 Review
Review

Affiliation:
1Polytechnic Institute of Coimbra, Coimbra Health School, Biomedical Laboratory Sciences, 3046-854 Coimbra, Portugal
†These authors contributed equally to this work.
Affiliation:
1Polytechnic Institute of Coimbra, Coimbra Health School, Biomedical Laboratory Sciences, 3046-854 Coimbra, Portugal
2Research Centre for Natural Resources Environment and Society (CERNAS), Polytechnic Institute of Coimbra, 3045-601 Coimbra, Portugal
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0002-0157-6648
Affiliation:
1Polytechnic Institute of Coimbra, Coimbra Health School, Biomedical Laboratory Sciences, 3046-854 Coimbra, Portugal
3Clinical Pathology Service, Unidade Local de Saúde de Coimbra, EPE, 3004-561 Coimbra, Portugal
Affiliation:
4Department of Life Sciences, University of Coimbra, CFE-Centre for Functional Ecology: Science for People & Planet, Marine Resources, Conservation and Technology-Marine Algae Lab, 3000-456 Coimbra, Portugal
Email: leonel.pereira@uc.pt
ORCID: https://orcid.org/0000-0002-6819-0619
Explor Cardiol. 2024;2:114–133 DOI: https://doi.org/10.37349/ec.2024.00026
Received: March 21, 2024 Accepted: May 10, 2024 Published: June 24, 2024
Academic Editor: Andrea Borghini, Institute of Clinical Physiology - National Research Council (IFC-CNR), Italy
The article belongs to the special issue Molecular Mechanisms of Cardiovascular Aging
Metabolic syndrome (MetS) is known as a non-communicable disease (NCD) that affects more and more individuals. MetS is closely related to type 2 diabetes mellitus (T2DM), cardiovascular disease (CVD), obesity and inflammation. It is associated with T2DM due to the disturbance in insulin secretion/effect, eventually leading to insulin resistance (IR). The link between MetS and CVD is due to accelerated atherosclerosis in response to chronic inflammation. This literature review was based on a search in the PubMed database. All selected articles are written in English and cover a period of approximately 10 years (January 2014 to May 2023). The first selection used MeSH terms such as: “metabolic syndrome”, “type 2 diabetes mellitus”, “obesity”, “inflammation”, and “insulin resistance” and different associations between them. Titles and abstracts were analyzed. In the end, 44 articles were selected, 4 of which were meta-analysis studies. Currently, an individual is considered to have MetS if they present 3 of the following changes: increased waist circumference, increased triglycerides (TG), reduced high-density lipoprotein cholesterol (HDL-C), increased fasting blood glucose and hypertension. We believe this can often lead to a false diagnosis. The objective of this paper is to compile what we consider to be an appropriate panel of MetS indicators. The markers that stand out in this review are the lipid profile, anti- and pro-inflammatory function and oxidative stress. Considering the research, we believe that a complete panel, to correlate the most characteristic conditions of MetS, should include the following markers: TG/HDL-C ratio, small dense low-density lipoprotein cholesterol (SdLDL-C), lipid peroxidation markers, leptin/adiponectin ratio, plasminogen activator inhibitor-1 (PAI-1), activin-A and ferritin levels. Finally, it is important to expand research on the pathophysiology of MetS and confirm the most appropriate markers as well as discover new ones to correctly diagnose this condition.
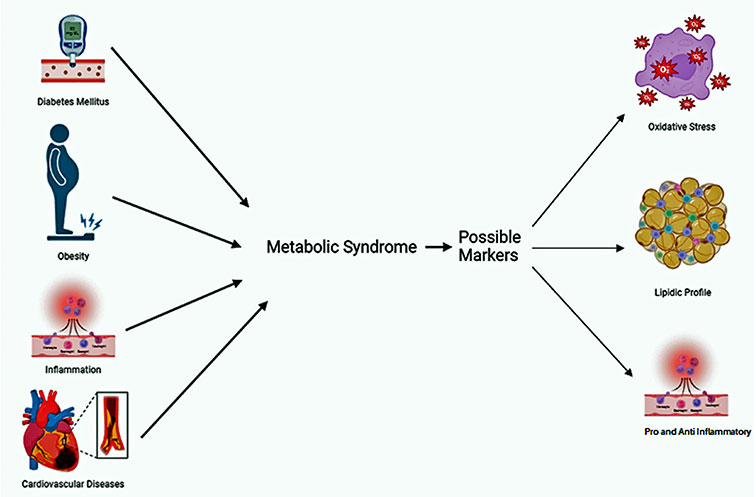
Factors associated with metabolic syndrome and possible biomarkers. Created in Biorender.com
The metabolic syndrome (MetS) was first introduced in 1988 as “Syndrome X” by Reaven [1]. MetS is the combination of several interconnected disorders characterized by hypertension, hyperinsulinemia, hyperglycemia, dysglycemia, dyslipidemia, obesity, and insulin resistance (IR) [2, 3]. When these conditions coincide and act together, they can result in the emergence of vascular and neurological complications, non-alcoholic fatty liver disease, and an elevated risk of cardiovascular disease (CVD). Those with MetS face a doubled risk of mortality and a tripled risk of experiencing a heart attack or stroke compared to those not afflicted by this syndrome. Moreover, individuals with MetS are five times more prone to developing type 2 diabetes mellitus (T2DM) [4–7].
This pathological condition is becoming increasingly common, and it is estimated that 20–30% of adults worldwide are affected by this syndrome, making it a global health problem [8]. However, the frequency and distribution of MetS vary according to the diagnostic criteria applied, which can be different according to factors such as ethnicity, culture, and geographical location [9].
MetS is characterized as a non-communicable disease (NCD) and it is affecting more and more people and for this reason it has been increasingly studied with the aim of understanding its pathophysiology and possible markers so that its screening can be done earlier and earlier. The World Health Organization (WHO) has set as a goal, by 2030, to “reduce premature mortality from NCD by one third through prevention and treatment” [10].
This review will focus on markers that can be studied in the laboratory’s routine, such as: lipid profile, markers of inflammatory function, as well as other types of molecules orientated to the pathology under investigation (Table 1).
Summary of laboratory markers (established or candidates) cited in the article, as marker of MetS
| Marker | Class | Variation | Type | Eligible | References |
|---|---|---|---|---|---|
| Waist circumference | Physical | Increase | Establish | Yes | Fahed et al. [3], 2022 |
| TG | Lipidic profile | Increase | Establish | Yes | Fahed et al. [3], 2022 |
| HDL-C | Lipidic profile | Decrease | Establish | Yes | Fahed et al. [3], 2022 |
| Fasting glucose | Lipidic profile | Increase | Establish | Yes | Fahed et al. [3], 2022 |
| Blood pressure | Lipidic profile | Increase | Establish | Yes | Fahed et al. [3], 2022 |
| TG/HDL-C | Lipidic profile | Increase | Candidate | Yes | Zhang et al. [17], 2022 |
| SdLDL-C | Lipidic profile | Increase | Candidate | No | Hirano et al. [24], 2022 |
| ApoB/ApoA1 | Lipidic profile | Increase | Candidate | No | Reynoso-Villalpando et al. [25], 2019 |
| OMEN | Inflammatory function | Decrease | Candidate | No | Makiel et al. [27], 2023 |
| Resistin | Inflammatory function | Increase | Candidate | No | Kim et al. [34], 2022 |
| Visfatin | Inflammatory function | Increase | Candidate | No | Berezin et al. [29], 2020 |
| Activin-A | Inflammatory function | Increase | Candidate | Yes | Peng et al. [42], 2018 |
| Leptin/Adiponectin | Inflammatory function | Increase | Candidate | Yes | Adejumo et al. [32], 2019 |
| TNF-α | Inflammatory function | Increase | Candidate | No | Berezin et al. [29], 2020 |
| IL-6 | Inflammatory function | Increase | Candidate | No | Berezin et al. [29], 2020 |
| PAI-1 | Inflammatory function | Increase | Candidate | Yes | Kim et al. [34], 2022 |
| AngII | Inflammatory function | Increase | Candidate | No | Ragino et al. [35], 2020 |
| NLRP3 | Inflammatory function | Increase | Candidate | No | Lu et al. [39], 2023 |
| TBARS, MDA, 4-HNE, and F2 isoproteins | Oxidative stress | Increase | Candidate | Yes | Masenga et al. [13], 2023 |
| Protein carbonyls, advanced glycation, ox-LDL, and advanced oxidation proteins | Oxidative stress | Increase | Candidate | No | Masenga et al. [13], 2023 |
| 8-Oxo-Dg, 5-chlorouracil, and 5-chlorocytosine | Oxidative stress | Increase | Candidate | No | Masenga et al. [13], 2023 |
| Xanthine oxidase, GGT, MPO, NOX, and NOS | Oxidative stress | Increase | Candidate | No | Masenga et al. [13], 2023 |
| Serum ferritin | Oxidative stress | Increase | Candidate | Yes | Masenga et al. [13], 2023 |
AngII: angiotensin II; GGT: gamma-glutamyl transferase; HDL-C: high density lipoprotein-cholesterol; IL: interleukin; MDA: malonyldialdehyde; MPO: myeloperoxidase; NLRP3: NOD-like receptor family, pyrine domain containing 3; NOS: nitric oxide synthase; NOX: NADPH oxidase; OMEN: omentin; ox-LDL: oxidized-low-density lipoprotein; PAI-1: plasminogen activator inhibitor-1; SdLDL-C: small dense low-density lipoprotein cholesterol; TBARS: thiobarbituric acid reactive substances; TG: triglycerides; TNF-α: tumor necrosis factor α; 8-oxo-Dg: 8-oxo-2-deoxyguanosine; 4-HNE: 4-hydroxy-2-nonenal; ApoA1: apolipoprotein A1; ApoB: apolipoprotein B
The writing of this review article was based on a search using PubMed as a database. A total of 636 articles were selected, all of them written in English and within a time frame of 10 years. This first selection used MeSH terms such as: “metabolic syndrome”, “inflammation”, “insulin resistance”, “type 2 diabetes mellitus”, and “obesity”.
Afterward, the titles were analyzed, resulting in a selection of 171 articles, followed by the analyze of the abstracts, resulting in a selection of 51 articles, 7 of which were eventually excluded since they still showed pertinent doubts about the possible use of a given marker and/or because they were studies that lacked scientific robustness. In the end, 44 articles were used, 4 of which are meta-analysis studies, with a time frame of 9 years (June 2015 to May 2023) as illustrated in the flow diagram (Figure 1).
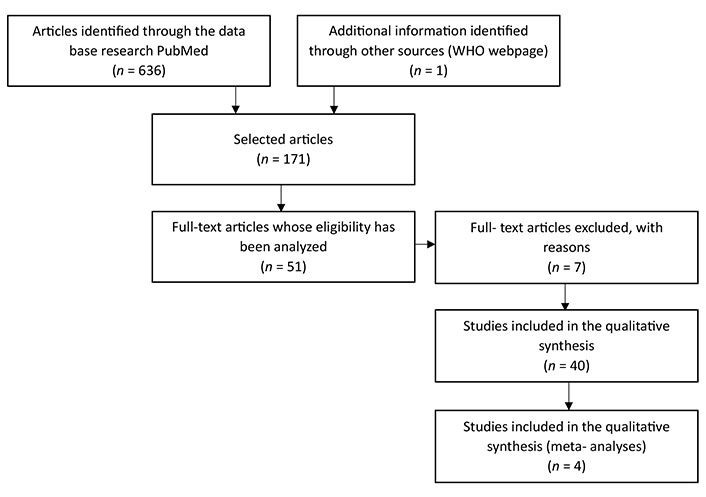
Flowchart—representative flowchart of the method and selection criteria used in the search and selection of articles. It was used the database PubMed. On the first search were used the following MeSH terms: “metabolic syndrome”, “type 2 diabetes mellitus”, “obesity”, “inflammation”, and “insulin resistance” and different associations between them. On the search were used the following criteria: articles in English with a tame frame of 10 years
The low number of articles selected, and the long-time frame are due to the fact that the search was specific to laboratory markers that could guide to the correct diagnosis of MetS. This specificity meant that the time frame had to be extended and that all the articles relating MetS to another type of illness (for example) had to be excluded.
MetS is the name given to the set of the following conditions: hyperglycemia, atherogenic dyslipidemia, IR, systemic arterial hypertension, and central/abdominal obesity. MetS was classified as a NCD by the WHO [10].
This syndrome has been linked to CVD, whereas these are associated with accelerated atherosclerosis in response to chronic inflammation and vascular endothelial dysfunction, and to T2DM, mainly due to a disturbance in insulin secretion or its effect and IR [3, 5].
An individual is considered to be suffering from this syndrome when presenting alterations in 3 of the following aspects: waist circumference [≥ 102 cm and ≥ 88 cm (to European men and women, respectively)], elevated triglycerides (TG; ≥ 150 mg/dL), reduced HDL-C [< 40 mg/dL and < 50 mg/dL (to European men and women, respectively)], elevated fasting glucose (≥ 100 mg/dL) and high blood pressure (≥ 130/85 mmHg) (Table 1) [3, 4, 11, 12].
Even though the pathophysiology of MetS is not fully discovered, there are some conditions that have been described as risk factors for the development of this syndrome: genetic and epigenetic factors, smoking, increasing age, obesity, low socioeconomic status, physical inactivity, excessive alcohol consumption, poor diet, as well as other types of lifestyle factors and behaviors [4, 5, 7].
Moreover, the gut microbiome is entangled in the pathophysiology of numerous chronic conditions, such as obesity, diabetes, dyslipidemia, and hypertension, all of which can contribute to the onset of MetS. Serving as the most diverse microbial assembly in the human body, the gut microbiome performs a pivotal role in sustaining physiological equilibrium by regulating host nutrition, energy generation, epithelial stability, immune responses, and drug metabolism, thereby ensuring overall homeostasis [13].
Circadian rhythms consist of physiological, cognitive, and behavioral changes that adhere to a 24-h cycle. These natural processes primarily react to light and darkness, influencing the majority of living organisms. It constitutes an inherent molecular timekeeping system found in virtually all cells and tissues. It is an important element in homeostatic regulation, controlling a large variety of genes involved in cellular metabolism. An example, of a light-related circadian rhythm is sleeping at night and being awake during the day. Diseases like obesity, T2DM, mental illness, among others, have been associated with circadian clock disturbances [14, 15].
Alkhulaifi and Darkoh [7] demonstrated that the timing and frequency of meals—eating frequent meals and eating in the morning can have a protective effect on this syndrome; increasing physical activity [16], which has been shown to be an effective way of reducing body weight and the accumulation of visceral fat, controlling blood pressure, improving HDL-C and TG values and weakening IR; eating a healthy diet (e.g., Mediterranean diet) and good circadian rhythms can reduce the risk of MetS [7].
The global prevalence of obesity has surged over the last five decades, prompting the classification of this rise as an “epidemic” and solidifying obesity and MetS as major public health challenges. Obesity has evolved into a worldwide health crisis due to its substantial elevation of the risk for various diseases, including T2DM, CVD, hypertension, fatty liver disease, and various forms of cancer. Consequently, it detrimentally impacts both quality of life and life expectancy [17, 18]. Evidence suggests that the gut microbiome significantly contributes to obesity development through its interaction with host metabolism, given its crucial role in maintaining physiological function. When there’s a disruption in the composition of the intestinal microbiome, it can lead to a proliferation of pathogenic species, triggered by factors like infections, antibiotic usage, diseases, dietary habits, and lifestyle choices. This imbalance heightens the risk of MetS by fostering inflammation and escalating levels of reactive oxygen species (ROS) and oxidative stress [13]. Obesity and MetS share the common feature of excessive body fat accumulation, yet they are distinct entities [15]. Experimental studies in both animals and humans have demonstrated that obesity primarily arises from overconsumption of macronutrients and/or diminished energy expenditure. This, in turn, leads to disturbances in lipid and glucose regulation [19], in other words, the accumulation of excess body fat. This accumulation can lead to changes in gut microbiome function. These studies also suggest that the intestinal microbiome regulates the accumulation of fat in the host, thereby influencing obesity. This is a metabolic condition in which calorie intake exceeds consumption, resulting in fat cell hypertrophy and visceral fat accumulation. Adipose tissue primarily consists of lipids, such as TG and free cholesterol. The principal secretion from adipose tissue is free fatty acids (FFA), whose elevation contributes to various metabolic disorders [13]. Conversely, MetS is a condition involving disturbances in energy accumulation and utilization, driven by low-grade systemic inflammation. This leads to central adiposity, hypertension, dyslipidemia, and/or IR [19].
Thus far, studies have consistently shown a strong correlation between obesity and blood lipid levels. As a result, circulating lipid components serve as readily accessible biomarkers for predicting obesity and other metabolic disorders [17].
Insulin, a peptide hormone, is released by pancreatic beta cells when blood glucose levels are elevated. It functions by exerting anabolic effects, inhibiting lipolysis and hepatic gluconeogenesis, and simultaneously enhancing glucose uptake in the liver, muscles, and adipose tissues [3].
The main potential risk factors for MetS are abdominal obesity (AO) and IR. Pre-diabetes is also considered part of MetS, due to it association with IR and is highly predictive of new-onset T2DM [4]. DM comprises a cluster of metabolic disorders marked by elevated blood glucose levels resulting from disruptions in insulin secretion, action, or both. Globally, approximately 415 million individuals are affected by diabetes, and projections indicate that this figure could escalate to 642 million by 2040. Persistent hyperglycemia associated with T2DM is linked to enduring harm and debilitating, potentially life-threatening health complications, encompassing CVD, neuropathy, and nephropathy [20]. When IR manifests in adipose tissues, the ability of insulin to inhibit lipolysis is impaired. This leads to a rise in circulating FFA, triggering alterations in the insulin signaling pathway across multiple organs. Consequently, this exacerbates IR, setting off a detrimental cycle of worsening IR [3].
In muscles, FFA disrupt insulin receptor substrate-1 (IRS-1)-associated PI3K activity, hindering the translocation of GLUT-4 to the cell surface, thus reducing glucose uptake. Additionally, FFA stimulate gluconeogenesis and lipogenesis in the liver. This prompts a state of hyperinsulinemia to sustain normal glucose levels. However, this compensatory mechanism eventually falters, resulting in decreased insulin levels. FFA’s lipotoxic effect on pancreatic beta cells exacerbates this decline. Furthermore, elevated FFA concentrations promote the synthesis of cholesterol esters and TG, leading to the production of very low-density lipoprotein (VLDL) abundant in TG. These VLDL activate cholesterol ester transfer protein (CETP), facilitating the transfer of TG from VLDL to HDL-C, thereby reducing HDL-C concentrations. Moreover, TG-rich low-density lipoprotein (LDL), formed after the exchange for LDL cholesterol ester, undergo hydrolysis by lipoprotein or hepatic lipase (HL), generating cholesterol-depleted, small dense LDL (SdLDL) particles. These alterations in lipoprotein levels characterize the atherogenic dyslipidemia associated with IR in MetS [3, 21].
IR contributes to MetS by promoting the development of hypertension. This occurs partly due to the loss of insulin’s vasodilatory effect and FFA-induced vasoconstriction, which leads to the production of ROS and subsequent elimination of nitric oxide (NO). Other mechanisms involve heightened sympathetic stimulation and renin-induced sodium reabsorption in the kidneys. Additionally, IR elevates serum viscosity, induces a pro-thrombotic state, and triggers the release of pro-inflammatory cytokines from adipose tissue. These factors collectively elevate the risk of CVD and T2DM [3].
For many years, IR has been predominantly linked to its insufficient impact on glucose metabolism, overlooking its broader actions. However, insulin is a multifaceted hormone with diverse effects on lipid and protein metabolism, ion and amino acid transport, cell cycle regulation, proliferation, differentiation, and NO synthesis. In the vascular system, insulin stimulation prompts vasodilation via NO production, but an IR state disrupts NO synthesis and vasodilation. IR contributes to elevated blood pressure through various mechanisms, including heightened tissue angiotensin II (AngII) and aldosterone activities, increased sympathetic nervous system activation, and oxidative stress [22].
In 2020, Mancusi and colleagues [22] documented the correlation between hypertension, IR, and subsequent hyperinsulinemia. They found that untreated individuals with hypertension exhibit elevated fasting and postprandial insulin levels compared to normotensive individuals. Moreover, there exists a direct correlation between plasma insulin concentrations and blood pressure [22].
The connection between IR and hypertension is intricate and influenced by various factors, encompassing genetic predisposition and environmental elements. Sedentary lifestyles and excessive caloric intake prevalent in Western countries are foundational in fostering IR, primarily through epigenetic alterations. Specifically, mechanisms such as DNA methylation, histone modifications, and the activity of non-coding RNA, such as micro-RNA (miRNA), play pivotal roles in modifying protein transcription and expression, thereby influencing cellular phenotype [22].
The translocation of GLUT-4 to the cell membrane stands as a crucial step in insulin-triggered glucose uptake. In the state of IR, there’s a notable reduction in GLUT-4 expression levels and impaired translocation. Specifically, within myocytes, miRNA-106b disrupts insulin signaling by diminishing the insulin-stimulated translocation of GLUT-4. Additionally, epigenetic alterations can impact mitochondrial function, which is also pivotal in the development of IR. Methylation of the gene encoding peroxisome proliferator-activated receptor alpha (PPAR-α) has been observed in obese individuals. This data underscores the notion that epigenetic modifications likely contribute, at least in part, to the association between lifestyle habits and IR [22].
CVD is a group of disorders of the heart and blood vessels that includes pathologies, such as: coronary artery disease, cerebrovascular disease, congestive heart failure, and peripheral vascular disease. The primary cause of vascular disease worldwide is atherosclerotic CVD (ASCVD) [4]. In 2019, it is estimated that 17.9 million people died from CVD, which represents 32% of all deaths worldwide, and out of the 17 million premature deaths (under the age of 70) due to NCD, 38% were caused by CVD [23].
Although genetics plays a significant role in CVD, the main risk factors result from lifestyle, such as: dyslipidemia, hypertension, smoking, and altered glucose metabolism [4]. Hypertension stands as one of the primary cardiovascular risk factors associated with MetS. Numerous factors contribute to its onset, encompassing genetics, environmental influences, adaptive, and endocrine factors, as well as hemodynamic forces. Contemporary research suggests that oxidative stress plays a substantial role in driving the development of hypertension [2, 22]. Oxidative stress and persistent inflammation have been associated with endothelial impairment and vascular dysfunction, cardiovascular restructuring, renal dysfunction, heightened activation of the sympathetic nervous system, stimulation of immune cells, and systemic inflammation, all contributing to hypertension and CVD. In hypertension, key sources of ROS encompass hyperactivation of non-phagocytic NADPH oxidase (NOX), uncoupling of NO synthase (NOS), activity of xanthine oxidase, mitochondrial stress, and endoplasmic reticulum stress [2, 22].
MetS is associated with a range of cardiovascular disorders, including microvascular dysfunction, coronary atherosclerosis and calcification, cardiac dysfunction, myocardial infarction, and heart failure. The risk and severity of CVD escalate when MetS components are present in combination [2, 22]. In contrast to other risk factors associated with MetS, hypertension is not only recognized as a significant risk factor for CVD but is also acknowledged as a central characteristic of MetS itself. Elevated blood pressure not only exacerbates cardiovascular cellular damage but also poses a threat to the functionality of vital organs like the kidneys and lungs, crucial in the progression of CVD and ultimately, MetS [13].
Inflammation plays a key role in metabolic diseases involving IR, such as: obesity, MetS, and T2DM [18]. Chronic low-grade inflammation plays a significant role in the development of obesity and IR, thus closely linking it to the metabolic abnormalities seen in MetS. Intestinal permeability’s pivotal role in chronic low-grade inflammation positions the microbiome as a key contributor to the inflammatory processes underlying metabolic defects. Inflammation is characterized as a sequence of reactions by vascularized tissue in response to injury or infection [14].
MetS is characterized by a distinctive clinical feature: a persistent low-grade inflammatory condition resulting from the clustering of various metabolic risk factors [22]. T2DM is a metabolic disorder with multiple etiologies, characterized by chronic hyperglycemia arising from deficiencies in insulin secretion and/or action [20]. Additionally, CVD may be associated with heightened levels of oxidative stress biomarkers and diminished antioxidant capacity, indicating the connection between MetS, a pro-inflammatory state, and adverse health outcomes [12]. MetS is acknowledged as a state characterized by inflammation and increased blood clotting, with adipose tissue playing a central role in its development. Adipose tissue is now recognized as a biologically active organ, capable of both endocrine and paracrine functions. In response to excessive nutrition, adipocytes undergo hypertrophy and hyperplasia, potentially outgrowing their blood supply and inducing a state of hypoxia. Hypoxia can lead to cell necrosis with macrophage infiltration and high production of adipocytokines and other mediators [5], such as: interleukin-6 (IL-6), C-reactive protein (CRP) and tumor necrosis factor alpha (TNF-α). Significant changes in these levels are evident in individuals with MetS. As previously mentioned, IR, and obesity-induced systemic oxidative stress trigger inflammatory processes, culminating in tissue fibrosis and, consequently, CVD [3].
Diabetes stands as a significant contributor to ASCVD, with diabetes-related dyslipidemia closely linked to a heightened prevalence of ASCVD, irrespective of hyperglycemia. Diabetic dyslipidemia manifests through elevated TG, reduced HDL-C, and an abundance of SdLDL particles. Furthermore, individuals with diabetes exhibit a preference for triglyceride-enriched lipoproteins across all lipoprotein spectrums, including LDL-C [24]. HDL-C exerts protective effects against atherosclerosis, apoptosis, and inflammation, while also inhibiting the oxidation of LDL-C. Additionally, HDL-C facilitates reverse cholesterol transport, aiding in the removal of cholesterol from arteries and its transport to the liver. Apolipoprotein A1 (ApoA1), the primary constituent of HDL-C, plays a pivotal role in maintaining cellular cholesterol balance, as well as contributing significantly to immune function and possessing anti-platelet properties. Conversely, apolipoprotein B (ApoB) is present on the surface of atherogenic proteins like LDL-C and VLDL cholesterol (VLDL-C) [25].
Zhang et al. [17], exposes the atherogenic index of plasma (AIP), which is calculated by the log10 (TG/HDL-C), as a simple biomarker for predicting the risk of CVD or MetS. They, also, concluded that IR and hyperlipidemia could be identified in patients at high risk of CVD, that is, they would have a high TG/HDL-C ratio (depending on the value established for each population) [17]. The AIP has demonstrated superior predictive capabilities for CVD and obesity when compared to conventional biomarkers. Elevated levels of LDL-C typically signal a heightened risk of coronary artery disease events. However, it’s noteworthy that a considerable number of CVD patients do not display elevated LDL-C levels [17]. The TG/HDL-C ratio has a good diagnostic accuracy in predicting the presence of IR, dyslipidemia, and MetS, as demonstrated by Lechner et al. [20]. Lipoprotein particles circulating in the blood can be categorized based on their size and density. SdLDL represents a distinct subclass of LDL that can be isolated from other LDL particles through methods such as ultracentrifugation or gradient gel electrophoresis. Unlike larger LDL particles, SdLDL is particularly prone to oxidation and plays a significant role in the formation of arterial plaque, contributing to the development of atherosclerotic lesions. Consequently, SdLDL has been recognized as a cardiovascular risk factor by the National Cholesterol Education Program [17]. Numerous studies have consistently shown that SdLDL cholesterol (SdLDL-C) outperforms LDL-C in predicting CVD. A recent report from the ARIC (Atherosclerosis Risk In Communities) study suggests that LDL-C, TG may serve as a sensitive marker for predicting both coronary heart disease and cerebrovascular disease [24]. HL plays a role in the conversion of triglyceride-rich LDL into SdLDL, suggesting a precursor-product relationship between LDL, TG and SdLDL. While SdLDL-C has been extensively researched, the characteristics of LDL-TG remain less understood. Nevertheless, given the derivation of SdLDL from triglyceride-rich LDL, a significant correlation between LDL-TG and SdLDL-C is unsurprising. Despite their proposed precursor-product relationship, these risk factors exhibit distinct properties. SdLDL-C shows a strong correlation with factors related to MetS, such as increased visceral fat area, elevated systolic blood pressure, high blood glucose levels, fatty liver, and a high triglyceride/low HDL-C ratio. In contrast, LDL-TG demonstrates limited associations with these MetS-related factors. SdLDL are primarily derived from large triglyceride-rich VLDL1, with VLDL1 production boosted by IR in the liver and excess FFA supplied by adipose tissue [24]. Hirano and colleagues [24] noted a strong association between LDL-TG levels and variation in the HL region. They also found that alleles associated with elevated LDL-TG were linked to low HL activity. Previous studies have shown that HL activity is boosted by IR and is elevated in T2DM. Consequently, it’s speculated that there exists an inverse relationship between SdLDL-C and LDL-TG with HL activity; low HL activity could lead to increased LDL-TG levels but decreased SdLDL-C. This phenomenon could partially explain why SdLDL-C correlates with MetS-related factors while LDL-TG does not [24].
The ApoB/ApoA1 ratio serves as an indicator of the equilibrium between atherogenic and anti-atherogenic elements in plasma. These risk factors are intertwined with the components of MetS, potentially leading to cardiovascular complications. Nevertheless, there remains debate regarding the insights garnered from this marker. While some studies have suggested an independent association between the ApoB/ApoA1 ratio and T2DM, others have posited it as an independent predictor of IR, advocating for its inclusion in forthcoming clinical guidelines [25]. In the European population, researchers discovered that the ApoB/ApoA1 ratio, as well as ApoB and ApoA1 individually, served as highly predictive markers of cardiac risk. Recent findings indicate that the ApoB/ApoA1 ratio outperforms ApoB, ApoA1, cholesterol, and HDL-C individually in predicting MetS. This aligns with previous investigations conducted in the Caucasian population. However, further studies are necessary to validate the prognostic significance of this ratio in both MetS (Table 1) and cardiovascular risk [25].
MetS is characterized as a condition that triggers inflammation and thrombosis, with adipose tissue playing a central role in its development. In individuals with a lean physique, adipose tissue releases anti-inflammatory adipokines like omentin (OMEN)-1, transforming growth factor beta (TGF-β), IL-10, IL-4, IL-13, IL-1 receptor antagonist (IL-1Rα), adiponectin, and apelin. Conversely, in obese individuals, adipose tissue predominantly releases pro-inflammatory substances such as TNF-α, IL-6, leptin, resistin, visfatin, plasminogen activator inhibitor-1 (PAI-1), and AngII (Table 1) [26].
OMEN exists in two forms: OMEN-1 and OMEN-2, with OMEN-1 being the predominant isoform in the human body. Primarily produced in visceral fat tissue, OMEN-1 exhibits anti-inflammatory properties, with its circulating concentration showing negative correlations with waist circumference, IR, body mass index (BMI), and glucose intolerance. Gender differences also influence OMEN-1 levels, with women typically having higher concentrations than men. The OMEN level holds potential for predicting metabolic outcomes or diseases associated with obesity. Additionally, OMEN demonstrates positive associations with serum adiponectin levels and negative correlations with leptin levels. In vitro studies have revealed that OMEN-1 promotes vasodilation by upregulating endothelial NO synthase while downregulating TNF-α [27, 28]. In addition to the observed low serum concentrations of OMEN in patients with T1DM and T2DM, Makiel et al. [27] discovered that serum levels of OMEN-1 were notably diminished in individuals with coronary atherosclerosis. This suggests that OMEN-1 levels could serve as a biomarker for vascular dysfunction and atherosclerosis. Furthermore, lower levels of OMEN-1 were correlated with increased cardiac events, such as death and re-hospitalization, as well as conditions including carotid disease, coronary artery disease, and heart failure. Given these findings, serum OMEN-1 is increasingly recognized as a biomarker for coronary artery disease, obesity, cancer, MetS, inflammatory diseases, atherosclerosis, and DM [27, 28].
Adiponectin is an adipokine that is anti-atherogenic, anti-inflammatory, and anti-diabetic [27]. Adiponectin, secreted by adipocytes, exists in several isoforms. Its biological role involves enhancing the β-oxidation of FFA, promoting glucose transport activation, and inhibiting gluconeogenesis in target organs like the liver, heart, and skeletal muscle. This occurs through the activation of AMP-activated protein kinase (AMPK), p38 mitogen-activated protein kinase, and peroxisome PPAR-α, consequently mitigating IR. To ensure the biological response, specific receptors (AdipoR1 and AdipoR2) are present. Adiponectin’s ability to boost the expression of anti-inflammatory IL-10 suggests its role in suppressing the NF-κB signaling pathway, thereby downregulating TNF-α and curtailing inflammatory responses [16, 29].
It’s been proposed that elevated adiponectin levels serve as a compensatory mechanism, aiding in overcoming metabolic dysregulation and adiponectin resistance to thwart the progression of heart failure. This is particularly significant given the compromised perfusion in target organs linked to heart failure, encompassing skeletal muscles, liver, heart, vasculature, and kidneys. This compromised perfusion is associated with the uncoupling G-protein, which integrates into the structure of adiponectin receptors AdipoR1 (present in skeletal muscles, heart, vasculature, and kidneys) and AdipoR2 (found in the liver). AdipoR1 serves as a potent regulator of peroxisome PPAR-γ coactivator-1-α and mitochondrial Ca2+-dependent ionic channels, as well as the AMPK/sirtuin 1 (SIRT1) signaling pathway [29]. On the contrary, AdipoR2 facilitates the transmission of tissue-protective signals via the ubiquitin-proteasome pathway and insulin receptor tyrosine phosphorylation. Moreover, miRNA-150, which opposes the G-subunit of AdipoR2, could potentially contribute to adiponectin resistance in heart failure. Consequently, the tissue expression of AdipoR2 was notably reduced in cases of advanced heart failure [29].
Adiponectin plays a crucial role in MetS, DM, obesity, and atherosclerosis. The interrelation among these conditions emerges when disruptions occur in both adiponectin receptors, impairing adiponectin’s capacity to bind and enhance glucose and lipid metabolism. Consequently, there’s a transition from aerobic glucose metabolism to anaerobic pathways of glucose oxidation, altering the uptake of FFA by skeletal muscle and inducing lipid peroxidation and mitochondrial dysfunction. This cascade ultimately leads to IR. While adiponectin typically promotes FFA uptake by skeletal muscle and mitochondrial biogenesis, oxidative and mitochondrial stress can diminish its efficacy in glucose utilization and fatty acid enhancement [29].
The primary functional antagonists of adiponectin include leptin and resistin, both of which exert biological effects on energy homeostasis and target tissue metabolism that are diametrically opposed to those of adiponectin. Notably, circulating levels of adiponectin demonstrate an inverse correlation with glucose, insulin, TG concentrations, liver fat content, and BMI [29].
TNF-α is a widely recognized adipocytokine synthesized by vascular smooth muscle cells, adipocytes, and antigen-presenting cells. It plays a pivotal role in modulating both local and systemic inflammation, as well as influencing the immune response and IR [29]. TNF-α synthesis correlates directly with adipose tissue mass, and it has been observed to act as a paracrine mediator, diminishing IR in adipocytes. However, TNF-α exerts its pathogenic effects by impairing insulin signaling in both adipocytes and hepatocytes, leading to diminished metabolic responses to insulin. Furthermore, TNF-α contributes to IR by promoting hepatic lipolysis, consequently elevating circulating levels of FFA [30]. IL-6, a potent inflammatory cytokine, plays a crucial role in the development of IR and T2DM. Macrophages and adipocytes release IL-6, which regulates lipid and glucose metabolism and contributes to IR through intricate mechanisms. Elevated IL-6 levels are observed in the adipose tissue of individuals with DM and obesity, as well as in those with MetS. Epidemiological research links increased IL-6 concentrations with conditions like hypertension, atherosclerosis, and cardiovascular events. In murine studies, chronic exposure to IL-6 resulted in IR accompanied by high blood sugar levels. Moreover, elevated IL-6 levels stimulate the production of acute phase reactants such as CRP. Notably, high CRP levels exhibit the strongest correlation with cardiac events, DM, and MetS [5, 18, 30].
Adipokines play a pivotal role in regulating critical biological functions across various organs, including the endocrine pancreas, skeletal muscle, brain, immune system, cardiovascular system, and liver. This intricate network sheds light on the profound association between obesity and its metabolic as well as cardiovascular complications. The dysregulation of numerous adipokines in obesity can disrupt appetite and satiety mechanisms, alter adipose tissue distribution and homeostasis, impair endothelial function, insulin secretion, and sensitivity, provoke inflammation, affect energy expenditure and angiogenesis, elevate blood pressure, and influence osteoarticular functions and reproduction [26]. Leptin, originating from adipocytes, is a versatile hormone with a primary function of regulating glucose homeostasis and insulin sensitivity. It achieves this by curbing food intake and boosting energy expenditure. Leptin resistance emerges in individuals with obesity and MetS. Serum leptin levels correspond directly to obesity levels and mirror the body’s energy status. The resistance to leptin is a pivotal factor driving the advancement of MetS, necessitating deeper investigation for comprehensive understanding. Insufficient leptin levels may contribute to the onset of T2DM, CVD, and certain cancers [27, 30]. Numerous studies have indicated a significant positive correlation between leptin levels and IR, AO, T2DM, and heart failure. Additionally, a robust positive relationship exists between leptin concentration and adipose tissue mass, suggesting that elevated leptin levels (hyperleptinemia) and tissue leptin tolerance could serve as potential markers for the onset of MetS [13, 31]. Leptin operates as a physiological counterforce to adiponectin by binding to specific receptors and initiating the JAK-STAT3 signal transduction pathway. Structurally akin to IL-2 and growth hormone 1, leptin orchestrates both innate and adaptive immune responses and augments the pro-inflammatory potential of TH1 lymphocytes and macrophages. By stimulating the JAK2-STAT3 pathway, leptin heightens the production of various pro-inflammatory cytokines, including IL-2, IFNγ, TNF-α, and CC-chemokine ligands (CCLs, CCL3, CCL4, and CCL5). Consequently, leptin significantly enhances the migratory and proliferative capabilities of mononuclear cells and monocytes, while also triggering the release of free radicals, exacerbating oxidative stress, and ultimately culminating in cellular damage and clinical manifestations of MetS [13, 29]. Falahi et al. [30] and more recently, Adejumo et al. [32] proposed that the ratio of leptin and adiponectin is a better marker for predicting the risk of MetS than leptin and adiponectin alone. High levels of adiponectin and low levels of leptin could offer some protection against developing MetS. However, further studies are needed to ascertain this [32].
Resistin belongs to a group of small, secreted cysteine-rich proteins with hormone-like functions that trigger inflammatory pathways. It serves as an adipokine and has shown a positive correlation with fat mass and IR. Recent findings indicate that increased levels of resistin are linked to the occurrence and seriousness of coronary artery disease, heart failure, and adverse cardiovascular events. Investigations into the mechanisms by which resistin might promote atherogenesis and contribute to unfavorable cardiovascular outcomes have been conducted in both animal models and human subjects [9, 33].
Resistin reduces glucose uptake in skeletal muscle cells independently of insulin-activated pathways. Additionally, it promotes hepatic gluconeogenesis and is associated with decreased insulin sensitivity, fostering inflammation in adipose tissue, heightened lipolysis leading to increased serum FFA and glycerol, and the accumulation of FFA in skeletal muscle. Resistin has been implicated in the formation of foam cells by enhancing lipid accumulation in macrophages. Elevated resistin levels have been detected in human atherosclerotic lesions and the serum of individuals with premature coronary artery disease, suggesting its significant role in CVD and MetS. More recently, a potential mechanism for resistin’s pro-atherogenic role has emerged. It’s proposed that resistin diminishes the expression of the LDL receptor (LDLR) in human hepatocytes in a PCSK9-dependent manner. Consequently, resistin influences serum lipid metabolism and CVD by modulating PCSK9-induced LDLR expression. Additionally, a positive correlation has been observed between resistin levels and various clinical parameters including waist circumference, systolic and diastolic blood pressure, plasma glucose, serum triglyceride level, serum cholesterol level, serum VLDL level, plasma insulin level, and IR [34].
Visfatin, also known as nicotinamide phosphoribosyl transferase (NAMPT), functions as an anti-inflammatory enzyme with recognized growth factor activity. It plays a crucial role in the biosynthesis pathway of NAD+. In humans, plasma levels of visfatin escalate during the progression of obesity, T2DM, MetS and acute myocardial infarction [13, 34].
A direct relationship exists between plasma visfatin levels and TG, while an inverse correlation is observed with HDL-C levels and OMEN-1 [29]. Visfatin is actively secreted by both macrophages and adipocytes, circulating throughout the body where it regulates oxidative stress, the immune response, apoptosis, and inflammation via SIRT1-dependent and MAP kinase ERK1/2-related pathways. It’s established that visfatin binds to insulin receptor 1, exerting an insulin-like effect. As adipocytokines with diverse effects, visfatin contributes to IR, inhibits oxidative stress and inflammation in adipose tissue, fosters vascular repair, and suppresses ischemia-induced apoptosis of cardiac myocytes, primarily by up-regulating pro-inflammatory cytokines like TNF-α. Nonetheless, further research is necessary for a comprehensive understanding of visfatin’s role in MetS [13, 34].
PAI-1 is a serine protease inhibitor and acts to inhibit tissue plasminogen activator and vitronectin. It is pro-thrombotic and synthesized in adipose tissue, playing a role in atherothrombosis and in adipose tissue development, as well as in controlling insulin signaling. Elevated levels of circulating PAI-1 are observed in obese individuals with MetS and patients with T2DM. A positive correlation exists between the severity of MetS and PAI-1 plasma concentration. Moreover, PAI-1 levels are positively associated with various MetS components such as BMI, blood pressure, LDL-C, and IR measured by the homeostatic model assessment (HOMA-IR). Previous research indicates that plasma PAI-1 levels rise with obesity and decrease with weight loss. The precise mechanism underlying adipocyte dysregulation remains unclear, although obesity-induced oxidative stress is suggested to play a role. Human and animal studies demonstrate a positive correlation between fat accumulation and oxidative stress, characterized by increased production of ROS and upregulation of NOX, alongside reduced expression of antioxidant enzymes. Experiments on cultured adipocytes from both PAI-1+/+ and PAI-1–/– mice reveal that adipocyte-specific deletion of PAI-1 enhances IR by fostering glucose uptake and adipocyte differentiation. Furthermore, in obese mice, PAI-1 mRNA expression in visceral fat rises proportionally with obesity severity. In a mouse model of diet-induced obesity, inhibiting plasma PAI-1 activity led to improvements in hyperlipidemia. As previously noted, PAI-1 plays a role in atherothrombosis as well. Deleting PAI-1 inhibited carotid artery atherosclerosis, while pharmacological PAI-1 inhibition mitigated atherosclerosis in an obese mouse model of MetS by suppressing macrophage accumulation and cellular senescence within atherosclerotic plaques [5, 21, 34].
The association between the renin-angiotensin system and IR is widely recognized. Dysregulation of this system, commonly seen in hypertension, is believed to hinder insulin signaling, thus promoting IR. Proposed pathophysiological mechanisms include systemic vasoconstriction triggered by renin-angiotensin system activation, resulting in reduced blood flow that impacts glucose and insulin delivery to skeletal muscle cells. Additionally, this activation may induce oxidative stress via ROS production and directly impede insulin signaling at the cellular level [19]. Certain observations indicate that the pathophysiological mechanisms driving IR in hypertensive individuals may differ somewhat from those contributing to decreased insulin sensitivity in diabetics. AngII, via the angiotensin type 1 receptor, hampers insulin activity by eliciting ROS production through NOX. These ROS serve as significant intracellular second messengers [22]. The production of ROS is implicated in AngII-induced IR. Over recent decades, it has become apparent that IR is not simply a metabolic anomaly but rather a multifaceted syndrome that can impact blood pressure regulation. Dysregulation of the neurohumoral and neuroimmune systems contributes to the pathophysiology of both IR and hypertension, fostering chronic low-grade inflammation that disrupts insulin signaling. Molecular abnormalities associated with IR include deficiencies in insulin receptor structure, number, binding affinity, and/or signaling capacity. For instance, hyperglycemia hampers insulin signaling by generating ROS, which hinder insulin-induced tyrosine autophosphorylation in the insulin receptor. Other mechanisms inhibiting insulin signaling include proteasome-mediated degradation of IRS-1/2, phosphatase-mediated dephosphorylation, and kinase-mediated serine/threonine phosphorylation of both the insulin receptor and IRSs. IR also plays a pivotal role in the pathogenesis and advancement of target organ damage induced by hypertension, such as left ventricular hypertrophy, atherosclerosis, and chronic kidney disease [2].
Taken together, these abnormalities substantially elevate the risk of developing T2DM. In vascular smooth muscle cells extracted from the rat thoracic aorta, AngII notably diminishes IRS-1 protein levels via ROS-mediated phosphorylation of IRS-1 at Ser307, followed by proteasome-dependent degradation. ROS has also been validated by in vivo investigations as a pivotal player in the pathogenesis of AngII-induced IR. Specifically, chronic infusion of AngII in rats induces hypertension, diminishes insulin-triggered glucose uptake during hyperinsulinemia-euglycemic clamp, and elevates plasma cholesteryl ester hydroperoxide levels, indicating heightened oxidative stress [2]. Conversely, in endothelial cells, AngII triggers IR by phosphorylating IRS-1 at Ser312 and Ser616 via JNK and ERK1/2-dependent pathways, respectively. This disrupts the interaction between IRS-1 and the p85 regulatory subunit of PI3K, compromising the insulin-mediated vasodilatory signaling pathway involving PI3K/Akt/eNOS. Another mechanism underlying the role of IR in hypertension etiology is the upregulation of AngII subtype 1 receptors (AT1R) induced by hyperinsulinemia. This amplifies the adverse effects of AngII on the cardiovascular system. Taken together, these findings offer clear insights into the pathogenic mechanisms of AngII-induced IR in hypertension development [2, 35].
NLRP3 (NOD-like receptor family, pyrine domain containing 3), an inflammasome complex [36] that it widely exists in immune cells, including macrophages, monocytes, dendritic cells and neutrophils. The canonical NLRP3 inflammasome comprises the NLRP3 receptor protein, the apoptotic adaptor protein, and pro-caspase-1. This inflammasome identifies diverse danger signals both in vivo and in vitro, participating in inflammatory responses that contribute to pathological processes across various diseases [37]. While at rest, cellular expression of the NLRP3 protein remains below the threshold necessary for activating the NLRP3 inflammasome. Exogenous stimuli (such as intense ultraviolet light, cholesterol crystals, uric acid crystals, etc.), endogenous stimuli (high concentration of ATP released after cell injury [38], ROS, mitochondrial oxidized DNA and lysosomal destabilization [37], high mobility group B1 protein, purine metabolites, perforin, saturated fatty acid palmitate, serum amyloid A, etc.), pathogens-related irritants (lipopolysaccharide, bacteria, viruses, etc.), as well as the presence of harmful substances, increase the expression of the NLRP3 protein [39].
It can also be triggered by low intracellular potassium levels or high calcium concentrations, which occur in response to cellular stress. Once activated, NLRP3 facilitates the cleavage of pro-ILs by the caspase 1 subunit of its complex, resulting in mature IL-1β and IL-18, pivotal markers of low-grade inflammation. NLRP3 is recognized as a crucial factor in the initiation and progression of chronic inflammation, implicating it in the pathogenesis of various chronic inflammatory conditions, including gout, rheumatoid arthritis, T2DM, and atherosclerosis. Particularly relevant to IR, MetS, and T2DM, the NLRP3 inflammasome seems to orchestrate an inflammatory response to nutrient surplus and mitochondrial dysfunction [36, 37]. IR and chronic low-grade inflammation are hallmark features of T2DM. Recent studies suggest that chronic inflammation might be intricately linked to the activation and regulation of the NLRP3 inflammasome due to irregular glucose metabolism. Glucose can activate the NLRP3 inflammasome through recognition of the ATP/P2X purinergic receptor 4 pathways. Additionally, glucose can induce and enhance the expression of thioredoxin-interacting protein (TXNIP), a key cofactor of the NLRP3 inflammasome, thereby amplifying its activation. Islet cells exposed to chronic hyperglycemia respond to elevated extracellular glucose levels by activating the NLRP3 inflammasome, leading to the production and secretion of IL-1β, which exacerbates IR. In the presence of a severe metabolic disorder imbalance, there is an escalation of ROS and mitochondrial damage due to chronic inflammation, triggering the activation of the NLRP3 inflammasome. Some studies have demonstrated that NLRP3 inflammasome activation disrupts the interplay among glucose tolerance, insulin sensitivity, and intestinal microorganisms, contributing to the onset and progression of T2DM. Conversely, as T2DM advances, multiple organ dysfunctions ensue, further stimulating the NLRP3 inflammasome. In essence, T2DM initiates the activation of the inflammasome, or vice versa, establishing a detrimental cycle. While extensive research has elucidated the mechanisms of inflammasome action and its correlation with IR (see Table 1), further studies are warranted to deepen our understanding of these mechanisms [38–40].
The prevalence of AO and DM continues to rise globally, leading to a greater burden of CVD due to accelerated atherosclerosis, endothelial dysfunction, and microvascular inflammation. Some observational studies have demonstrated that AO and DM are linked to a heightened risk of heart failure, regardless of other traditional cardiovascular risk factors. Even mild increases in fasting glucose levels and IR abnormalities, even in the absence of diagnosed diabetes, significantly elevate the risk of heart failure. Individuals with diabetes face an elevated risk of heart failure, and evidence suggests an increased risk even within the prediabetic blood glucose range among those with AO [29].
Broadly, activins are expressed across diverse tissues, with particularly elevated levels in adipose tissue. These molecules have been demonstrated to govern a range of biological processes, including the regulation of apoptosis, proliferation, and differentiation in various cell types. Specifically, activins contribute to several stages of both the normal and abnormal development of adipose tissue, suggesting that heightened activin-A activity could contribute to IR and an inflammatory milieu [41].
Given the association among CVD, T2DM, and MetS, it’s essential to address serum activin-A as a potential biomarker for MetS. Multiple studies have demonstrated significantly elevated levels of activin-A in individuals with MetS, particularly in obese patients presenting with increased waist circumference, elevated systolic blood pressure, and impaired glucose metabolism [38].
Activin-A belongs to the TGF-β superfamily and serves various cytokine functions, encompassing the regulation of cell proliferation, differentiation, fibrosis, inflammation, immune responses, wound repair, apoptosis, and embryogenesis. Furthermore, activin-A stimulates insulin secretion in pancreatic islet cells and enhances insulin sensitivity in hepatocytes. Serum activin-A levels may serve as a predictive indicator for cardiovascular events and mortality in patients with T2DM [42].
From a CVD perspective, elevated levels of circulating activin-A may serve as a biomarker indicating increased cardiometabolic risk, stemming from factors such as hyperglycemia, inflammation, adipogenesis, and atherosclerosis, whether individually or collectively. Notably, activin-A has been linked to both acute and chronic inflammation, acting as an anti-inflammatory mediator in atherosclerosis. In patients with heart failure, heightened expression and blood levels of activin-A, a component of the TGF-β cytokine, have been observed. Conversely, concerning T2DM, activin-A plays a significant role in modulating insulin secretion and potentially influences inflammatory responses, angiogenesis, vascular remodeling, and atherosclerosis (Table 1). Moreover, activin-A exerts a critical adipogenic function via the Smad2 pathway, contributing to the development of obesity [42, 43].
Oxidative stress biomarkers encompass molecules affected by ROS within the microenvironment, as well as antioxidant system molecules that undergo alterations in response to redox stress [21]. Risk factors disrupt cellular signaling pathways, leading to elevated levels of inflammatory markers, lipid peroxides, and free radicals, resulting in cell damage and clinical manifestations of MetS. Oxidative stress in MetS can be assessed through the measurement of total antioxidant capacity. Isoprostanes (IsoP), specifically 8-iso prostaglandin-F2alpha (8-isoPGF2), are derived from arachidonic acid [13]. Increased urinary excretion of IsoP has been observed in obese children and adolescents, with a notable association with visceral obesity, rather than BMI and blood pressure [44]. However, it could serve as a valuable tool for concurrently investigating oxidative stress and inflammation in conditions where both are believed to be involved. Studies have been conducted in individuals with MetS, simultaneously assessing levels of oxidative stress biomarkers and antioxidant enzyme activity. The findings indicate that the presence of MetS correlates with elevated oxidative stress biomarkers and reduced antioxidant capacity. This suggests an association between MetS and pro-inflammatory conditions, indicating poor health, as part of a multifaceted process leading to CVD. Markers of oxidative stress encompass biomarkers of lipid peroxidation, protein and amino acid oxidation, and DNA oxidation [13].
The presence of lipid peroxidation can be assessed using various markers, including thiobarbituric acid reactive substances (TBARS), malonyldialdehyde (MDA), 4-hydroxy-2-nonenal (4-HNE), and F2-IsoP. Protein oxidation is indicated by markers such as protein carbonyls, advanced glycation end products, oxidized-LDL (ox-LDL), and advanced oxidation protein products. Markers of DNA oxidation include 8-oxo-2-deoxyguanosine (8-oxo-dG), 5-chlorouracil, and 5-chlorocytosine. Markers associated with ROS production include xanthine oxidase, gamma-glutamyl transferase (GGT), myeloperoxidase (MPO), NOX, and NOS. GGT plays a role in recycling glutathione (GSH) precursors, which are antioxidants and metabolic substrates. Elevated GGT levels have been linked to the prediction of MetS, DM, and hypertension. Non-enzymatic markers encompass glycoprotein A (GPA), CRP, ferritin, and uric acid. Individuals with MetS often exhibit elevated serum ferritin levels without transferrin saturation. Serum ferritin levels have been positively correlated with indicators of oxidative stress and IR, as well as liver damage [13]. Serum ferritin levels are, therefore, important in the diagnosis of MetS (Table 1) [13, 16].
MetS is a NCD that affects people all over the world. This review highlights markers of: lipid profile, inflammatory function, and oxidative stress (Figure 2).
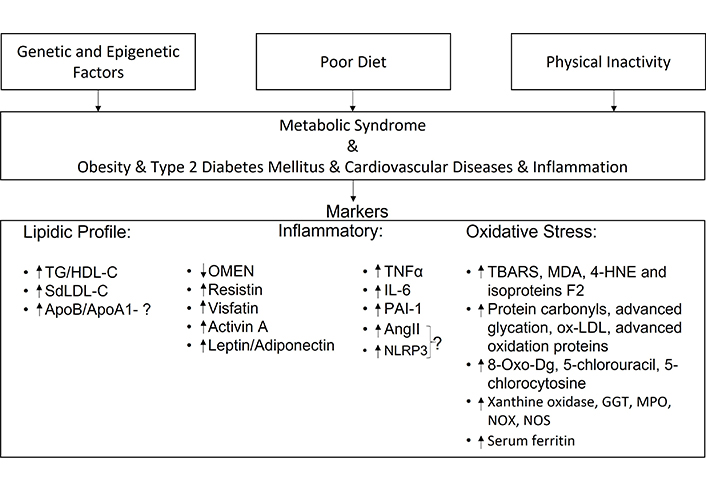
Summary of the review article—the article describes the main causes that can lead to MetS and its associated diseases. This research has allowed us to select possible markers for the diagnosis of MetS. AngII: angiotensin II; GGT: gamma-glutamyl transferase; HDL-C: high density lipoprotein-cholesterol; IL: interleukin; MDA: malonyldialdehyde; MPO: myeloperoxidase; NLRP3: NOD-like receptor family, pyrine domain containing 3; NOS: nitric oxide synthase; NOX: NADPH oxidase; OMEN: omentin; ox-LDL: oxidized-low-density lipoprotein; PAI-1: plasminogen activator inhibitor-1; SdLDL: small dense low-density lipoprotein; TBARS: thiobarbituric acid reactive substances; TG: triglycerides; TNF-α: tumor necrosis factor α; 4-HNE: 4-hydroxy-2-nonenal; 8-oxo-Dg: 8-oxo-2-deoxyguanosine
According to the research carried out, lipid profile markers include: an increase of the TG/HDL-C ratio and an increase in SdLDL-C. The increase of the ApoB/ApoA1 ratio could also be a marker, but further studies are needed to confirm this. As for markers of inflammatory function: OMEN has already been used as a marker of MetS and has been shown to be decreased in the presence of this syndrome, as well as the adiponectin; however, leptin has been found to be increased. However, the leptin/adiponectin ratio has proved to be more valuable as a marker than assessing leptin and adiponectin values individually. Resistin, which is more closely related to CVD, and visfatin, which is more closely related to T2DM, have both been shown to be increased in the presence of MetS.
Alterations in TNF-α, IL-6 and PAI-1 have been shown to be increased in MetS, since it is a syndrome characterized by chronic inflammation. AngII is altered in IR, but it has not yet been proven that this alteration is closely linked to MetS.
The NLRP3 inflammasome is elevated in cases of inflammation and it has been proven that DM leads to its activation, but more studies are needed before it can be considered a marker of MetS. Given the relationship with cardiometabolic risk, inflammation, and DM, among other pathologies, the increase in activin-A in the circulation may be important for indicating MetS. Oxidative stress markers include lipid peroxidation (TBARS, MDA, 4-HNE, and F2 isoproteins), protein oxidation (protein carbonyls, advanced glycation, ox-LDL and advanced oxidation proteins) and amino acid and DNA oxidation (8-oxo-Dg, 5-chlorouracil, and 5-chlorocytosine). The markers associated with ROS are xanthine oxidase, GGT, MPO, NOX, and NOS. Serum ferritin levels are also important in the diagnosis of MetS.
However, given the research conducted, we believe that a complete panel, in order to correlate the conditions most characteristic of MetS, should include the following markers: the TG/HDL-C ratio, SdLDL-C and lipid peroxidation markers in the lipid profile, the leptin/adiponectin ratio more closely related to T2DM, PAI-1 more closely related to CVD and activin-A as a marker of both T2DM and CVD and ferritin levels which are related to IR.
In any case, more research into the pathophysiology of this syndrome is needed to definitively confirm these markers and select/discover new ones.
4-HNE: 4-hydroxy-2-nonenal
8-isoPGF2: 8-iso prostaglandin-F2alpha
8-oxo-dG: 8-oxo-2-deoxyguanosine
AIP: atherogenic index of plasma
AngII: angiotensin II
AO: abdominal obesity
ApoA1: apolipoprotein A1
ApoB: apolipoprotein B
ASCVD: atherosclerotic cardiovascular disease
AT1R: angiotensin II subtype 1 receptors
BMI: body mass index
CCLs: CC-chemokine ligands
CETP: cholesterol ester transfer protein
CRP: C-reactive protein
CVD: cardiovascular disease
DM: diabetes mellitus
FFA: free fatty acids
GGT: gamma-glutamyl transferase
GPA: glycoprotein A
GSH: glutathione
HDL-C: high-density lipoprotein cholesterol
HL: hepatic lipase
IL: interleukin
IR: insulin resistance
IRS-1: insulin receptor substrate-1
IsoP: isoprostanes
LDL: low-density lipoprotein
LDLR: low-density lipoprotein receptor
MDA: malonyldialdehyde
MetS: metabolic syndrome
miRNA: micro-RNA
MPO: myeloperoxidase
NAMPT: nicotinamide phosphoribosyl transferase
NCD: non-communicable disease
NLRP3: NOD-like receptor family, pyrine domain containing 3
NO: nitric oxide
NOS: nitric oxide synthase
NOX: NADPH oxidase
OMEN: omentin
ox-LDL: oxidized-low-density lipoprotein
PAI-1: plasminogen activator inhibitor-1
PPAR-α: peroxisome proliferator-activated receptor alpha
ROS: reactive oxygen species
SdLDL: small dense low-density lipoprotein
SdLDL-C: small dense low-density lipoprotein cholesterol
T2DM: type 2 diabetes mellitus
TBARS: thiobarbituric acid reactive substances
TG: triglycerides
TGF-β: transforming growth factor beta
TNF-α: tumor necrosis factor alpha
TXNIP: thioredoxin-interacting protein
VLDL: very low-density lipoprotein
WHO: World Health Organization
FM: Conceptualization, Investigation, Writing—original draft. AV: Conceptualization, Writing—review & editing, Validation, Supervision. JM: Writing—review & editing, Validation. LP: Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Jonica Campolo ... Maria Grazia Andreassi
Abdullatif Taha Babakr
Michael Li ... Robert M. Lust
Muhammet Cihat Çelik ... Yusuf Karavelioğlu
