Cholestasis associated to inborn errors in bile acid synthesis
Several metabolic pathways are involved in the biotransformation of C27 neutral cholesterol to C24 primary bile acids (BAs), mainly cholic acid (CA) and chenodeoxycholic acid (CDCA), which are then conjugated with glycine or tauri
[...] Read more.
Several metabolic pathways are involved in the biotransformation of C27 neutral cholesterol to C24 primary bile acids (BAs), mainly cholic acid (CA) and chenodeoxycholic acid (CDCA), which are then conjugated with glycine or taurine. This process can start with the modification of the steroid ring or the shortening of the side chain and involves enzymes present in different subcellular compartments. Inborn errors affecting the biogenesis of organelles, such as peroxisomes, or the expression or function of specific enzymes of these convergent routes result in: i) the lack of mature C24-BAs, with the subsequent impairment in digestion and absorption of dietary fat and liposoluble vitamins, such as vitamin K, which may account for a deficient hepatic synthesis of several coagulation factors; ii) the accumulation of intermediate metabolites, which may affect hepatocyte physiology, causing cholestasis as a commonly shared alteration besides other deleterious hepatic events; and iii) extrahepatic clinical manifestations due to accumulation of toxic metabolites in other territories, such as the nervous system, causing neurological disorders. In general, diseases whose primary alteration is a genetic defect in BA synthesis are diagnosed in children or young individuals with a very low incidence. The symptomatology can markedly vary among individuals, ranging from mild to severe conditions. Oral therapy, based on the enrichment of the BA pool with natural C24-BAs, such as CA, CDCA, glyco-CA, or ursodeoxycholic acid (UDCA), depending on the exact deficiency causing the disease, may be beneficial in preventing life-threatening situations. In contrast, in other cases, a liver transplant is the only option for these patients. This review describes the updated information on the genetic and molecular bases of these diseases and the current approaches to achieve a selective diagnosis and specific treatment.
Ricardo Espinosa-Escudero ... Maria J. Monte
View:1712
Download:65
Times Cited: 0
Several metabolic pathways are involved in the biotransformation of C27 neutral cholesterol to C24 primary bile acids (BAs), mainly cholic acid (CA) and chenodeoxycholic acid (CDCA), which are then conjugated with glycine or taurine. This process can start with the modification of the steroid ring or the shortening of the side chain and involves enzymes present in different subcellular compartments. Inborn errors affecting the biogenesis of organelles, such as peroxisomes, or the expression or function of specific enzymes of these convergent routes result in: i) the lack of mature C24-BAs, with the subsequent impairment in digestion and absorption of dietary fat and liposoluble vitamins, such as vitamin K, which may account for a deficient hepatic synthesis of several coagulation factors; ii) the accumulation of intermediate metabolites, which may affect hepatocyte physiology, causing cholestasis as a commonly shared alteration besides other deleterious hepatic events; and iii) extrahepatic clinical manifestations due to accumulation of toxic metabolites in other territories, such as the nervous system, causing neurological disorders. In general, diseases whose primary alteration is a genetic defect in BA synthesis are diagnosed in children or young individuals with a very low incidence. The symptomatology can markedly vary among individuals, ranging from mild to severe conditions. Oral therapy, based on the enrichment of the BA pool with natural C24-BAs, such as CA, CDCA, glyco-CA, or ursodeoxycholic acid (UDCA), depending on the exact deficiency causing the disease, may be beneficial in preventing life-threatening situations. In contrast, in other cases, a liver transplant is the only option for these patients. This review describes the updated information on the genetic and molecular bases of these diseases and the current approaches to achieve a selective diagnosis and specific treatment.
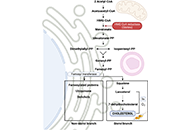 Hypoxia signaling and cholesterol/steroidogenic acute regulatory protein 1 axis: interplay and role in alcohol and non-alcohol-related liver diseasesOpen AccessPerspectiveMetabolic zonation in the liver carries out the maintenance of organ and body homeostasis. Hypoxia is an inherent physiological feature of the liver and contributes to the zonal properties of the he [...] Read more.Sandra Torres ... Carmen Garcia-RuizPublished: December 30, 2022 Explor Dig Dis. 2022;1:170–186
Hypoxia signaling and cholesterol/steroidogenic acute regulatory protein 1 axis: interplay and role in alcohol and non-alcohol-related liver diseasesOpen AccessPerspectiveMetabolic zonation in the liver carries out the maintenance of organ and body homeostasis. Hypoxia is an inherent physiological feature of the liver and contributes to the zonal properties of the he [...] Read more.Sandra Torres ... Carmen Garcia-RuizPublished: December 30, 2022 Explor Dig Dis. 2022;1:170–186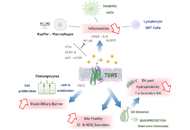 The bile acid receptor TGR5 and cholestasisOpen AccessReviewMetabolic zonation in the liver carries out the maintenance of organ and body homeostasis. Hypoxia is an inherent physiological feature of the liver and contributes to the zonal properties of the he [...] Read more.Grégory Merlen ... Thierry TordjmannPublished: December 30, 2022 Explor Dig Dis. 2022;1:154–169
The bile acid receptor TGR5 and cholestasisOpen AccessReviewMetabolic zonation in the liver carries out the maintenance of organ and body homeostasis. Hypoxia is an inherent physiological feature of the liver and contributes to the zonal properties of the he [...] Read more.Grégory Merlen ... Thierry TordjmannPublished: December 30, 2022 Explor Dig Dis. 2022;1:154–169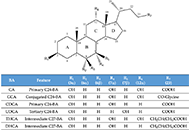 Cholestasis associated to inborn errors in bile acid synthesisOpen AccessReviewSeveral metabolic pathways are involved in the biotransformation of C27 neutral cholesterol to C24 primary bile acids (BAs), mainly cholic acid (CA) and chenodeoxycholic acid (CDCA), which are then conjugated with glycine or tauri [...] Read more.Ricardo Espinosa-Escudero ... Maria J. MontePublished: December 7, 2022 Explor Dig Dis. 2022;1:137–153
Cholestasis associated to inborn errors in bile acid synthesisOpen AccessReviewSeveral metabolic pathways are involved in the biotransformation of C27 neutral cholesterol to C24 primary bile acids (BAs), mainly cholic acid (CA) and chenodeoxycholic acid (CDCA), which are then conjugated with glycine or tauri [...] Read more.Ricardo Espinosa-Escudero ... Maria J. MontePublished: December 7, 2022 Explor Dig Dis. 2022;1:137–153