that are important for the recognition of specific antigenic epitopes [30]. Specific IGHD genes (IGHD3-3 and IGHD3-10) and IGHJ genes, along with longer heavy complementarity determining region 3 (VH CDR3) sequences, are also observed. Recombinant monoclonal antibodies from IGHV1-2*04 SMZL demonstrated reactivity with various human cell antigens and human serum, indicating both poly- and self-reactivity [35]. Taken together, cases carrying the IGHV1-2*04 allele are believed to represent a distinct molecular variant of SMZL in the context of immune-driven initiation, leading to lymphoproliferation associated with autoimmunity and/or ongoing SHM [36].
Copy number alterations (CNAs) and karyotypic complexity are key features with clinical utility in other B-cell neoplasms such as CLL [37, 38]. In SMZL, a G-banding study of 330 cases found that 50% of cases harboured complex karyotypes (as defined by having ≥ 3 cytogenetic aberrations per metaphase cell), while 30% of cases harboured single aberrations, and 18% of cases harboured two [39]. The most common abnormalities include 7q22-q32 deletions and chromosome 3/3q gains (approximately 25%, Figure 2A, Table 2). Complex karyotypes involving chromosomes 1, 3, 7, 8, and 14 are also frequent [39–41]. TP53 deletions, occurring within the del(17p) minimally deleted region (MDR), are found in 8–20% of SMZL cases and are associated with adverse outcomes [39, 42].

(A) Ideogram of recurrent copy number alterations in SMZL. Yellow indicates a trisomy, while red and green indicate common regions of deletions and gains, respectively; (B) schematic diagram of the recurrent 7q deletion in SMZL. The chromosome 7 ideogram indicates the common breakpoints of deletion found among SMZL cases. The panel below shows an example of a copy number plot of a case with clear lower segmentation means indicating the 7q deletion. A total of 38 coding genes and 6 microRNAs (miRNAs) are found in the 7q32 minimally deleted region (MDR)—those that were found to be transcriptionally under-expressed are indicated by the yellow highlight [43]. SMZL: splenic marginal zone lymphoma
Recurrent molecular lesions in SMZL
| Lesion | Prevalence | Genomic location, target genes, functions, and associations | References |
|---|---|---|---|
| Copy number alterations | |||
| del(7q) | 14–44% | Under-expression of 18 genes and 8 miRNAs in the 7q32.1-32.2 MDR, rare mutations in the FLNC gene. | [29, 43–47] |
| dup(3q) | 15–34% | Breakpoints typically span 3q26-q29, associated with gains in chromosome 12, common to several MZLs. | [39, 44] |
| +12 | 8–25% | +12 is associated with worse overall survival in univariate analysis, but no correlation with event-free survival. | [43] |
| +18 | 8–23% | Mutually exclusive in samples with a 3q gain. | [39] |
| del(17p) | 8–24% | TP53 gene. Faster clearance and elimination of rituximab. | [39, 42] |
| del(13p) | 5–18% | Same as MDR in CLL. del(13q) presented a slower rate of elimination of rituximab. | [48] |
| del(14q) | 3–10% | 30–40% harbour translocations involving BCL3. | [49, 50] |
| del(6q) | 8–24% | No candidate genes were found in a small series of transformed SMZL. | [51] |
| dup(8q) | 2–20% | Associated with poor clinical outcomes if inclusive of the MYC gene locus. | [52] |
| del(8p) | 4–15% | Associates with poor outcomes. Amongst MZLs, is only common in SMZLs. | [48] |
| Somatic mutations | |||
| NOTCH2 | 10–25% | Crucial for MZ B-cells differentiation, associated with 7q deletions, KLF2 somatic mutations, and IGHV1-2*04 usage, an independent risk factor for TTFT (multivariate analysis). | [29, 53–59] |
| NOTCH1 | Approximately 5% | Mutations cause NOTCH1 activation in several mature B-cell tumours. | [60–62] |
| SPEN | Approximately 5% | Negative regulator of B lymphocyte differentiation into MZ B cells. | [63] |
| TNFAIP3 | 7–15% | Mutations resulted in a loss of NF-κB cascade inhibition in other NHLs. | [64] |
| KLF2 | 12–42% | Most frequent mutation in SMZL, KLF2 knockout murine B-cells have impaired migration and MZ differentiation, through NF-κB deregulation. | [28, 29, 56, 59, 65–68] |
| CARD11 | Approximately 5% | Involved in the BCR and NF-κB signaling pathways. | [54, 69] |
| TRAF3 | Approximately 8% | TRAF3 inactivation leads to B-cell lymphomagenesis in mice, through a non-canonical NF-κB pathway. | [70] |
| TP53 | Approximately 15% | Key tumour-suppressor that promotes cell-cycle arrest and apoptosis. Often deleted. Associated with unfavourable overall survival. | [29, 52, 71] |
| MYD88 | 5–15% | A small proportion of MYD88 mutations in SMZL are L265P (6%). Role in MZ B-cell differentiation, and its absence leads to a deficiency in their availability. Promotion of B-cell proliferation and survival by activation of NF-κB. | [72–79] |
| KMT2D | 11–15% | Frequent in all lymphoid tumours, mutants have delayed GC formation. | [80, 81] |
| CREBBP | Approximately 5% | Mutations were found to be fully clonal and likely early initiating events, like that in FL. Frequent in DLBCL and FL. Regulates enhancer networks with a crucial role in GC/post-GC cell fate decisions. | [53, 82, 83] |
BCR: B-cell receptor; SMZL: splenic marginal zone lymphoma; MZLs: marginal zone lymphomas; MDR: minimally deleted region; CLL: chronic lymphocytic leukaemia; MZ: marginal zone; NF-κB: nuclear factor kappaB; GC: gastric cancer; FL: follicular lymphoma
In SMZL, the most prominent cytogenetic feature is del(7q), detected in 14–44% of patients [39, 44, 45, 48]. The breakpoints have been shown to vary from 7q21 to q36, but a 3.04 Mb MDR at 7q32.1-32.2 (127.03–130.07 Mb) has been identified, with another less common deletion region at 7q22 [43, 45, 46] (Figure 2B). A study comparing cases with or without del(7q) identified 38 coding genes within the 7q MDR [43, 46], and of those genes, 18 were significantly under-expressed in cases with the deletion and some of these had further evidence for hypermethylation [46]. Additionally, a cluster of six miRNAs in the del(7q) MDR exhibited reduced expression, including miR-593, miR-129, miR-182, miR-96, miR-183, miR-335, miR-29a, and miR-29b1 [46]. Integrated CNA and whole-exome sequencing (WES) analysis identified three mutated putative candidate genes of this deletion, CUL1, EZH2, and FLNC, with FLNC located within the mapped MDR [53].
The second most common aberration is the dup(3q), which is present in up to 34% of patients and is sometimes the result of unbalanced translocations [44, 48]. In contrast to other MZLs, where whole trisomy 3 is predominant, SMZL frequently exhibits a gain in 3q23, often in conjunction with a complex karyotype [39]. The breakpoints and partners in dup(3q) rearrangements vary, indicating secondary events [84]. Compared with the more aggressive diffuse large B-cell lymphoma (DLBCL), SMZL genomes infrequently harbour balanced chromosomal translocations, though IgH locus translocations can occur (9% of cases) in t(14;19)(q32;q13), t(6;14)(p21;q32), t(9;14)(p13;q32), and t(1;14)(q21;q32), targeting BCL3, CCND3, PAX5, and BCL9, respectively [39, 85]. The (14;19)(q32;q13) translocation was first discovered in CLL, with the identification of BCL3 as the translocation partner of the IgH locus [49]. A cluster of breakpoints in the 3’ region of BCL3 was shown to be associated with cases of atypical SMZLs with mutated IGHV status and complex karyotypes-this breakpoint did not, however, result in changes in expression of the BCL3 [86]. Hence, in such cases, it is still not clear what the relevant target genes of these translocations may be [50].
Genome-wide studies, employing unpaired whole-genome sequencing (WGS), WES, and targeted sequencing technologies, have revealed recurrently mutated genes and pathways in SMZL development. However, these approaches have limitations in exploring non-coding genome regions and in investigating mutational signatures contributing to disease. KLF2 (12–42%), NOTCH2 (10–25%), and TP53 (15%) are among the most frequently mutated genes [28, 32, 53–55] (Table 2). Recurrent truncating mutations are observed in TNFAIP3, KMT2D, and MYD88, where the latter results in the promotion of B-cell proliferation and survival by activation of NF-κB [72]. Additionally, other genes within pathways that relate to crucial B-cell functions are also mutated, including the NOTCH pathway, NF-κB signaling, chromatin remodeling, cytoskeleton organization, and toll-like receptor (TLR) signaling pathways.
In B-cells, NF-κB activation influences signaling pathways and downstream genes fundamental to development, differentiation, and survival [87, 88]. NF-κB is typically sequestered in an inactive state in the cytoplasm by inhibitory IκB proteins. There are two pathways of NF-κB signaling: canonical and non-canonical. These pathways involve distinct NF-κB family members and are triggered by specific receptor-proximal signals, such as those emanating from the BCR, tumour necrosis factor-alpha (TNF-α), TLRs, and interleukin-1 (IL-1). It involves phosphorylation and degradation of IκB proteins by IκB kinase α (IKKα) and IKKβ, releasing and activating NF-κB heterodimers (e.g., p65/RelA) that translocate to the nucleus to regulate gene expression. Conversely, the non-canonical pathway is activated by stimuli like B-cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL), which bind to receptors such as BAFF-receptor (BAFF-R) and CD40, leading to activation of IKKα. This results in the processing of p100 to p52-REL-B heterodimers, which regulate gene expression. The TNFAIP3 (A20) protein and other negative feedback mechanisms modulate its activation [89].
Constitutive NF-κB activation has been implicated in the growth of various B-cell leukaemia and lymphomas, with mutations in multiple genes playing a part. Constant NF-κB activation in MZ B-cells models results in abnormal B-cell expansion [90], and enhanced signaling is observed in other lymphoid malignancies including Hodgkin lymphomas, DLBCL, and multiple myeloma [91]. Genetic abnormalities in genes involved in this pathway including CARD11, TNFAIP3, TRAF3, and BIRC3 [53, 54, 92] have also previously been reported (Figure 3A).
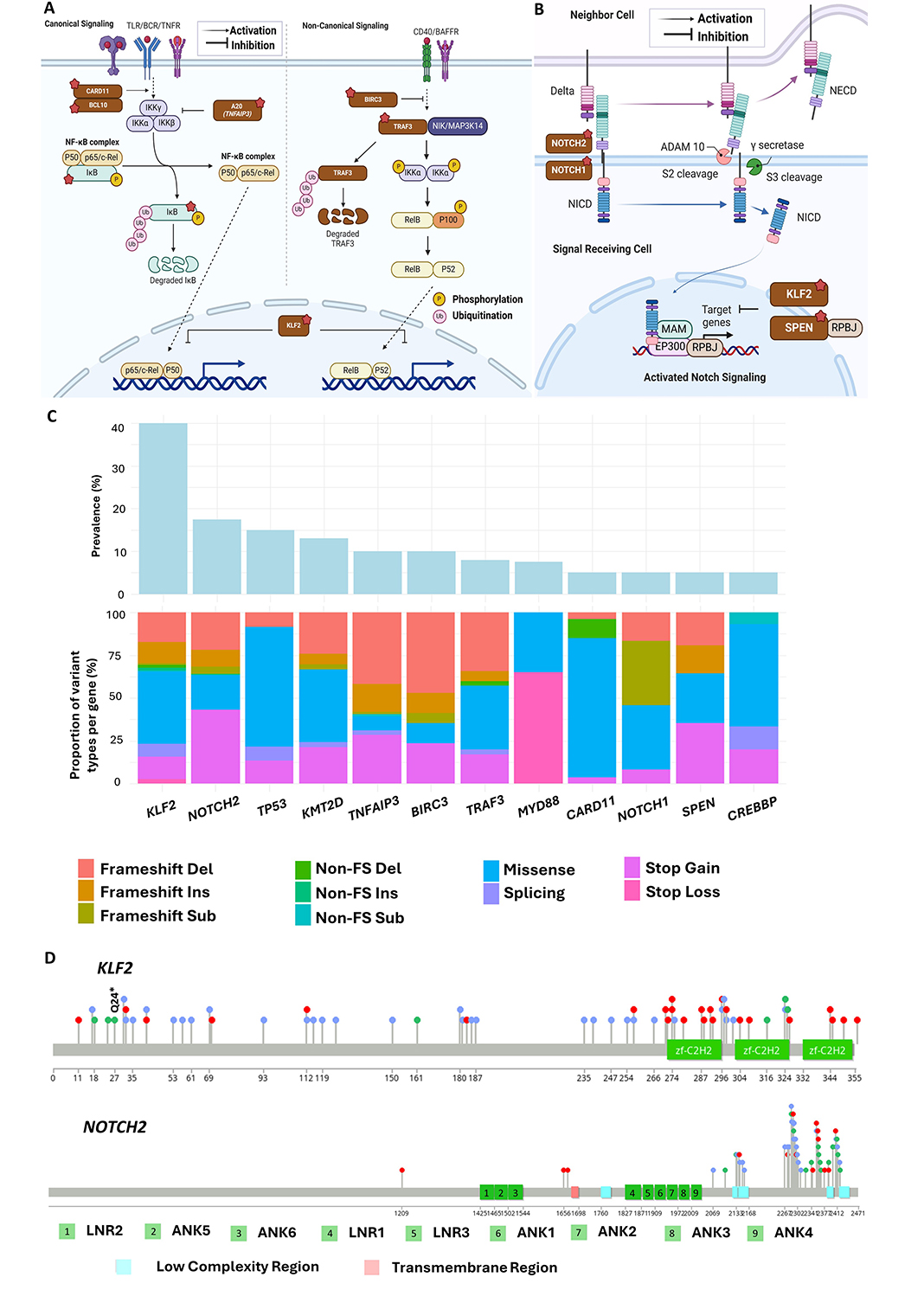
Recurrently mutated genes and pathways in splenic marginal zone lymphoma (SMZL). (A) Illustration of the NF-κB signaling pathway. Response to diverse stimuli, including ligands of B-cell receptors (BCRs) and toll-like receptors (TLRs) leads to the primary mechanism of nuclear factor kappa B (NF-κΒ) activation through the inducible degradation of the IκB, which is triggered by the site-specific phosphorylation of the multi-subunit IκB kinase (IKK) complex. Recurrent coding mutations in KLF2, TRAF3, TNFAIP3, BIRC3, CARD11, and BCL10 are recurrently found across SMZL; (B) illustration of the NOTCH signaling pathway. When NOTCH2 is activated by the binding of ligands such as Jagged ligands and delta-like ligands, its intercellular domain [Notch intracellular domain (NICD)] is cleaved and released into the cytosol, where it then traffics into the nucleus to act as a transcriptional regulator. Mutations in SPEN, NOTCH1, and NOTCH2 are recurrently found across SMZL; (C) types of unique mutations found across recurrently aberrant genes in SMZL; (D) distribution of mutations found across KLF2 (top) and NOTCH2 (bottom) in SMZL. TNFR: tumour necrosis factor receptor; BAFFR: B-cell activating factor receptor; NICD: Notch intracellular domain; NECD: Notch receptor extracellular domain; MAM: mastermind; TRAF3: tumour necrosis factor-receptor associated factor; FS: frameshift; RPBJ: recombination signal binding protein for immunoglobulin kappa J region. Figure 3A and 3B were created with Biorender.com
Note. Figure 3A was adapted from “Notch Signaling Pathway”, by BioRender.com (2020). Retrieved from: https://app.biorender.com/biorender-templates/figures/all/t-5f6d097dbedf1200aa63bc9c-notch-signaling-pathway; “NF-KB Signaling Pathway”, by Iwasaki A (2020). Retrieved from: https://app.biorender.com/biorender-templates/figures/all/t-5f5b7c3514d40300aa942984-nf-kb-signaling-pathway
The TNF-receptor associated factor (TRAF) proteins act as docking molecules for the interaction between the cytosolic regions of various tumour necrosis factor receptors (TNFRs) and TLRs. Studies have shown the action of the A20 protein, encoded by the TNFAIP3 gene, inhibits NF-κΒ activation in the TNFR1 signaling pathway with the help of several binding partners including TRAF1 and TRAF2 [93, 94]. TRAF3 has recurrently mutated in a variety of B-cell malignancies, including SMZL (21%) [95]. Inactivation of TRAF3 has been found to increase the survival of mature B-cells, leading to the development of B-cell lymphomas in murine models [70]. Mutational clusters were found within the C-terminal domain of the protein, which is required for BIRC3 protein recruitment. Alternatively, truncating mutations within the RING domain of BIRC3 occur in 10% of SMZLs and result in the loss of ubiquitin ligase activity, further stabilizing NF-κB-inducing kinase (NIK) and resulting in the constitutive upregulation of NF-κB targets [93, 96]. Mutations that result in the activation of positive regulators of the NF-κB pathway, such as in CARD11 and IKBKB, have also been identified in a proportion of SMZL cases [93, 97].
The TLR pathway plays a significant role in the development of SMZL by acting as a critical activator of the NF-κB pathway itself [98]. Of particular importance is the MyD88 adaptor protein, which is essential for transmitting signals through the TLRs (other than TLR3) as well as the IL-1 receptor family. The MYD88 gene is found to be mutated in up to 15% of SMZL cases [73, 69]. Functional studies have demonstrated that MyD88 is necessary for MZ B-cell differentiation [74]. Common mutations within the Toll/interlukin-1 receptor (TIR) domain common to TLR and IL-1 family of receptors result in structural changes resulting in MyD88-dependent recruitment of IRAK-1/4 and enhanced NF-κB signaling [99]. The L265P mutation has been observed in various lymphoid malignancies, particularly including lymphoplasmacytic lymphoma (LPL) [73, 75]. This gain-of-function alteration has also been found in SMZL, associated with improved disease survival [76–78].
KLF2 emerges as the most recurrent mutation in SMZL, present in 12–40% of cases and strongly associated with usage of the IGHV1-2*04 allele and del(7q). KLF2 acts as a transcription factor, regulating key genes, including those in the NF-κB and NOTCH signaling pathways [65, 100] (Figure 3A and 3B). Its DNA binding and transcriptional control play vital roles in the differentiation and maintenance of mature peripheral B-cells, acting as a quiescence factor [66, 67]. In vivo studies show that KLF2 deficiency results in marked MZ B-cell expansion in murine models [68]. Impaired KLF2 also affects the migration of germinal cells to splenic MZ B-cells, influencing the trafficking and differentiation of MZ B-cells [66, 101].
Most KLF2 mutations occur in the N-terminus and middle inhibitory domain, with missense mutations clustering in the zinc-finger 1 (ZF1) domain, impairing protein function [28] (Figure 3C and 3D). Truncating mutations disrupt the entire protein or remove ZF domains, including putative nuclear localization signal sequences [32, 56]. The recurrent nonsense mutation Q24* suggests a potential hotspot [29]. In vitro SMZL studies demonstrate that KLF2 mutants result in significant loss of suppression in the NF-κB pathway, impacting various activating signaling pathways, such as those associated with the BCR, TLR, TNFR, and BAFF-R [28].
NOTCH2 has an important role in MZ B-cell development, differentiation, and homing [57, 58, 102]. It is a cell-surface receptor important in determining cell fate, proliferation, differentiation, and apoptosis [103] (Figure 3B). In MZ B-cells, conditional NOTCH2 knockout resulted in defects in MZ B-cells and their presumed precursors of type 2 transitional B-cells [58]. Whilst common in SMZL, NOTCH2 mutations are rare in CLL, MCL, and FL [60, 104]. In SMZL, frameshift indels and nonsense mutations predominate and cluster near the C-terminal PEST (proline, glutamic acid, serine, threonine)-rich domain [105] (Figure 3C and 3D). A recurrent p.R2400* nonsense mutation within this domain represents approximately 24% of NOTCH2 mutations in SMZL [54]. Mutations in NOTCH2, like those targeting KLF2, are enriched in cases with 7q deletion and IGHV1-2*04 usage and are associated with adverse clinical outcomes [29, 55]. Immunohistochemical analysis using antibodies that recognize active nuclear NOTCH2 in situ supports NOTCH2 activation in SMZL, but shows an expression of NICD2 in both wild-type and NOTCH2 mutant tumours [106]. This suggests that akin to NOTCH1 mutations in CLL, NOTCH2 mutations in SMZL are secondary events that stabilize prior ligand-dependent Notch activation through selective pressure on this signaling pathway.
Whilst TP53 mutations are present in up to 15% of SMZL cases, concomitant with del(17p) in most cases, other genes are also recurrently mutation that are implicated in cell cycle control, such as CCND3 (5%) and ATM (4%) [29, 107]. In SMZL, TP53 disruptions primarily occur via missense mutations within its DNA binding domain, compromising its tumour suppressor function. Mutant p53 proteins lose the ability to activate canonical p53 target genes, leading to unchecked cell proliferation and the accumulation of genomic mutations, ultimately fostering tumour growth. TP53 mutations signify a poor prognosis and are more prevalent in cases with unmutated IGHV genes, showing acquisition of del(17p) in a handful of transformed cases [29, 39, 48, 108].
Several genes involved in chromatin remodeling are frequently mutated in SMZL, highlighting the significance of epigenetic processes in disease initiation and development. These genes include KMT2D (11–15%), ARID1A (4–6%), EP300 (2%), and CREBBP (5%) [29, 54, 109]. It should be noted that these gene mutations are found across several mature B-cell tumours, so have limited utility for differential diagnosis. KMT2D is a histone lysine methyltransferase that modifies lysine-4 of histone 3 (H3K4) marker, where mutant proteins have a functional role in DLBCL and FL [80]. The protein genacts as a bona fide tumour suppressor, and loss of its function has been shown to result in delayed germinal centre formation and overall promotion of lymphoma development in mouse models [81]. In SMZL, CREBBP mutations are clonal and appear to be early initiating events, like those found in FL [29]. The recurrent hotspot variant Y1450C has been shown to compromise its acetylation ability on Bcl-6 and p53 in DLBCL. ARID1A is crucial in maintaining chromatin accessibility at enhancer regions, and recurrent mutations have been observed in various cancer types [110, 111].
In SMZL, several molecular classification systems have been used to help identify poor-risk patients. However, despite these advancements, further validation and investigations are needed to integrate these findings into clinical practice effectively. Overall refinement of SMZL sub-classifications and their clinical utility could conceivably help tailor treatments and improve outcomes for patients. In this section, we discuss some of the useful molecular sub-types that have emerged from recent research and how they contribute to our understanding of SMZL biology.
Bonfiglio et al. [59] identified subgroups of SMZL based on the enrichment of particular gene mutations clustering into specific biological pathways. The authors studied 303 splenic-derived tumour specimens from an international multicentre study (IELSG46) using a multi-omic approach. This study used a pathology expert panel, confirming the WHO classification for all cases. Using genetic and phenotypic disease features, they were able to identify a number of genetic subtypes, with differing survival outcomes. Most importantly, of the four total clusters, two prominent subgroups were proposed, termed NNK (mutations in NF-κB, NOTCH, and KLF2, 58.2% of all cases), and DMT [with DNA-damage response, mitogen-activated protein kinases (MAPK), and TLR modules, 32% of cases]. These sub-types exhibited different patterns of chromosomal lesions, immunogenetic features, transcriptional signatures, immune microenvironments, and clinical outcomes, with inferior survival in the NNK group.
The co-occurrence of mutations within the NNK cluster, which accounts for the majority of cases, alludes to a possible role of KLF2 as a master regulator of both NOTCH and NF-κB signaling, suggesting the possible cooperation between these lesions in dysregulating MZ B-cell differentiation. As KLF2, an important transcription factor, is mutated in the immune-suppressive NNK SMZL subgroup, future studies might focus on how deregulation of KLF2 or downstream targets results in immune cell reprogramming, recruitment, or evasion, as has been shown for CREBBP and EZH2 in other B-cell lymphomas [82].
Recent studies have shown that epigenetic factors, such as changes in DNA methylation and histone modifications can play a critical role in establishing B-cell identify [112, 113]. In SMZL tumours, DNA methylation can contribute to the disruption of cell survival and proliferation pathways [114]. Arribas et al. [115] demonstrated that promotor DNA methylation patterns can be used to identify two distinct epi-clusters, based on the level of cytosine-guanine (CpG) methylation. They identified a high genome-wide promoter methylation subgroup (High-M) in 21% of their cohort, that displayed inferior overall survival, high risk of histologic transformation, and was enriched for IGHV1-2*04 usage, NOTCH2 mutations, and del(7q). Gene promoters showing elevated methylation in the High-M group including PRC2 target genes, those harbouring tri-methylation marks (H3K27me3 and H3K4me3), those involved in chromatin remodeling, and a variety of tumour suppressor genes (including KLF4, DAPK1, CDKN2A, WT1, and GATA4). Hypomethylated promotors included those in PRC2 component genes, and those involved in cell cycle control and proliferation [115]. This study supports the presence of an aggressive tumour sub-group carrying established poor-risk molecular features with fundamental deregulation of EZH2 and broader PRC2 perturbation.
The global impact of DNA methylation can extend beyond the regulation of gene expression, and it has recently been shown that hypo-methylation in low CpG-content heterochromatin and hyper-methylation in high CpG-content polycomb regions is concomitant with ongoing cell division in B-cell tumours. This measure, termed epigenetically determined cumulative mitoses (epiCMIT) reflects the mitotic history of B cell tumours, both prior and subsequent to malignant transformation [116]. In addition to the initial paper which showed that epiCMIT scores can predict more aggressive disease in CLL, MCL, and DLBCL, there is recently emerging evidence for its relevance in SMZL as well. Parker et al. [117] profiled 142 SMZL patients drawn from international leaders in the management of the disease, using a variety of molecular approaches including DNA methylation analysis. In doing so, the authors made the following important observations; (1) that epiCMIT scores are highly heterogeneous in SMZL, suggesting that the quantity of tumour cell proliferation is highly variable between patients; (2) that epiCMIT scores negatively correlate with telomere length, supporting the association between cellular growth and telomere attrition; (3) that patients with high epiCMIT were more likely to carry other poor-risk features, such as IGHV1-2*04 usage, del(7q), mutations in key driver genes (KLF2 and NOTCH2), and be classified in the High-M or NNK disease subgroups; and (4) that survival analysis selected epiCMIT as a strong independent marker of treatment free survival in univariate and multivariate models that account for other established clinical and molecular classification systems.
Genome-wide transcriptional analysis of SMZL highlights the deregulation of genes within the BCR, TNFR signaling pathways, and those associated with NF-κB activation [118]. One specific example is the link between elevated NOTCH2 expression and a more aggressive clinical course [119]. Bonfiglio et al. [59], in addition to their aforementioned genomic analysis, also performed transcriptional analysis and identified elevated expression of NOTCH pathway genes in the NNK genetic sub-class, supporting deregulation of non-canonical NF-κB transcription factors in this subtype. Conversely, the DMT-SMZL sub-class exhibited impaired p53 and apoptotic functions [59]. Other expression studies have implicated the deregulation of signal transduction, immunity, metabolic, and TLR pathways [82]. From the perspective of differential diagnosis, RNA analysis of a spectrum of B-cell lymphomas has identified a 135-gene SMZL-specific gene expression signature (SSGES) [120], including three specific genes that appear to be particularly important for distinguishing SMZL from other small cell lymphomas (ILF1, SETX, and CD40) [118]. In the High-M epigenetic cluster defined by Arribas and co-workers [115], the authors showed that several tumour suppressor genes were downregulated due to promoter methylation deregulation, including KLF4, DAPK1, CDKN1C, and CDH1/2. The authors propose that these genes might offer a survival advantage to tumour cells, with the potential of therapeutic targeting [115].
miRNAs are a subclass of non-coding RNA molecules varying in length, that play critical roles in gene regulation. At the genomic level, miRNA loci are often located within introns, frequently within clusters of multiple miRNAs that are expressed as a result of host gene transcription. miRNAs have been broadly implicated in the pathogenesis of other mature B-cell malignancies, with perhaps the most notable example being the analysis of del(13q14) in CLL, that down-regulated miR15a and miRNA16-1, negative regulators of MCL-1 and BCL2 [121, 122]. Again, data in SMZL is limited, but several miRNAs have been found to be dysregulated, including the down-regulation of miR-29a and miR-29b-1 [123]. A study comparing miRNA profiles of SMZL to reactive spleens revealed differential expression of 30 miRNAs, with nine of them also distinguishing SMZL from other B-cell lymphomas [124]. The hepatitis C virus has also been shown to downregulate miR-26b in SMZL [125]. Down-regulation of miR-593, miR-129, miR-182, miR-96, miR-183, miR-335, miR-29b1, and miR-29a is seen in 7q deleted patients, with TCL1A, BCL2, RB1, and BCL-w as a putative target genes [126, 127].
Although molecular studies have provided insights into the biology of SMZL they are yet to be incorporated into routine practice. This reflects, in part, the rarity of the disease and paucity of clinical trials, the difficulty in making a definitive diagnosis and accessing suitable samples in some cases, and the limited availability of appropriate investigations. However, advances have been made to bridge current molecular understanding to emergent clinical hypotheses. This section aims to summarize the current understanding of the molecular landscape of SMZL, draw comparisons with other B-cell tumours, and discuss the implications for patient management and care in clinical settings.
No single genomic or immunogenetic marker is pathognomonic for SMZL. However, in the context of the differential diagnosis of a primary splenic lymphoma, the use of IGHV1-2*04 and/or del(7q) are very strong pointers for the diagnosis of SMZL, while a combination of these markers with or without mutations of genes within the NF-κΒ pathway is not seen in other splenic lymphomas. As discussed earlier in this review, transcriptomic studies may also have diagnostic potential. DNA methylation studies of normal B-cell maturation and more common B-cell tumours have identified both disease-specific signatures and differing patterns within diseases that have clinical implications. A recent study, published as an abstract, investigated DNA methylation profiles in splenic lymphomas with the pathological diagnoses of HCL, HCL-V, SDRPL, or SMZL, and identified 5 distinct methylation subgroups [128]. While the majority of the 170 SMZL cases formed a distinct subgroup, 29 were included in subgroups encompassing the new WHO classification of SBLPN. These cases were depleted in NOTCH2, KLF2 mutations, and IGHV1-2*04 usage, suggesting either that they could be reassigned to the new WHO subgroup or there were differences in the criteria used to diagnose SMZL.
Early studies indicated that both cytogenetic abnormalities and genomic mutations may have prognostic significance although the results were not always concordant among studies [54–56]. A cytogenetic study identified karyotypic complexity, 14q abnormalities and del(17p) as markers of a shorter overall survival in univariate, but not in multivariate analysis which included standard clinical and laboratory parameters and IGHV mutational status [39]. A multivariate analysis of 175 patients showed that NOTCH2 mutations and 100% IGHV identity were associated with a shorter time to first treatment, while TP53 mutations were associated with shorter overall survival [29]. It was, however, unclear whether TP53 abnormalities impacted the survival of most patients with SMZL who received treatments that act through p53-independent mechanisms.
As lymphoma-specific deaths account for less than one half of total deaths in SMZL, Bonfiglio et al. [59] used a classifier and survival relative to a matched general population to assess excess mortality in patient subgroups based on genomics. After adjusting for the effects of age, sex, and year of diagnosis, the 10-year life expectancies of patients with NNK-, DMT-, and “immune-suppressive”-SMZLs were reduced by 21.0%, 14.5%, and 20.4% respectively. Cases with both the NNK genotype and an “immune-suppressive” microenvironment, had a relative survival of 70.8% at 10 years. Furthermore, 99.4% of mutations detected in the spleen were also identified in blood or marrow samples, offering less invasive avenues for biopsy procedures.
Additionally, epigenetic alterations have emerged as valuable clinical indicators, serving either as a metric for cellular proliferation [117], or as a global measure of hypermethylation (High-M) [115]. Both these studies emphasise how global changes in the epigenome, such as those correlated with cell proliferation, may have greater prognostic significance than any individual genomic abnormality. A recent study by Hopper and colleagues [129] employed bulk RNA-sequencing on a cohort of indolent lymphomas, including 48 SMZL tumours. They identified five putative patient clusters, three of which were strongly enriched for an SMZL diagnosis. One cluster, which the authors termed LGBCL5, was enriched for genes involved in cell cycle control, T-follicular helper cells, mutations in TNFAIP3 and BCL2, and M1 macrophages in the tumour microenvironment (TME) inferred from the CIBERSORTx software. This cluster was also associated with a higher incidence of transformation to a high-grade lymphoma [129].
Histological transformation to a DLBCL is associated with a poor outcome in SMZL. In murine models, concurrent inactivating mutations of BLIMP1 and members of the NF-κB pathway can promote lymphomagenesis, leading to DLBCL with similar characteristics to human disease [130]. Genes involved in a proliferation signature were also found to be some of the most overly expressed upon the transformation of FL into DLBCL in human samples [131]. A study of 36 cases identified karyotypic complexity as the only risk factor for transformation in a multivariate analysis [21]. A more recent study [51] employed copy number arrays and a next-generation sequencing (NGS) panel to investigate genomic abnormalities in 41 SMZL patients with high-grade transformation, of whom 18 were studied at both diagnosis and at transformation (SMZL-T). The median time from diagnosis to transformation was 2.4 years (range 0–17) and the median survival from transformation was 6.55 years. SMZL-T clones arose by divergent evolution from a common altered precursor cell. Compared to SMZL, SMZL-T had higher genomic complexity. The most frequent abnormalities in SMZL-T were TNFAIP3 (59.4%), KMT2D (46.9%), TP53 (34.4%), ARID1A (31.3%), and KLF2 (31.3%) mutations, losses of 9p21.3 (CDKN2A/B, 40.6%) and 7q31-q32 (34.4%), and gains of 1q (40.7%). Four cases with a TP53 mutation at diagnosis, acquired a del(17p) at transformation. The main genomic variables predicting shorter survival from transformation were KLF2 mutations, a complex karyotype, and a trend for MYC gains. These results identify a subgroup of patients in whom more regular follow-up could be indicated, although it is unclear whether earlier treatment would improve outcomes.
Despite advancements in understanding and treating B-cell malignancies, research into SMZL lags behind. This is due to its rarity compared to other lymphomas, making it a lower research priority. The scarcity of cases also hinders large-scale studies, leading to combining MZL subtypes in recent trials of novel agents. Although we have made recent advancements in understanding SMZL’s deregulated pathways and molecular subtypes, actionable biomarkers and definitive genetic/transcriptional signatures for clinical use remain lacking. Diagnosing SMZL without splenic histology remains challenging, often relying on multiple factors that may not be readily available. This leads to variability in treatment and outcomes. Highlighting key knowledge gaps, we propose questions to better understand disease pathogenesis and suggest approaches to address them.
The research community is yet to generate the genome-wide data on coding and non-coding mutations in SMZL, from matched WGS of tumour and germline duos. The previous WGS study by Kiel et al. [55] did not include matched germ-line material from the SMZL patients, which is critical to generating a catalogue of the mutational landscape of any tumour, particularly when it comes to the non-coding space. This is because comparison to germline sequencing is required for adequate accuracy and sensitivity for the detection of high-confidence somatic variants. In contrast, in other B-cell tumours, such as CLL, larger cohorts with matched germline material have identified an expansive panel of genomic regions and genes targeted by non-coding mutations, such as PAX5 [132] and BACH2 [133]. Further insights into the key mutational signatures from paired WGS would be crucial in understanding the mechanisms involved in disease development and could potentially yield insights into malignant transformation. From a patient perspective, mutational signatures could provide insight into exposure to potential genotoxins helping to manage cancer risk, and might identify suitable treatment options for patient subgroups, such as poly- adenosine diphosphate (ADP)-ribose polymerase (PARP) or check-point inhibitors in patients with mutational signatures that suggest a deficiency in homologous recombination and mismatch repair, respectively.
This review outlines several studies highlighting the molecular features of poor-risk SMZL. Taking these studies together, a key aggressive subset can be defined, harbouring distinct, partially overlapping clinico-biological characteristics. Immunogenetically, these cases are defined by a highly biased usage of the IGHV1-2*04 allele, with intermediate levels of SHM that appear to persist throughout tumour development. At the genomic level, these cases harbour del(7q), genome complexity, short telomeres and mutations within key regulators of marginal-zone development and NF-κΒ signaling (NOTCH2 and KLF2). This group of cases also harbours high levels of global hypermethylation, including deregulation of the PCR2 complex, and evidence of elevated historical tumour proliferation with the capacity for future cellular growth. However, not all cases carry all these features, so it will be important to understand which has the greatest impact on disease biology and clinical outcome; these studies will require high-quality biological material for omics and functional analysis, from well-characterised patients with extensive follow-up, drawn from a large international consortium.
In humans, the MZ is located at the interface between the red and white pulp, where macrophages and resident B-cells can rapidly respond to antigens in the circulating blood supply. How the development of SMZL in the spleen might deregulate normal interactions between different cell populations remains poorly studied. In other tumour types, single-cell sequencing (SCS) technology has revealed insight into cellular components, functions, and interactions by circumventing noise caused by genetic and phenotypic variations across individual samples. In SMZL, it could enable direct measurement of molecular characteristics in thousands or even millions of individual cells, facilitating the characterization of cell types and revealing insights into sub-clonal dynamics, gene regulatory networks, transcription dynamics, and proteomic profiles. The precise identification of sub-clones could be crucial for understanding shifts in gene expression and for gaining insights into the SMZL TME and inflammatory cell milieu, where preliminary studies implicate a high-risk proinflammatory TME [134]. The importance of the TME and its contribution to different disease sub-types is shown in the study by Bonfiglio et al. [59], showing an “immune-suppressive” class encompassing macrophage, cytokine, and T-cell signatures, and an “immune-silent” class dominated by B-cell signatures. The presence of an ‘immune-suppressive’ subgroup opens the possibility of checkpoint inhibitor treatment in these patients.
The analysis of IGHV1-2*04 tumours in the spleen could further confirm the presence of ongoing SHM by using VDJ sequencing, and documenting interactions with T-cells by using TCR single-cell tracking. This type of immune repertoire analysis should maximize the development and deployment of future immunotherapies. SCS information could be powerful in predicting tumour progression, and transformation and for predicting drug sensitivity or resistance. As tumour cells evolve under treatment pressure, spontaneous mutations can confer drug resistance to a subset of cells, where single-cell proteomic analysis could enable a more accurate correlation between gene mutations and protein expression, elucidating diverse clinically relevant mechanisms. The application of spatial biology technologies could provide further insight into the organization and cell-to-cell interactions between tumour, immune and stromal cells, as well as their distributions within the SMZL TME. Cellular confirmation will ultimately be required, and whilst these studies are also rare, due to the paucity of variable material for study, they have already shown impaired T-cell and myeloid cell function in a proportion of patients [135–137].
The findings outlined in this review have significant clinical relevance and promise to enhance diagnosis and patient stratification in the future, provided that standardized testing protocols can be established. Achieving this goal presents challenges, necessitating the use of assays that capture the most clinically informative immunogenetic and (epi)genomic data. A single platform could be developed to perform: IGHV analysis, identification of gene mutations and CNAs linked to survival (del(7q), KLF2, NOTCH2, TP53, TNFAIP3), as well as those aiding in the differential diagnosis (e.g., MYD88 for WM, BRAF for HCL, NOTCH1 and SF3B1 for CLL, PTPRD for NMZL, and MAP2K1 for HCL-V), and assessment of proliferative history using DNA methylation data. As genomic knowledge expands, these platforms could be augmented with additional prognostically relevant and therapeutic target lesions, potentially facilitating treatment monitoring. While comprehensive multi-omics analysis, including epigenetic profiling, may offer the most precise patient identification for precision medicine, its adoption must be economically justified by selecting optimal treatment approaches. Regardless of the technical approach, each molecular marker must undergo rigorous validation across multiple cohorts, retrospective studies, and prospective trials to establish robust associations with disease outcomes. Analytical and clinical validation, followed by international harmonization, regulatory approval, and ongoing accreditation, are essential steps. Furthermore, guidelines for interpretation and reporting, along with appropriate training for clinical referral and patient communication, are imperative for effective implementation in clinical practice.
Addressing the knowledge gaps in SMZL requires concerted efforts from the scientific community to prioritize research into this rare lymphoma subtype. Collaborative initiatives that leverage multi-institutional partnerships and integration of comprehensive omics approaches hold promise in advancing our understanding of SMZL biology and identifying novel therapeutic targets. Additionally, the development of both pre-clinical models and clinical trials specifically designed for patients with SMZL is essential for evaluating the efficacy of targeted therapies and refining risk stratification algorithms. This will be particularly important in SMZL, as the potential to employ molecular information to improve risk-adapted stratification and guide therapy choice has not, as yet, been realised. In conclusion, despite the remarkable progress in the field of B-cell malignancies, the lack of comprehensive study into SMZL highlights the need for increased research efforts and resource allocation toward understanding the biology, genetics, and optimal management of this rare lymphoma subtype. By addressing these knowledge gaps, we can improve outcomes for patients with SMZL and contribute to the broader understanding of lymphomagenesis and improve treatment regimens.
BCR: B-cell receptor
CBL-MZ: clonal B-cell lymphocytosis of marginal zone origin
CLL: chronic lymphocytic leukaemia
CNAs: copy number alterations
CpG: cytosine-guanine
DLBCL: diffuse large B-cell lymphoma
epiCMIT: epigenetically determined cumulative mitoses
HCL: hairy cell leukaemia
HCL-V: hairy cell leukaemia variant
High-M: high genome-wide promoter methylation subgroup
IGHV: immunoglobulin heavy variable
IL-1: interleukin-1
MCLs: mantle cell lymphomas
MDR: minimally deleted region
MZLs: marginal zone lymphomas
NF-κΒ: nuclear factor kappaB
PRC2: Polycomb repressor complex 2
SBLPN: splenic B-cell lymphoma/leukaemia with prominent pucleoli
SDRPL: splenic diffuse red pulp lymphoma
SHM: somatic hypermutation
SMZL: splenic marginal zone lymphoma
TLR: toll-like receptor
TME: tumour microenvironment
TNFRs: tumour necrosis factor receptors
TRAF: tumour necrosis factor-receptor associated factor
WGS: whole-genome sequencing
WHO: World Health Organization
AM: Conception, Data curation, Methodology, Project administration, Writing—original draft, Writing—review & editing. HP: Writing—original draft, Writing—review & editing, Supervision. MA, RW, KS, and DB: Writing—original draft, Writing—review & editing. BS: Data curation, Methodology, Writing—original draft, Writing—review & editing. DGO: Conception, Writing—original draft, Writing—review & editing. JG and JCS: Conception, Project administration, Writing—original draft, Writing—review & editing.
Jonathan C. Strefford is the Editorial Board Member of Exploration of Targeted Anti-tumor Therapy, but he had no involvement in the journal review process of this manuscript. The other authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Amatta Mirandari is funded by Cancer Immunology Talent Fund, University of Southampton. Jonathan C. Strefford received funding from the Kay Kendall Leukemia Fund [873, 1104]; and Cancer Research UK ECRIN-M3 accelerator award [C42023/A29370]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

