 Review
Review

Affiliation:
1Division of Chemical and Systems Biology, Department of Integrative Biomedical Sciences, Faculty of Health Sciences, University of Cape Town, Cape Town 7925, South Africa
2Medical Biotechnology and Immunotherapy Research Unit, Institute of Infectious Diseases and Molecular Medicine, Faculty of Health Sciences, University of Cape Town, Cape Town 7925, South Africa
Email: gael.siwe@uct.ac.za
ORCID: https://orcid.org/0000-0002-1771-1939
Affiliation:
1Division of Chemical and Systems Biology, Department of Integrative Biomedical Sciences, Faculty of Health Sciences, University of Cape Town, Cape Town 7925, South Africa
2Medical Biotechnology and Immunotherapy Research Unit, Institute of Infectious Diseases and Molecular Medicine, Faculty of Health Sciences, University of Cape Town, Cape Town 7925, South Africa
ORCID: https://orcid.org/0000-0002-6278-2808
Affiliation:
1Division of Chemical and Systems Biology, Department of Integrative Biomedical Sciences, Faculty of Health Sciences, University of Cape Town, Cape Town 7925, South Africa
2Medical Biotechnology and Immunotherapy Research Unit, Institute of Infectious Diseases and Molecular Medicine, Faculty of Health Sciences, University of Cape Town, Cape Town 7925, South Africa
ORCID: https://orcid.org/0000-0002-1834-072X
Affiliation:
1Division of Chemical and Systems Biology, Department of Integrative Biomedical Sciences, Faculty of Health Sciences, University of Cape Town, Cape Town 7925, South Africa
2Medical Biotechnology and Immunotherapy Research Unit, Institute of Infectious Diseases and Molecular Medicine, Faculty of Health Sciences, University of Cape Town, Cape Town 7925, South Africa
ORCID: https://orcid.org/0000-0002-2790-1963
Affiliation:
1Division of Chemical and Systems Biology, Department of Integrative Biomedical Sciences, Faculty of Health Sciences, University of Cape Town, Cape Town 7925, South Africa
2Medical Biotechnology and Immunotherapy Research Unit, Institute of Infectious Diseases and Molecular Medicine, Faculty of Health Sciences, University of Cape Town, Cape Town 7925, South Africa
ORCID: https://orcid.org/0000-0001-9589-653X
Explor Asthma Allergy. 2024;2:373–398 DOI: https://doi.org/10.37349/eaa.2024.00052
Received: May 03, 2024 Accepted: June 18, 2024 Published: August 09, 2024
Academic Editor: Somasundaram Arumugam, National Institute of Pharmaceutical Education and Research, India
The article belongs to the special issue Atopic Dermatitis – Pathology and Treatment modalities
Atopic dermatitis (AD) represents the most common inflammatory skin disease with a highly intricated immune fingerprint. Until recently, AD management mostly relied on topical corticosteroids, calcineurin inhibitors, and systemic immunosuppressants, with a range of safety and tolerability concerns including toxicity, drug interactions, and contraindications. With the onset of biologics, safer and more targeted therapeutics have become available, displaying various degrees of success in treating AD, but not yet able to meet all the needs of AD patients. Some of the challenges encountered included variability of responses among patients, long-term safety, and limited access due to prohibitive costs. As the pathophysiology of AD has been increasingly understood within the last years, new approaches are explored, leading to an unprecedented diversification of therapeutic options to address these hurdles. This review highlights current immunotherapeutic strategies developed towards AD, whether already in the clinical pipeline or still in preclinical exploration.
Atopic dermatitis (AD), also referred to as atopic eczema, is a skin disease that is a very diverse in terms of ethnic-based phenotypic variability, age of onset, dynamic course of disease, risk of various cutaneous infections, atopic and non-atopic comorbidities [1]. AD, characterized by eosinophilic/spongiotic inflammation of the skin with characteristic age-dependent distribution patterns and morphology of lesions, is a chronically relapsing and intensely pruritic, Th2-driven skin disease often occurring in families with atopic diseases (AD, food allergy, bronchial asthma or allergic rhinoconjunctivitis). Although the etiology of AD is yet to be fully understood, it is commonly accepted that the pathophysiology of AD is driven by a combination of factors comprising epidermal barrier defects, genetic predisposition, immune dysregulation, imbalance of microbiome, and extrinsic stimuli like allergens, irritants, and microbes [2, 3]. According to the latest Global Burden of Disease study [4], AD represents 36.17% of all new immune-mediated inflammatory diseases reported across the world.
The current therapeutic regimen of AD incorporates different pharmacological approaches including topical anti-inflammatory drugs (topical corticosteroids, topical calcineurin inhibitors, phosphodiesterase-4 inhibitors), immunosuppressants (ciclosporin A, azathioprine, methotrexate, mycophenolate mofetil), targeted monoclonal antibodies and oral Janus kinase (JAK) inhibitors [5, 6]. Clinical management of AD is generally guided by disease severity (based on scoring); mild AD can usually be controlled with topical treatments, whereas systemic immunomodulatory therapies (with topicals as adjuvants) are recommended for moderate-to-severe forms of AD. For AD patients whose disease course is not satisfactory or whose quality of life is substantially lowered despite appropriate topical therapy, ultraviolet phototherapy might be considered alone or in combination with systemic therapy [7–9]. While substantial progress has been made to meet the therapeutic needs of AD patients, considerable limitations remain associated to these conventional approaches. For instance, the action of existing systemic immunomodulators is hampered by poor tolerability, the need for ongoing bloodwork monitoring, and transient relief [10]. The limitations of biologics as a class include difficulty in personalizing the dosage, repeated injections, conjunctivitis following interleukin IL-13 or IL-4/IL-13 blockade, possible anti-drug antibody formation, and decreased potency over time [1, 11]. As for phototherapy, major concerns have arisen including long-term cumulative adverse effects (photodamage), skin carcinogenesis, and melanoma induction, particularly for ultraviolet A1 radiations [12].
Over the last decade, novel immunotherapeutic strategies have been developed based on upraised understanding of AD mechanisms [13–18]. Furthermore, a broad range of immunotherapeutic options is currently under investigation for the management of other skin or immune-mediated diseases, with increasing evidence of their applicability in AD [19, 20]. This review provides insights upon current immunotherapy options, including the latest developments on biologics and targeted therapies by small molecules (JAK inhibitors, phosphodiesterase-4 inhibitors, etc.), with emphasis towards emerging therapies either at their exploratory phase (antibody-drug conjugates, antibody-cytokine fusion proteins), or in clinical development (allergen immunotherapy and cell-based therapy) (Figure 1). The literature search was conducted during the month of April 2024 using PubMed, PubMed Central, Embase, MEDLINE, Web of Science, and ClinicalTrials.gov. The search terms were specifically related to the fields of “immunotherapy of atopic dermatitis”, “allergen immunotherapy”, “cell therapy” and included studies published in English from 2000 to 2024.
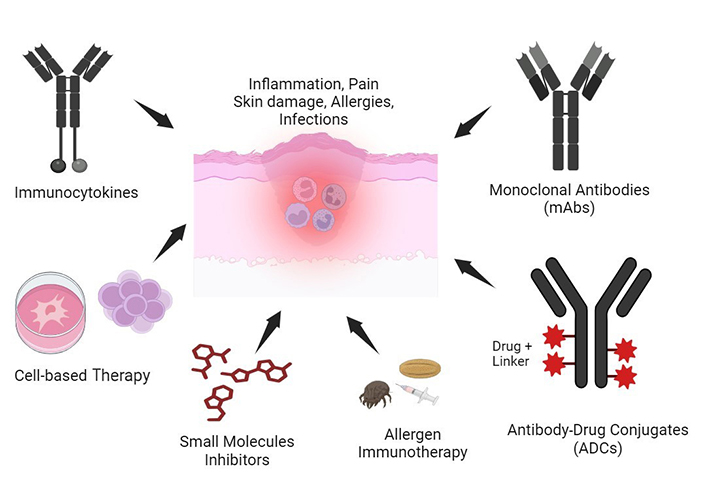
Immunotherapeutics developed for atopic dermatitis (AD) treatment. AD is characterized by chronic inflammation, immune dysfunction, and severe skin impairment. Several immunotherapeutics developed towards an inflammation/allergic reaction resolution include allergen immunotherapy, monoclonal antibodies, antibody-drug conjugates, immunocytokines, small molecule inhibitors, and cell-based therapies. These immunotherapy approaches have shown positive preclinical and clinical efficacy in AD. Created with Biorender.com
Antibody-drug conjugates (ADCs) represent a special class of immunotherapeutics composed of a cell-specific monoclonal antibody linked to cytotoxic small molecules (toxins or drugs) via a chemical (cleavable or non-cleavable) linker [21]. They have been extensively applied in cancer and inflammatory disease therapy [21, 22]. To date, 14 ADCs have been approved by the United States Food and Drug Administration (US FDA) and approximately 300 ADCs are undergoing preclinical/clinical evaluations for various cancers [21, 23]. Aside from cancers, there is a growing interest in the use of ADCs for the treatment of non-oncological conditions, especially chronic inflammatory and allergic diseases [22] like AD.
In a preclinical study, anti-inflammatory phosphodiesterase 4 (PDE4) inhibitor ADC was generated to target CD11a antigen on primary human monocytes and mouse peritoneal cells. The results showed both human and mouse αCD11a-PDE4 ADCs significantly inhibit tumor necrosis factor-alpha (TNF-α) secretion in dysfunctional immune cells in vitro and in vivo [24]. In another preclinical in vitro study targeting membrane immunoglobulin E B-cell receptor (IgE-BCR) fragment, an ADC containing a 15c112 antibody conjugated to monomethyl auristatin E (2:1) demonstrated highly selective binding and cytotoxicity to BCR-positive immune cells, which provides evidence for therapeutic potential in allergic conditions like AD, via IgE secretory cells depletion [25]. Furthermore, anti-E Selectin Dex (Dexa-AbhEsel) ADC, anti-TNF-α glucocorticoid ADC, and anti-CD163 Dex (Cymac-001) ADC, were each used to deliver dexamethasone as an anti-inflammatory payload in vitro and in vivo [23, 26, 27], with all studies demonstrating positive preclinical outcomes in depletion of dysfunctional immune cells and proinflammatory cytokines.
Aside from chemical moieties, cytotoxic molecules like toxins and cytolytic proteins can be chemically or genetically fused to cell-specific antibodies or antibody fragments for the development of recombinant immunotoxins (rITs) [28], and targeted human cytolytic fusion proteins (hCFPs) [29]. So far, these cytotoxic agents have been explored for cancer immunotherapy and treatment of cutaneous inflammation and lesions, including AD [29–31].
A study by Thepen et al. [32] reported the therapeutic potential of a rIT for the treatment of cutaneous inflammation in transgenic mice expressing human CD64 receptors. The immunotoxin consisted of an anti-CD64 single-chain variable fragment [H22(scFv)] chemically conjugated to ricin A and was used to treat sodium lauryl sulfate (SLS)-induced chronic skin inflammation in human CD64+ transgenic (hCD64tg) mice. The results from intradermal injection at the site of chronic inflammation showed selective elimination of activated M1 pro-inflammatory macrophages, downregulation of pro-inflammatory cytokines like IL-1 and IL-6, and subsequent resolution of chronic cutaneous inflammation after 24 hours. Also, clinical symptoms of inflammation, such as local skin temperature and capillary vasodilation, drastically decreased [32]. Another study from the same group later reported that a rIT containing H22(scFv), genetically fused to truncated Pseudomonas aeruginosa exotoxin A (ETA’) [H22(scFv)-ETA’], demonstrated selective elimination of dysregulated CD64+ pro-inflammatory macrophages and resolution of cutaneous inflammation in hCD64tg mice and patient biopsies [33]. In a different study that took advantage of the ability of cross-linked CD64 to rapidly internalize IgG, bivalent H22(scFv)2-ETA’ was constructed and evaluated for increased uptake and cytotoxicity in CD64+ monocytic cells in cutaneous inflammation. The results demonstrated that H22(scFv)2-ETA’ exerts more cytotoxicity against IFN-γ stimulated CD64+ monocytes compared to monovalent H22(scFv)-ETA’, hence providing evidence that increased valency could remarkably improve the efficacy of anti-CD64 rIT therapy in cutaneous inflammation [31]. Jiemy et al. [34] also developed a rIT containing an anti-CD64 (scFv) antibody fused to the cytolysin HALT-1 (sourced from Hydra magnipapillata) at the C- and T- terminus generating both HALT-1-scFv and scFv-HALT-1 rITs. The study showed that both rITs demonstrate selective binding and significant cytotoxic killing/potency on CD64+ in vitro and M1-like macrophages irrespective of HALT-1 positioning, which provides preliminary evidence for HALT-1 potential as a toxin moiety in therapeutic cell targeting for CD64+ chronic inflammatory disease models [34].
Besides rITs, humanized enzymes have been used to develop hCFPs that overcome the limitations of immunogenicity associated with toxins. A full human microtubule-associated protein (MAP tau) fused to an H22(scFv) antibody [H22(scFv)-MAP-tau] efficiently killed proliferating HL-60 pro-monocytic cells in vitro and specifically eliminated polarized M1 macrophages in hCD64tg mice in a cutaneous model of chronic inflammation [35]. This result further provides evidence that hCFPs specifically targeted CD64+ M1 proinflammatory macrophages, which are major players in chronic inflammatory diseases, including AD.
Taken together, these preliminary results clearly demonstrate the anti-inflammatory potency of these ADCs, making some of them serious candidates for first-in-human clinical trials.
Cell-based therapy explores the use of living cells as therapeutics to combat hard-to-treat diseases. It extensively involves the transfer of autologous and/or allogeneic cellular materials into a patient for medical purposes [36]. In parallel with pharmacological agents, cell-based therapies are now developed for AD treatment. A novel and effective cell-based therapy for AD treatment is mesenchymal stem cells (MSCs) therapy [3]. MSCs are multipotent stem cells able to differentiate into different cell lineages. MSCs are isolated from different sources and include human umbilical cord blood MSCs (hUCB-MSCs), human adipocytes MSCs (hAD-MSCs), and human bone marrow MSCs (hBM-MSCs) [37]. They express cell surface markers (CD73, CD90, CD105) but lack CD45 and CD34 available in hematopoietic stem cells (HSCs) [37].
Allogeneic MSCs function as an immunomodulatory therapy (immunosuppressive and immunotolerant), hence their use for the treatment of immunopathological disorders like AD [3, 38]. In AD treatment, MSC-based therapy inhibited T-cell and B-cell activation, and therefore upregulated the production of anti-inflammatory cytokines (IL-10 and TGF-β) and downregulation of proinflammatory cytokines (IL-4 and IFN-γ), as well as allergic IgE levels [39]. Some MSC-based therapies investigated in preclinical AD models are summarized below (Table 1).
MSC-based therapies in mouse models of AD
| MSC source | AD model | Mouse strain | Route | Mechanisms of action | References |
|---|---|---|---|---|---|
| hBM-MSCs | Ovalbumin-induced AD | BALB/c or C3H/HeN | IV | Suppression of T cells function via NO and suppression of B-cell function via class switch recombination | [40] |
| hUCB-MSCs | Dermatophagoides farinae-induced AD | Nc/Nga | SC | Inhibition of mast cells degranulation through PGE2 and TGF-β1 | [41] |
| hAD-MSCs | Dermatophagoides farinae-induced AD | Nc/Nga | IV | B-cell suppression via cyclooxygenase 2 regulation | [42] |
| hAD-MSCs | Dinitrochlorobenzene-induced AD | BALB/c | IV | Regulation of MIP-2 and miR-122a-SOCS1 expression, as well as Th1/Th2 responses | [43] |
| hSOD3-MSCs | Ovalbumin-induced AD | BALB/c | SC | Suppression of response elicited by keratinocytes, mast cells, neutrophils, dendritic cells, and T cells through multiple mechanisms | [44] |
| hUCB-MSCs | Dermatophagoides farinae-induced AD | Nc/Nga | SC | Pre-conditioning of MSC with mast cells granules optimizes suppression of mast and B cells | [45] |
| hUCB-MSCs | Dermatophagoides farinae-induced AD | BALB/c | SC | Control of both eosinophil-associated Th2 immunity and neutrophil-related Th17 | [46] |
| Primed T-MSCs | DNFB-induced AD | C57BL/6J | SC | Regulation of B cell-mediated inflammatory responses, which are dependent on CD40 expression on primed T-MSCs mediated through the non-canonical NF-κB pathway | [47] |
| HA-SH/hAD-MSCs (3D-Hydrogel) | 1-chloro-DNCB-induced AD | BALB/c | SC | Immunomodulatory action on immune cells via downregulation of major inflammatory cytokines (IL-13, CCL11, and CCL24), reduced epidermal thickness, and mast cell penetration | [48] |
| hAD-MFSCE | Dermatophagoides farinae-induced AD | NC/Nga | Topical | Decrease of IgE and inflammatory cytokines levels. Inhibition of epidermal thickness, mast cell infiltration, and expression of expression of IL-4, IL-10, IFN-γ, TNF-α, activation-regulated chemokines | [49] |
| hAD-MSCs | Ovalbumin-induced AD | BALB/c | SC | Decrease of serum IgE levels, and mast cells infiltration skin lesions. Suppression of Th17 cells proliferation and proinflammatory cytokines (IL-17A and RORγT) expression. Upregulation of PD-L1, TGF-β and PGE2 levels | [50] |
| hUCB-MSCs | 1-chloro-DNCB-induced AD | BALB/c | SC + IV | Anti-inflammatory and Immunomodulatory function resulting in amelioration of skin lesions, through inhibition of JAK-STAT pathway and different cytokines (IL-4, IL-13, IL-17, and IgE) receptors | [51] |
CCL11: eotaxin 1; CCL24: eotaxin 2; DNCB: 2,4-dinitrobenzene; DNFB: 2,4-dinitrofluorobenzene; IL: interleukin; IV: intravenous; MIP-2: macrophage inflammatory protein 2; NF-κB: nuclear factor kappa B; PGE2: prostaglandin E2; SC: subcutaneous; TGF-β1: transforming growth factor beta 1
Immunocytokines, also referred to as antibody-cytokine fusion proteins, are molecules that consist of a cytokine moiety fused to a monoclonal antibody or an antibody fragment [52], with antibody and cytokine serving as vehicle and payload, respectively. The development of antibody-cytokine fusion proteins emerged as a novel immunotherapy strategy to circumvent the detrimental effects associated with the systemic administration of cytokines as a monotherapy [53]. In fact, while cytokines may display a potent therapeutic activity in preclinical models of cancer and other conditions, their clinical use is often limited by severe systemic toxicities that prevent a dose escalation to therapeutically active regimes. The antibody-mediated delivery of cytokines to the site of disease might be a step forward in increasing therapeutic activity, while reducing side effects [54]. Immunocytokines have been extensively investigated for cancer treatment and a recent review describes their engineering formats and clinical benefits [55]. Immunocytokines also attract huge interest in the field of chronic inflammatory conditions, including inflammatory skin diseases.
In inflamed tissue, the lymphatic vasculature undergoes extensive remodeling and expansion, including lymphangiogenesis (formation of new lymphatic vessels) and enlargement of preexisting vessels. Vascular endothelial growth factor C (VEGF-C) was found to potently reduce inflammation in a mouse model of oxazolone-induced psoriasis-like skin inflammation [56]. Later on, Schwager et al. [57] generated and intravenously administered a human VEGF-C fused to the F8 diabody (F8-VEGF-C) to two other mouse models of psoriasis-like skin inflammation (induced by oxazolone in transgenic VEGF-A overexpressing animals or repeated application of imiquimod). F8 antibody is highly specific for the angiogenesis-marking extradomain A (EDA) of fibronectin, a protein selectively expressed in inflamed tissues. F8-VEGF-C treatment significantly reduced leukocyte infiltration in inflamed tissue, including CD4+ and γδ T cells. In addition, the treatment reduced ear skin edema and significantly improved lymphatic drainage function [57]. These results were further confirmed by the same group, who investigated the long-term effects of F8-VEGF-C using the oxazolone-mediated contact hypersensitivity (CHS) model in mice overexpressing VEGF-A in their epidermis. Authors reported that F8-VEGF-C treatment resulted in a long-term anti-inflammatory activity that persisted for more than 70 days, by reducing the severity of edema and immune cell (CD4+ T cells, macrophages, and dendritic cells) infiltration upon hapten re-challenge [58]. As psoriasis and AD are two inflammatory skin diseases sharing some common features, this promising approach, though at its early stage, could be exploited in the near future for AD treatment.
Targeted monoclonal antibodies were first introduced in dermatology for the treatment of psoriasis [59] before their extension to AD.
AD involves predominately Th2-mediated inflammation and a key feature of the disease is the upregulation of IL-13 and IL-4 in lesional and non-lesional skin [60]. The two key Th2 cytokines, IL-4/IL-13 are suggested to drive important pathologic processes in AD including; keratinocyte differentiation, tight junction formation, recruitment of inflammatory cells to skin by upregulating Th2-associated chemokines (CCL17 and CCL26), antimicrobial peptides activity, and inhibition of important physiological processes like lipid synthesis [61]. Dupilumab is a monoclonal antibody targeting both IL-4 and IL-13 receptors, through IL-4Rα, and has been the first FDA-approved (March 2017) monoclonal antibody for use in AD [62]. Dupilumab’s success paved the way in exploring the individual contribution of IL-4 and IL-13 in AD pathophysiology, leading to the development of two monoclonal antibodies targeting IL-13, namely tralokinumab and lebrikizumab. Tralokinumab acts by binding to IL-4Rα and IL-13Rα1 (the two subunits of the IL-13 receptor), as well as to the IL-13Rα2 decoy receptor [63], whereas, lebrikizumab specifically binds soluble IL-13 at an epitope that strongly overlaps with the binding site of IL-4Rα [64]. Thymic stromal lymphopoietin (TSLP) is a cytokine produced by keratinocytes that has recently been postulated to play a key pathogenic role in AD. In proinflammatory states, damaged keratinocytes produce TSLP that acts on myeloid dendritic cells and Langerhans cells, leading to a Th2-skewed inflammatory response with the release of IL-4, IL-5, and IL-13 [65]. In a recent phase 2b clinical trial (NCT03809663), tezepelumab (an anti-TSLP monoclonal antibody) was investigated for its use as monotherapy in moderate-to-severe AD patients but could not achieve the primary endpoints for this patient population.
AD is not only Th2-centered among various disease subtypes, it also overexpresses Th22-related cytokines and chemokines, such as IL-22 which was found to positively correlate with disease severity [66]. IL-22 was proven to increase epidermal barrier dysfunction, induce epidermal hyperplasia, downregulate key genes (such as filaggrin and loricrin) involved in epidermal differentiation, and drive the epidermal acanthosis characterizing chronic AD lesions [67]. Fezakinumab (a fully human monoclonal antibody directed against IL-22) was investigated in adults with uncontrolled AD for 20 weeks in a phase 2a clinical trial and SCORAD 50 (proportion of patients achieving scoring atopic dermatitis improvement of ≥ 50% from the baseline) endpoint was achieved, at week 12, in severe AD subpopulation [68].
Th17-related cytokines, specifically IL-17 and IL-23, are the key drivers of the inflammatory process in psoriasis, and their potential use as therapeutic targets in the context of AD is supported by several studies [61]. IL-17 synergizes with IL-22 to promote tissue inflammation and barrier defects, while IL-23 activates both Th17 and Th22 pathways [69]. Ustekinumab is a fully human monoclonal antibody directed against the p40 subunit of IL-12 and IL-23, preventing them from binding to T cell-surface receptors and causing differentiation and clonal expansion of naïve T cells into specific Th subtypes [70]. A phase 2 double-blinded and placebo-controlled study revealed that ustekinumab administration only resulted in a numerical improvement of AD clinical scores, without statistical significance [71]. A summary of clinical trials investigating monoclonal antibody therapy in AD, irrespective of disease subtypes or immune axis explored, has been tabulated (see Table 2).
Monoclonal antibodies in advanced clinical trials for AD
| Target | Drug | Route | Status | Global endpoints achieved | Reported adverse effects | References |
|---|---|---|---|---|---|---|
| IL-1α | Bermekimab | SC | Phase II | EASI-75, SCORAD-50 | Wheezing, injection-site reaction, nausea | NCT03496974 |
| IL-4α | Stapokibart | SC | Phase III | Ongoing | Ongoing | NCT06277765NCT05265923 |
| Rademikibart | SC | Phase II | IGA 0/1 | Headache, vomiting, injection-site reaction, eye pruritus | [72] | |
| 611 | SC | Phase III | Ongoing | Ongoing | NCT06173284 | |
| AK120 | SC | Phase I/II | EASI-75 | Upper respiratory tract infection, headache, abdominal pain, mouth ulceration, injection-site reaction | [73]NCT05048056 | |
| MG-K10 | SC | Phase III | Ongoing | Ongoing | NCT06026891 | |
| Dupilumab | SC | Approved | EASI-75; EASI-90 | Conjunctivitis, injection-site reaction, nasopharyngitis, herpes viral infections, viral gastroenteritis, dental caries | [62, 74–79] | |
| IL-13 | Tralokinumab | SC | Approved | IGA 0/1; EASI-50; EASI-75; EASI-90 | Viral upper respiratory tract infection, injection-site reaction, conjunctivitis | [80–82] |
| Lebrikizumab | SC | Approved | IGA 0/1; EASI-75; EASI-90 | Headache, oral herpes, conjunctivitis, nasopharyngitis, dry eye, folliculitis, upper respiratory tract infection | [83, 84] | |
| Eblasakimab | SC | Phase IIb | EASI-75 | Not available | NCT05158023 | |
| IL-5Rα | Benralizumab | SC | Phase II | EASI-90 | Bronchitis, conjunctivitis, Covid-19, nasopharyngitis, upper respiratory tract infection | NCT04605094 |
| IL-12/IL-23p40 | Ustekinumab | SC | Phase II | No significant efficacy | Upper respiratory tract infection, musculoskeletal pain | [71] |
| IL-17A | Secukinumab | SC | Phase II | No efficacy | Orbital cellulitis, upper respiratory infection, streptococcal pharyngitis | [85] |
| IL-22 | Fezakinumab | SC | Phase IIa | SCORAD-50 in severe AD subpopulation | Viral upper respiratory tract infections, facial cellulitis | [68] |
| LEO138559 | SC | Phase IIb | Ongoing | Ongoing | NCT05923099 | |
| IL-23 | Rizankisumab | SC | Phase II | No efficacy | Cellulitis, nasopharyngitis, pruritus | [86] |
| IL-31 | Nemolizumab | SC | Approved (Japan) | EASI-90; IGA 0/1 | Upper respiratory tract infection, nasopharyngitis | [87–91] |
| IL-33 | Astegolimab | SC | Phase II | No efficacy | None recorded | [92] |
| Etokimab | IV | Phase IIa | EASI-75 | Headache, upper respiratory tract infection, conjunctivitis | [93] | |
| Tozorakimab | SC | Phase IIa | No efficacy | Oral herpes, cellulitis, conjunctivitis, nasopharyngitis, urinary tract infection | NCT04212169 | |
| REGN3500 | SC | Phase IIb | No efficacy | Peripheral oedema, nasopharyngitis | NCT03738423 | |
| IL-36R | Spesolimab | IV | Phase IIa | No significant efficacy | Upper respiratory tract infection, nasopharyngitis, depression | [94] |
| OX40 | GBR830 | SC | Phase IIb | No significant efficacy | Upper respiratory tract, myalgia, headache, postprocedural infection, fatigue | [95] |
| Amlitelimab | IV | Phase IIa/b | EASI-75; IGA 0/1 | Nasopharyngitis, Covid-19, headache, hyperhidrosis, upper respiratory tract infection, pyrexia, increased aspartate aminotransferase and iron deficiency anemia | [96, 97] | |
| Rocatinlimab | SC | Phase IIb | EASI-90 | Atopic cataract, small intestine ulcer, pyrexia, vertigo, aphthous ulcer, constipation, diarrhea, vomiting, nausea, chills, nasopharyngitis, arthralgia, oropharyngeal pain | NCT03703102 | |
| IgE | Omalizumab | SC | Phase II | No significant efficacy | Viral infection, stomach pain, sore throat, toothache, urticaria, headache | [98–100] |
| mIgE | FB825 | IV | Phase II | Not available | Not available | NCT04413942 |
| TSLP | Tezepelumab | SC | Phase IIb | No significant efficacy | Headache, diarrhea, injection-site erythema | [101]NCT03809663 |
| SCF | OpSCF | SC | Phase IIa | Ongoing | Ongoing | NCT06101823 |
EASI: eczema area and severity index; EASI 50/75/90: ≥ 50/75/90% improvement in EASI from baseline; IGA: investigator global assessment; IGA 0/1: “clear or almost clear” with ≥ 2-point improvement in IGA from baseline; IgE: immunoglobulin E; ILα: alpha chain of interleukin; ILRα: alpha chain of interleukin receptor; IV: intravenous; mIgE: membrane-bound immunoglobulin E; NCT: national clinical trial identifier consultable on www.clinicaltrials.gov (website consulted last time as of April 30, 2024), only clinical trials in advanced phases (Phase II onwards) are listed; SCF: stem cell factor; SC: subcutaneous; SCORAD: scoring atopic dermatitis; SCORAD 50: ≥ 50% improvement in SCORAD from baseline; TSLP: thymic stromal lymphopoietin
Besides antibody-based therapies that usually target cytokines or their receptors, small molecules exert their effects by interfering with specific intracellular signaling pathways. Various small molecules are being evaluated as targeted therapeutics for AD and given their potential in blocking several immune pathways, these drugs significantly contribute to the expanding toolbox for AD management. Moreover, their action can be further potentiated when fused to antibodies targeting specific receptors.
JAK inhibitors are intracellular enzymes mediating the signaling cascade from a cytokine receptor into the cell [102]. The JAKs are a family of tyrosine kinases, including JAK1, JAK2, JAK3, and TYK2. The intracellular domain of various cytokine receptors is phosphorylated by JAKs, which results in binding and activation of the signal transducer and activator of transcription (STAT). The JAK-STAT signaling then mediates a range of intracellular immune dysregulations, including robust Th2 responses [61, 103]. Upadacitinib and abrocitinib are two recent FDA-approved oral JAK inhibitors with high potency and good safety profiles.
Cyclic adenosine monophosphate (cAMP) was reported to play a role in the regulation of inflammatory and immune responses, with increased levels of cAMP being associated with monocytes and T cell suppression. PDEs are enzymes that break down cAMP or cyclic guanosine monophosphate (cGMP), with certain isoenzymes expressed only in selected tissues [104, 105]. PDE4 is found in immune and inflammatory cells (basophils, mast cells, eosinophils, B and T cells, monocytes, macrophages, neutrophils and endothelial cells) and PDE4 isoforms (PDE4A, PDE4B, PDE4C and PDE4D) have been observed to be increased in dermal fibroblasts of AD patients skin samples compared to dermal fibroblasts from healthy skin [106]. PDE4 inhibitors impair cytokine production by elevating cAMP levels, thereby directly or indirectly blocking the NFκB pathway and the subsequent cytokine and chemokine synthesis. Moreover, this mechanism of immunologic suppression has been suggested to also occur in keratinocytes, indicating a role in skin barrier protection [107]. To date, crisaborole and difamilast are the only approved PDE4 inhibitors for mild-to-moderate AD in adults and children, and the FDA recently accepted the supplemental new drug application (sNDA) for roflumilast cream 0.15% to treat AD in patients aged 6 and up.
Molecules targeting histamine 4 receptor (H4R) could be exploited for AD treatment, as histamine, in addition to inducing pruritus, was found to inhibit the terminal differentiation of keratinocytes and impair the skin barrier in AD [108]. Sphingosine1-phosphate (S1P) is a bioactive lipid mediator that impacts immunological processes by regulating migration of lymphocytes out of lymphatic tissues and dendritic cells migration to lymph nodes [109]. Therefore, S1P receptors (S1PRs) are a promising therapeutic target, with several clinical trials evaluating S1PRs modulators for the treatment of multiple inflammatory disorders, including AD. Etrasimod is an oral selective S1PR1/4/5 modulator in development for multiple immune-mediated inflammatory disorders. Its efficacy and safety have been confirmed in patients with moderate-to-severe AD in a Phase 2b clinical trial [110] and Pfizer® has recently announced ongoing Phase III clinical development of etrasimod for AD. Other immune pathways still in their exploratory phase for small molecules usage include neurokinin 1 receptor (NK1R), liver X receptor (LXR), purinergic receptor P2X (P2X3) or kappa opioid receptor (KOR). A comprehensive overview of small molecules in clinical trials is captured in Table 3.
Small molecules in advanced clinical trials for AD
| Target | Drug | Route | Status | Global endpoints achieved | Reported adverse effects | References |
|---|---|---|---|---|---|---|
| JAK1 | Upadacitinib | Oral | Approved | EASI-75; EASI-90; EASI-100; IGA 0/1 | Nasopharyngitis, acne, headache, oral herpes, diarrhea, eczema, herpes zoster | [111–114] |
| Abrocitinib | Oral | Approved | IGA 0/1; EASI-50; EASI-75 | Transient decreased platelet count, acne, dizziness, nasopharyngitis, acne, headache, oral herpes, vomiting, diarrhoea, eczema, herpes zoster, nausea, folliculitis | [115–120] | |
| LNK01001 | Oral | Phase III | Ongoing | Ongoing | NCT06277245 | |
| SHR0302 | Oral | Phase II; Phase II/III | IGA 0/1; EASI-75 | Blood pressure increased, upper respiratory tract infection, folliculitis, urinary tract infection, hyperlipidemia, hyperuricemia, headache | [121]NCT04717310 | |
| JAK1/2 | Baricitinib | Oral | Approved | IGA 0/1; EASI-75 | Headache, increased blood CPK, nasopharyngitis, herpes simplex, influenza, diarrhea, nausea, folliculitis, back pain, acne | [122–128] |
| Ruxolitinib | Topical | Approved | IGA 0/1; EASI-75; EASI-90 | Nasopharyngitis, headache | [129–131] | |
| JAK1/3 | Tofacitinib | Topical | Phase IIa | EASI-75 | Viral upper respiratory tract infection, gastroenteritis, bronchitis, nausea | [132] |
| ATI-1777 | Topical | Phase IIa/b | mEASI-75 | Blood CPK increased, headache | [133]NCT05432596 | |
| ATI-502 | Topical | Phase IIb | No significant efficacy | None reported | [134]NCT03585296 | |
| JAK1/TYK2 | Brepocitinib | Topical | Phase IIb | EASI-90 | Nasopharyngitis, erythema | [135] |
| JAK1/2/3/TYK2 | Delgocitinib | Topical | Approved (Japan) | mEASI-50; mEASI-75; mEASI-90 | Acne, folliculitis, gastroenteritis, herpes simplex, paronychia, nasopharyngitis | [136–140] |
| Pan-JAK | Jaktinib | Oral | Phase III | Ongoing | Ongoing | NCT05526222NCT05676242 |
| PDE4 | Crisaborole | Topical | Approved | IGA 0/1 | Application-site pain, upper respiratory tract infection, nasopharyngitis, headache, oropharyngeal pain, cough | [141–143] |
| PDE4 | Lotamilast | Topical | Phase II | Not available | Not available | NCT03394677 |
| PDE4 | DRM02 | Topical | Phase II | Not available | Not available | NCT01993420 |
| PDE4 | Difamilast | Topical | Approved (Japan) | IGA 0/1; EASI-75; EASI-90 | Nasopharyngitis, folliculitis, gastroenteritis | [144–146]NCT05571943NCT05372653 |
| PDE4 | Roflumilast | Topical | Phase III | IGA 0/1; EASI-75 | Headache, nausea, application-site pain, nasopharyngitis, Covid-19, upper respiratory tract infection, diarrhea, vomiting | [147]NCT04773587NCT04773600 |
| P2X3 | BLU-5937 | Oral | Phase II | Not available | Not available | NCT04693195 |
| NK1R | Serlopitant | Oral | Phase II | No significant efficacy | Nasopharyngitis, urinary tract infection | NCT02975206 |
| NK1R | Tradipitant | Oral | Phase III | WI-NRS ≥ 4; IGA 0/1; SCORAD 50 | Not available | [148]NCT04140695 |
| KOR | Difelikefalin | Oral | Phase II | I-NRS ≥ 4 | Abdominal pain/discomfort, nausea, dry mouth, headache, dizziness, hypertension, hyponatremia, nephrolithiasis, costochondritis | [149] |
| S1PR1 | Vibozilimob | Oral | Phase II | Ongoing | Ongoing | NCT04684485 |
| S1PR1 | Udifitimod | Oral | Phase II | No significant efficacy | Skin mass, depression, upper respiratory tract infection, pyrexia | NCT05014438 |
| S1PR1/S1PR4/S1PR5 | Etrasimod | Oral | Phase IIb | EASI-50 | Nausea, constipation, back pain, dizziness | [110] |
| H4R | Adriforant | Oral | Phase II | EASI-75 | Headache, somnolence, nasopharyngitis, eczema, urinary tract infection | [150] |
| GPCR19 | HY209 | Topical | Phase II | Not available | Not available | NCT04530643 |
| LXR | VTP-38543 | Topical | Phase I/II | No significant efficacy | Upper respiratory tract infection, fatigue, cellulitis, dizziness, pruritus | [151] |
| LXR | ALX-101 | Topical | Phase IIb | Not available | Not available | NCT03859986 |
CPK: creatine phosphokinase; EASI: eczema area and severity index; EASI 50/75/90: ≥ 50/75/90% improvement in EASI from baseline; GPCR19: G protein-coupled receptor 19; H4R: type 4 histamine receptor; IGA: investigator global assessment; IGA 0/1: “clear or almost clear” with ≥ 2-point improvement in IGA from baseline; I-NRS: itch numeric rating scale; JAK: janus kinase; KOR: kappa opioid receptor; LXR: liver X receptor; mEASI: modified eczema area and severity index; mEASI 50/75/90: ≥ 50/75/90% improvement in mEASI from baseline; NCT: national clinical trial identifier consultable on www.clinicaltrials.gov (website consulted last time as of April 30, 2024), only clinical trials in advanced phases (Phase II onwards) are listed; NK1R: neurokinin 1 receptor; PDE4: phosphodiesterase 4; Pan-JAK: multiple janus kinases inhibitor; P2X3: purinergic receptor P2X; SCORAD: scoring atopic dermatitis; SCORAD 50: ≥ 50% improvement in SCORAD from baseline; S1PR1/4/5: sphingosine 1-phosphate receptor 1/4/5; TYK: tyrosine kinase; WI-NRS: worst itch numeric rating scale; WI-NRS/I-NRS ≥ 4: ≥ 4-point change in WI-NRS/I-NRS from baseline
Over the last decade, MSCs therapy has grown to a position of serious alternative treatment for AD, following successful proof-of-concept preclinical studies.
In 2011, Ra et al. [152] reported case studies where intravenous injections of autologous human adipose-derived MSCs (hAdMSCs) where used as a compassionate treatment for 4 AD patients with exhausted therapeutic options. Patients were administered 6–10 × 108 hAdMSCs in total and followed up for 2 to 5 and half months after treatment. The treatment resulted in significant AD score improvement in treated patients with no adverse events recorded during the follow-up period, providing thus, the first evidence for clinical benefit of hAdMSCs in AD. The first-in-class clinical trial was published in 2016 [153], from a study that enrolled 34 moderate-to-severe AD adult patients with symptoms not adequately controlled by topical corticosteroids or systemic immunosuppressants. Patients received subcutaneous injections of low dose (2.5 × 107) or high dose (5 × 107) of hUCB-MSCs fortnightly for 4 weeks in Phase 1 trial, and 12 weeks in Phase 2. Half of the patients receiving the high dose of hUCB-MSCs experienced a 50% reduction of EASI score, while the investigator global assessment (IGA) and SCORAD index decreased by 33% and 50%, respectively. The treatment also reduced serum IgE and blood eosinophils levels, while appearing to be globally safe. In another study, conditioned media from hUCB-MSCs was investigated for its beneficial effects in 34 patients suffering from mild AD [154]. Topical application of conditioned media (contained in a cream formulation) twice daily for 4 weeks on patients’ lesions strengthened the skin barrier and reduced the trans-epidermal water loss. This outcome was attributed to the anti-inflammatory potency of the conditioned media, as it was found to inhibit the expression of TNF-α and IL-6 cytokines in HaCaT cells, as well as IL-4 and IL-13 levels in Th2 cells in vitro. Shin et al. [155] reported the use of allogenic bone marrow-derived clonal MSCs (bmMSCs), in a single-center investigator-initiated clinical trial, in five adult patients with moderate to severe AD. Patients received intravenous injections of bmMSCs (1 × 106 cells/kg) three times every 2 weeks during a 4 weeks period and were followed up the next 12 months. At week 16, 4 out of the 5 patients (80%) achieved EASI-50 after one or two treatment cycles and two patients maintained good clinical response for over 84 weeks, with significant improvement of IGA, SCORAD, and BSA. Minor adverse reactions such as cellulitis, otitis, and acute upper respiratory tract infection were recorded, but were only transient with full recovery of patients with, and often, without symptomatic treatment. Authors surmised that efficacy of bmMSCs would be related to increased levels of IL-17, associated to lower blood levels of CCL-17, IL-13, and IL-22. Other human clinical trials still in progress or which results are not yet published involve umbilical cord-derived MSCs (NCT01927705; NCT05004324; NCT03269773), adipose tissue-derived MSCs (NCT04137562; NCT02888704), bone marrow-derived MSCs (NCT04179760), or induced pluripotent stem cell-derived exosomes (NCT05969717).
Not only humans are affected by AD but dogs as well, with a prevalence of 10–15% worldwide [156]. The therapeutic use of MSCs has also been investigated in canine AD and to date, a handful of studies involving adipose tissue-derived MSCs treatment in canine AD (cAD) have been documented [38, 157–161]. Notwithstanding the positive outcomes conveyed, large scale placebo-controlled, long-term studies are necessary to further validate these results.
Also known as specific desensitization, hyposensitization or allergen-specific immunotherapy, allergen immunotherapy (AIT) involves the administration of gradually increasing quantities of relevant allergens in order to induce immunologic tolerance. This therapeutic option has yielded evidenced benefits in allergen-induced conditions like allergic asthma, allergic rhinitis and Hymenoptera venom allergy [162, 163]. Since AD is characterized by varied clinical presentations and heterogeneous phenotypes, AIT in AD is a controversial issue. Allergen-specific immunotherapy for AD has several indications, including exacerbation of symptoms following aeroallergen exposure, aeroallergen sensitization along with positive skin prick test (SPT) and/or sIgE production, moderate to severe SCORAD [25] and atopy patch testing (APT) positivity against the aeroallergen [164, 165]. Allergen extracts used for AIT primarily consist of allergenic proteins derived from grass/tree pollen, pet dander, dust mites, insect venom, and mold. House dust mite (HDM) extract, whose main trigger agents are Dermatophagoides pteronyssinus and Dermatophagoides farinae, is by far the most used allergen in AIT of AD [166]. Roughly, the proposed mechanism of long-term tolerance induction in AIT, even upon treatment discontinuation, involves desensitization of effector cells (basophils, mast cells, innate lymphoid cells, dendritic cells) through activation of allergen-specific regulatory T and B cells, downregulation of effector type 2 responses, decrease in the production of IgE and increase in production of allergen-specific blocking antibodies such as IgG2 and IgG4 [167]. AIT administration includes subcutaneous immunotherapy (SCIT) and sublingual immunotherapy (SLIT), both being well established as safe and effective treatments to address allergies to aeroallergens for allergic rhinitis and allergic asthma [2]. Since SCIT requires an injection-based administration, SLIT may offer a viable alternative to reluctant patients. Furthermore, SLIT reduces the utilization of healthcare resources and staff time, does not necessitate specialized expertise or facilities, and can be administered at home, making it a suitable option even for young children [166, 168]. However, both SCIT and SLIT can induce local and systemic reactions, requiring physicians and patients to be aware of the potential occurrence of such adverse events. Fortunately, manifestation of systemic life-threatening reactions, such as severe anaphylactic reactions, remains very uncommon [169]. Lately, the clinical efficacy of AIT in AD patients has been extensively investigated and the main outcomes of some recent studies are summarized in Table 4.
AIT studies in AD
| Study type | Allergen | Treatment duration | Number of patients enrolled | Complementary medication | Route | Global endpoints achieved | Adverse reactions reported | References |
|---|---|---|---|---|---|---|---|---|
| Open-label, controlled, randomized trial | HDM | 12 months | 60 | Moisturisers, topical and systemic drugs | SL | Significant increase of allergen-specific Ig4 | Oropharyngeal itching, localized lip swelling | [170] |
| Randomized controlled trial | HDM | 36 months | 440 | Topical glucocorticoids, skin moisturizers, oral antihistamines | SL | Significant improvement of SCORAD | Lip swelling, numbness, pruritus | [171] |
| Randomized controlled trial | HDM | 6 months | 14 | Moisturizers, antihistamine and mild-potency anti-inflammatory topical treatments | SL | Significant improvement of SCORAD and TEWL | Not available | [172] |
| Randomized, double-blind, placebo-controlled trial | HDM | 18 months | 66 | Topical corticosteroids, topical immunosuppressors, and systemic immunosuppressants | SL | Significant improvement of SCORAD | Vomiting, abdominal pain, aphthous ulcer, alopecia, dyspnea | [173] |
| Randomized, placebo-controlled, double-blind trial | HDM | 12 months | 37 | Oral antihistamines, topical medications | SC | Significant improvement of EASI | Mild local reaction, transient urticaria | [174] |
| Multi-centre, randomized, double-blind, placebo-controlled study | HDM | 36 weeks | 239 | Mometasone furoate cream, levocetirizine hydrochloride tablets, mupirocin ointment, clarithromycin tablets | SL | Significant improvement of SCORAD and SLA | Swelling in tough and lips, throat pain, diarrhoea, itching, numb in mouth | [175] |
| Randomized controlled study | HDM | 24 months | 96 | Oral levocetirizine hydrochloride tablets, topical fluticasone | SL | Significant reduction of SCORAD | Transient oral itching, gastrointestinal discomfort | [176] |
| Randomized controlled study | HDM | 12 months | 107 | Oral antihistamines and topical medications | SL | Significant increase of serum-specific IgG4 level | Transient oral itching, gastrointestinal discomfort | [177] |
| Randomized, double-blind, placebo-controlled study | HDM | 18 months | 56 | Short term use (3 days) of topical fluticasone propionate and/or oral hydroxyzine | SL | Significant reduction of SCORAD in mild-moderate patients | Tiredness, oral itching, headache, mouth, lips, and face swelling | [178] |
| Observational study | HDM; cat/dog dander | 2–58 months | 19 | Topical treatment and/or oral antihistamines | SL | Significant improvement of IGA and EASI | Not available | [179] |
| Multi-centre randomized, double-blind, placebo-controlled parallel group trial | HDM | 18 months | 168 | Topical and (as necessary) systemic medication | SC | Significant reduction of SCORAD in severe AD patients | Flare-ups of eczematous and urticarial lesions, symptoms of rhinitis, pruritus, transient headache, and asthma | [180] |
AD: atopic dermatitis; AIT: allergen immunotherapy; EASI: eczema area and severity index; HDM: house dust mite; IGA: investigator global assessment; SC: subcutaneous; SL: sublingual; SLA: skin lesion area; SCORAD: scoring atopic dermatitis; TEWL: transepithelial water loss
Besides immunotherapeutic strategies, harnessing improved formulations and delivery systems of existing molecules could go a long way to improve their clinical efficacy. Nanocarrier-based formulations for enhanced delivery of AD treatments have recently arisen as a promising approach prompted by positive deliveries in vivo. These alternate drug delivery systems include nanoemulsions (oil-in-water, water-in-oil, oil-in-water-in-oil, water-in-oil-in-water), liposomes, transfersomes, ethosomes, lipid nanoparticles (nanostructured lipid carrier, solid lipid nanoparticles), nanocrystals, polymeric nanoparticles (nanocapsules, nanospheres) and polymeric micelles [181, 182]. These nanocarriers have significant advantages over traditional formulations such as improved skin penetration and retention, controlled release, and reduced off-target effects, largely driven by optimized physicochemical features such as shape, size, structural design, and surface charge. Nevertheless, major drawbacks, including large-scale production, the scarcity of global regulatory guidelines, intellectual property rights, cost effectiveness [183], and lack of models that emulate the entire AD complexity, are still to be addressed on the path to clinical translation and commercialization.
A better stratification strategy of AD patients is also needed in achieving patient-specific treatments (precision medicine), by taking into account phenotypical features (as AD varies among different subpopulations), disease triggers, genetic hallmarks, and specific biomarkers. This would enable medical practitioners to identify AD endotypes more amenable to certain treatments (most probably in the form of combinatory therapies), consequently improving therapeutic efficacy while simultaneously limiting the issues of adverse side effects. This approach could be exemplified by a genuine use of the patient’s molecular profiles to identify appropriate therapeutic approaches for each patient. For instance, fezakinumab (an IL-22 antagonist) revealed significant clinical improvement, compared with placebo, especially in patients with higher IL-22 levels, through downregulation of Th1, Th2, and Th17-related biomarkers [184]. Developing easy-to-use and non-invasive techniques, such as skin tape strip sampling, would therefore facilitate diagnosis and then treatment based on the patient’s specific immune pathway [185]. Building such a sharp stratification strategy would further require the development of new and robust tools able to analyze a tremendous amount of highly complex data. Recent advances in artificial intelligence (AI) have shown promise in the diagnosis, classification, and predictive treatment of AD. Ghosh et al. [186], identified 89 genes expressed in AD to build a “signature” model, using a support vector machine, and achieved an AD predictive accuracy of 98%, compared to normal skin [186]. Furthermore, a recent machine learning tool successfully classified infant AD patients into various clusters, based on several chemokines and cytokines expression profiles [187]. Beyond diagnostic, scoring, and staging purposes, AI-based tools have been very recently applied in AD for drug repurposing, new drug eligibility, and prediction of the therapeutic response to biologics [188].
Understanding a complex disorder such as AD in its full phenotypic and mechanistic array remains a daunting goal, however, with the ever-increasing pathophysiology insights, improved patient management and outcomes are expected. Notwithstanding the limited armamentarium of approved targeted biological therapies at present, current trends in alternative diseases, particularly cancer, are certain to pave the way to address this unmet need for more well tolerated and narrow-targeted therapeutics. In addition to monoclonal antibodies which seem to offer safer effective systemic therapies, compared with the off-label use of broad-acting immunosuppressants, small molecules, AIT and cell-based therapy bring additional options addressing different causative factors within AD context. ADCs and immunocytokines, albeit still at their preclinical exploratory phase, also hold the promise of a possible breakthrough in AD therapeutic arsenal.
The advent of immunotherapy in the context of AD has marked a paradigm shift towards personalized medicine and has led to remarkable successes. However, major limitations remain, and ongoing efforts to further understand the complete picture of the complex pathophysiology of AD are expected to yield more effective immunotherapeutic agents. Integration of these immunotherapeutic strategies into a holistic approach involving optimized drug delivery systems and AI-based technologies has the potential to profoundly change the landscape of AD healthcare, with the promise of more customized treatments and a better quality of life for AD patients.
AD: atopic dermatitis
ADC: antibody-drug conjugate
AI: artificial intelligence
AIT: allergen immunotherapy
bmMSC: bone marrow-derived clonal mesenchymal stem cell
cAMP: cyclic adenosine monophosphate
ETA’: exotoxin A
FDA: Food and Drug Administration
hAdMSC: human adipose-derived mesenchymal stem cell
hCFP: human cytolytic fusion protein
hUCB-MSC: human umbilical cord blood mesenchymal stem cell
IgE: immunoglobulin E
JAK: janus kinase
MSCs: mesenchymal stem cells
PDE4: phosphodiesterase 4
rIT: recombinant immunotoxin
scFv: single-chain variable fragment
SCIT: subcutaneous immunotherapy
SCORAD: scoring atopic dermatitis
SLIT: sublingual immunotherapy
S1PR: sphingosine1-phosphate receptors
TNF-α: tumor necrosis factor-alpha
TSLP: thymic stromal lymphopoietin
VEGF-C: vascular endothelial growth factor C
GTS: Conceptualization, Writing—original draft, Writing—review & editing. EAF and MM: Writing—original draft. KN: Writing—review & editing. SB: Conceptualization, Writing—review & editing, Supervision, Funding acquisition. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This research was funded by the South African Research Chairs Initiative of the Department of Science and Technology (DST) and the National Research Foundation (NRF) [47904] of South Africa. The NRF and the South African Research Chair in Cancer Biotechnology have provided bursaries to support students contributing to this work. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Serap Maden
Perpetua U. Ibekwe ... Bob A. Ukonu
Luis Angel Hernández-Zárate ... Víctor González-Uribe
Antara Baidya, Ulaganathan Mabalirajan
