
Abstract
Hepatocellular carcinoma (HCC) is the most common type of liver cancer and its death rate is rising faster than that of any other cancer, while we still lack effective treatments. The increasing incidence of liver cancer in western countries is closely associated with the growing prevalence of metabolic dysfunction-associated steatohepatitis (MASH) linked to metabolic diseases. While the contribution of lipids in the progression of MASH pathogenesis and its progression to HCC is well recognized, the specific contribution of cholesterol is subject to controversy. The liver plays a central role in cholesterol metabolism, where the majority of its biosynthesis, storage, excretion, recycling, and conversion into bile acids occur. Moreover, cholesterol is implicated in numerous hepatocyte cellular processes, encompassing endoplasmic reticulum function, formation of lipid microdomains in the plasma membrane, metabolism of lipoproteins, and mitochondrial function and performance. Therefore, it is not surprising that cholesterol plays key roles in initiation, promotion, and survival of HCC cells and there are several lines of evidence pointing to that cancer cells are subverting cholesterol metabolism to foster their proliferation and survival through various mechanisms. This narrative review provides a concise overview of the physiological and pathological roles of cholesterol in the transition from healthy hepatocytes to HCC, in the context of MASH. Gaining further understanding of how hepatic cancer cells disrupt cholesterol homeostasis and how these perturbations impact cancer progression will facilitate the identification of novel and more effective cancer treatment strategies in this complex and devastating disease.
Keywords
Cholesterol, tumor metabolism, hepatocellular carcinoma, apoptosisIntroduction
Hepatocellular carcinoma (HCC) is a major public health problem with increasing incidence [1, 2] being the fourth-leading cause of cancer-related deaths worldwide [3]. Historically, the main risk factor for HCC has been the chronic infection with hepatitis B or C viruses. However, in the near future, the leading risk role of chronic viral hepatitis will be replaced by other etiologic factors such as ethanol abuse and metabolic disorders derived from the current obesity epidemic in western countries, given that both alcoholic steatohepatitis (ASH) and metabolic-associated steatohepatitis-related cirrhosis are clear risk factors for HCC development [4, 5]. The obesity epidemic in western populations has dramatically increased the incidence of metabolic syndrome and its associated pathologies such as type II diabetes, insulin resistance, and cardiovascular disease. More importantly, it is also increasing the prevalence of its hepatic manifestation, the metabolic dysfunction-associated fatty liver disease (MASLD, formerly known as NAFLD), which is affecting 30% of adults in the US [6] and an estimated 70–80% of obese diabetic patients have MASLD condition [7]. Further progression may lead to liver cirrhosis [8], end-stage liver disease, portal hypertension (PH), and ultimately may initiate HCC. Consequently, the incidence of MASH-driven HCC is expected to raise globally in association with the alarming increase of obesity, type II diabetes, metabolic syndrome, and the consequent raised incidence of MASH in western countries [5, 9, 10].
While MASLD is characterized by hepatic steatosis which may be clinically silent, approximately 15% to 20% of MASLD patients manifest liver damage, inflammation, and fibrosis which are hallmark comorbidities of MASH [11]. MASH development is thought to depend on an underlying endoplasmic reticulum (ER) stress and mitochondrial dysfunction although its origin and mechanism of action remain incompletely understood [12, 13]. An important factor implicated in MASH is the accumulation of non-esterified cholesterol in hepatocytes [14] that can cause mitochondrial dysfunction [15], synergize with tumor necrosis factor [13], and cause liver damage.
The liver injury induced by different factors induces a progressive inflammatory milieu resulting in a cycle of hepatocellular cell death and regeneration with an associated genetic instability. This multi-step process involves the gradual accumulation of genetic and epigenetic alterations in the presence of hepatocellular ER stress, heightened inflammation, reactive oxygen species (ROS), and fibrosis. However, the precise molecular events that drive the initiation and progression of HCC are not fully understood [16]. HCC predominantly occurs in individuals with long-term liver damage and cirrhosis caused by inflammation, and it is typically diagnosed at an advanced stage, limiting treatment options. The management of early-stage HCC relies on factors such as tumor size, stage, and grade, and various treatment modalities are available, including radiotherapy, liver transplantation, and local ablation therapies. Nevertheless, even early HCC cases that received surgical resection or percutaneous ablation suffer frequent relapse. Moreover, only one-third of patients are diagnosed at early stages, and among advanced cases only a few systemic therapies are available. The multi-target tyrosine kinase inhibitor sorafenib was the only systemic therapy approved for the advanced stages of HCC until recently [17], and it showed only a three-month improvement in survival time and it is only effective in 30% of patients [18], while other approved systemic chemotherapies have only moderate improved outcomes [19]. Currently, over 50% of patients with HCC are given systemic therapies that are barely effective and cause considerable toxic damage to the remaining normal liver, further limiting clinical outcomes [20].
Although there have been some advancements in the field [21, 22], HCC continues to present as a particularly resilient manifestation of cancer, often manifesting secondary or acquired chemoresistance through mechanisms that are not yet completely understood [23, 24].
Advances in the last decade showed that immunotherapy appears to be one of the most promising strategies for HCC treatment [25, 26]. However, despite the significant improvements in patient outcomes, it is noteworthy that only a maximum of 30% of patients exhibit an objective response to the current standard-of-care treatments, with complete responses observed in less than 10% [2]. Therefore, much effort is needed to identify strategies to overcome treatment resistance and enhance overall response rates [27, 28]. The lack of effective treatments explains why while modern therapies for other types of cancer have reduced mortality, in HCC the mortality rate still remains dramatically elevated [29].
HCC, like other cancer cells, displays a range of metabolic transformations derived mainly by mutations, genetic aberrations, and epigenetic events which promote high cell proliferation, growth, ability to escape programmed cell death, and evasion from immune surveillance, defining the acquired features of cancer cells [30]. Some of these malignant cell traits, such as increased cellular proliferation [31, 32] and tumor growth [33, 34] are closely associated with augmented cholesterol requirement. Particularly, it has been shown that HCC critically depends on cholesterol for growth [35], and observational studies show a protective association between the use of statins and the risk of developing liver cancer [36, 37], although this trend has been also observed in other cancer types, such as prostate and gastrointestinal cancers [38]. Cellular cholesterol is stored in lipid droplets as esterified cholesterol, but its unesterified form is an indispensable component of cellular membranes, contributing to their rigidity and the formation of local microdomains. Moreover, cholesterol is the precursor of steroidal hormones and bile acids which have important biological activities. Recent evidence revealed different mechanisms where cholesterol may play a role in the carcinogenic transformation of hepatocytes and in the survival of HCC cells [39, 40]; these findings are briefly discussed in the following sections.
Physiological cholesterol metabolism in the liver
The liver plays a key role in the cholesterol metabolism for the entire organism accomplished through the synthesis in hepatocytes via the mevalonate pathway and subsequent esterification and storage into lipid droplets. Additionally, the liver receives dietary cholesterol and redistributes cholesterol across the body through lipoprotein transport, and facilitates its transformation into cholesterol derivatives via the bile acid pathways [41]. Cholesterol is an essential lipid for cellular homeostasis and is present in all mammalian cells, mainly localized in plasma membrane when is not in its non-esterified form, comprising about 30% of the lipid bilayer. Hence, cholesterol is an essential building block of the plasma membrane playing a pivotal role in structural integrity and regulating fluidity and permeability of cell membranes. Indeed, cholesterol plays a crucial role in the determination of fluidity and membrane curvature [42, 43] which determines lipid bilayer physical properties and functions. Within the membrane lipid bilayer, cholesterol is segregated in discrete domains called lipid rafts associated with glycosphingolipids, acting as scaffold for proteins that originate an array of signaling pathways, membrane trafficking, cytoskeletal organization, apoptosis, and cell adhesion [44]. In lipid rafts cholesterol is modulating the action of specific membrane protein receptors and carriers by altering membrane physical properties. As a consequence, depletion of cellular cholesterol leads to lipid raft disruption and redistribution of protein receptors and carriers into the plasma membrane, altering signal transduction [45]. In addition to its role in cellular organization and membrane biogenesis, cholesterol is the precursor of several molecules that play a central role in physiology, including bile acids, steroid hormones, oxysterols, and vitamin D, which all can act as ligands for different nuclear receptors with pleiotropic effects on physiology and cancer. Therefore, given the crucial role of cholesterol in cell integrity and organismal physiology it is not surprising its implication in the generation and survival of certain types of cancer [40, 46, 47], particularly in cancer arising from hepatocytes where cholesterol have a central role in its physiology.
De novo cholesterol synthesis and regulation in hepatocytes
The predominant mechanism to provide the need of cellular cholesterol is its de novo synthesis from acetate in the form of acetyl-CoA in an energy-consuming metabolic route called known as the mevalonate pathway. In addition to cholesterol, several other non-sterol components are generated through the mevalonate pathway. This pathway is initiated in the ER by the conversion of acetyl-CoA into 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) and then converted to mevalonate by the rate-limiting enzyme, HMG-CoA reductase (HMGCR). Afterwards, a series of enzymatic reactions convert mevalonate into farnesyl pyrophosphate (FPP) which can be diverted either to the sterol synthesis or to the non-sterol pathway. In the sterol branch, squalene synthase commits the FPP to the sterol biosynthesis [48] by converting the FPP to squalene and afterwards to lanosterol and finally cholesterol by two related pathways. The Block pathway use desmosterol and 7-hydroxycholesterol as the immediate precursors of cholesterol through their reduction by the 24-dehydrocholesterol reductase (DHCR24) and the Kandutsch-Russel pathway use the the DHCR7. UV radiation can convert the 7-dehydrocholesterol into vitamin D3, which has an important effect on cancer biology [49]. The biotransformation of lanosterol to cholesterol involves several redox reactions with oxygen requirement. Consequently, an additional mechanism that regulates cholesterol synthesis is oxygen availability whereas hypoxia has been shown to stimulate HMGCR degradation through both accumulation of lanosterol and Insigs induction [50].
In the non-sterol pathway, FPP can generate by additional enzymatic steps important cellular components such as dolichol, ubiquinone, heme A and isoprenoids for protein prenylation. Ubiquinone and heme A are essential for the mitochondrial transport chain and dolichol is a transmembrane carrier for glycol units in the synthesis of glycated proteins and lipids [51]. Isoprenoid generation in the mevalonate pathway is an essential mechanism of posttranslational modification of certain proteins by anchoring target proteins to cell membranes. FPP is used to add a prenyl moiety to proteins of the Ras family, while geranylgeranyl pyrophosphate (GGPP) is used to prenylate those of the Rho family [52]. Prenylation of G-proteins is required for intracellular signalling between receptors and effector enzymes [53]. Prenylation of Ras protein is an important step in translocation and activation of Ras, so that it can regulate cell growth, while inhibition of Ras prenylation leads to cell growth blockade [54].
Given the importance of cholesterol for cell homeostasis and the high energy used for its biosynthesis, the de novo cholesterol synthesis is a highly-regulated process controlled by an efficient feed-back mechanism. Accumulation of sterols in the ER membrane triggers binding of the membrane domain of HMGCR to a Insig1 and Insig2, which carry a membrane-anchored ubiquitin ligase called GP78 which ubiquitinates HMGCR, marking it for proteasomal degradation [55]. Moreover, HMGCR gene expression is regulated by ER-bound membrane transcription factor sterol regulatory element protein 2 (SREBP2), whose activation depends on the sterol-sensitive SREBP cleavage activating protein (SCAP) which is the true sterol level sensor of the ER. When cholesterol levels are low in the ER, SCAP binds to SREBP2 inducing SREBP2 to leave the ER and move to the Golgi, where it is sequentially cleaved by the specific proteases S1P and S2P. This event releases the soluble N terminus domain of SREBP2, which travels to the nucleus to activate the transcription factor activity of SREBP2, triggering expression of HMGCR and several enzymes in the mevalonate pathway and cholesterol metabolism [41].
The de novo synthesized cholesterol leaves the ER rapidly is targeted to the plasma membrane, mostly by non-vesicular mechanisms that bypass ER-Golgi membrane transport thereby helping to maintain low ER sterol content [56]. Also, cholesterol equilibrates with the preexisting cholesterol pool and accesses other sites such as endosomal compartments [57]. An important buffering mechanism to maintain steady levels of sterols is its esterification. Cholesterol can be esterified with long chain fatty acyl-CoA to cholesterol esters in a reaction catalyzed acyl-CoA cholesterol acyltransferase (ACAT). Esterified cholesterol increases the hydrophobicity of cholesterol and is usually a neutral form of storage of cholesterol within lipid droplets along with triglycerides.
ACAT has been reported to be overexpressed in different types of cancers, including glioma, pancreatic and liver cancer [58, 59]; although its role in liver cancer is not well understood with reports indicating a lower expression of ACAT in tumor areas from patients with HCC [60]. These controversial results may indicate different requirements of the different types of tumors for non-esterified cholesterol for their survival.
Exogenous hepatic cholesterol input and redistribution
In addition to the de novo synthesis, there is an important cholesterol input from diet to the liver, which plays a key role in its redistribution. Enterocytes and hepatocytes package cholesterol and triglycerides into lipoproteins, circumventing the physical problem of transport of hydrophobic molecules in a hydrophilic media in the circulation. Dietary cholesterol is absorbed very efficiently by gut enterocytes which package it along with triglycerides and apolipoprotein B-48 into chylomicrons. Once in circulation, an important amount of chylomicron triglycerides is hydrolyzed within few minutes while new apoproteins, such as ApoE are added, generating chylomicron remnants that are taken up by hepatocytes in the liver through the low-density lipoprotein (LDL) receptor. These particles are endocytosed by clathrin-coated vesicles and transported to the lysosomal compartment, where cholesteryl esters are hydrolyzed by acid lipase rendering unesterified cholesterol. The multivesicular late endosomes harbor two transporters the Niemann-Pick type C protein 1 and 2 (NPC1 and NPC2) which play a crucial role in cholesterol trafficking out of the endosomal system. Indeed, deficiency of either of these proteins leads to the accumulation of LDL-derived unesterified cholesterol in late endocytic organelles, which is characteristic of the lysosomal genetic disorder NPC disease [61]. Another site in the endocytic pathway for active sterol exchange is the recycling compartment. Recycling endosomes are enriched in cholesterol and sphingolipids and function as acceptors for non-vesicular sterol flux through the cytosol [62]. Once released from the endolysosomal system, cholesterol is delivered mainly to plasma membrane, lipid droplets for its esterification or may be transferred to other membranous organelles such as ER, recycling endosomes, and mitochondria.
Hepatocytes, in turn, redistribute cholesterol throughout the body by the secretion of in very low-density lipoprotein (VLDL) particles which carry lipids packaged with apolipoprotein B-100. VLDL particles are processed in the circulation by addition of several apolipoproteins and the hydrolysis of their triglycerides into LDL, the main lipoprotein that delivers cholesterol to peripheral cells through its uptake by the LDL receptor. When peripheral cells have excess cholesterol, it can be released to high density lipoproteins (HDL), through a series of transmembrane receptors of the ATP-binding cassette (ABC) transporter family such as ABCA1 by a process known as cholesterol efflux. Indeed, nascent HDL particles carrying ApoA-I and ApoA-II are released by the liver, they uptake unesterified cholesterol exerting the cholesterol efflux from peripheral tissues and this cholesterol is esterified by the HDL-associated enzyme lecithin-cholesterol acyltransferase (LCAT). These HDL particles exchange lipids and apolipoproteins with other lipoproteins in the blood and hepatocytes take up the cholesterol from mature HDL particles, returning the cholesterol to the liver in a process called reverse cholesterol transport [41]. Moreover, LDL particles and VLDL remnants can be cleared by hepatocytes returning the cholesterol and cholesterol esters to the liver for its redistribution or transformation in bile acids.
Bile acid synthesis
The pool of nonesterified cholesterol, both the newly synthesized in the liver and the one derived from the intestinal absorption or from circulating lipoprotein uptake, can be excreted from the hepatocyte directly to the bile by the function of the heterodimer of ABC transporters ABCG5/G8 found on the canalicular membrane of the hepatocytes. However, one essential pathway of cholesterol metabolism in hepatocytes is its conversion to bile acids. This complex process in which either CYP7A1 or CYP27A1 mediate the first limiting reaction generates an array of bile acid derivatives with diverse physical properties and physiological actions [63]. The microsomal CYP7A1 catalyzes the first step of the main classical bile acid synthesis pathway also known as the neutral pathway, performing the rate-limiting 7α-hydroxylation of cholesterol. Afterwards, the microsomal sterol 12α-hydroxylation by CYP8B1 is specifically required for cholic acid synthesis and its derivatives, after a series of enzymatic transformations. Conversely, the mitochondrial CYP27A1 starts the alternative and quantitatively minor bile acid synthesis (acidic pathway). In hepatocytes, mitochondrial cholesterol is metabolized by CYP27A1 to 27-hydroxycholesterol followed by CYP7B1, which then feeds the alternative mitochondrial pathway of bile acid synthesis leading mainly to chenodeoxycholic acid generation [64–66]. In mouse liver, but not in humans, chenodeoxycholic is metabolized to muricholic acids (α-MCA and β-MCA) [67]. This specie-specific difference has important implications given that chenodeoxycholic acid acts as a farnesoid X receptor (FXR) agonist, while muricholic acids display FXR antagonistic activity. Nevertheless, there are evidences showing that the transport of cholesterol to mitochondria, mediated by the cholesterol transporter steroidogenic acute regulatory protein (STAR) rather than CYP27A1 is the real rate-limiting step of the mitochondrial pathway of bile acid synthesis. Indeed, in primary hepatocytes or HepG2 cells, the overexpression of STAR resulted in a 5-fold increase in the rate of bile acid synthesis, while transfection with CYP27A1 upregulated bile acid synthesis only by 2-fold [68, 69]. In addition to the bile acid synthesis, STAR-mediated trafficking of cholesterol to mitochondria has important implications in its functionality and as a consequence, in MASLD pathogenesis and HCC development [40, 70], as will be discussed later. In humans, the mitochondrial acidic bile acid synthesis pathway of bile acid synthesis is considered to contribute approximately to 10% of the total bile acid pool and its physiological relevance is not well understood.
The primary cholic acid and chenodeoxycholic acids bile acids are quickly conjugated with the amino acids, taurine or glycine, by the action of amino acid N-acyltransferase (BAAT), changing their physical properties and lowering their cell toxicity. These conjugated bile acids are secreted into bile via bile salt export pump (BSEP) to the bile that along with phospholipids and cholesterol where they have critical roles in lipid digestion. In the ileum, conjugated bile acids are secreted into portal blood via organic solute transporter α/β and circulated back to hepatocytes via sodium-taurocholate co-transport peptide (NTCP). Furthermore, conjugated bile acids are deconjugated and 7α-dehydroxylated in the colon by specific bacterial flora. As a result, part of cholic acid and chenodeoxycholic acid are converted to deoxycholic acid and lithocholic acid, respectively. These secondary bile acids, which show higher toxicity, are absorbed and circulated to the liver like the conjugated and unconjugated primary bile acids. Approximately 95% bile acids are recovered and circulated back to the liver (enterohepatic cycle) and the 5% of bile acids that are lost in feces which can be compensated by de novo synthesis in the liver from cholesterol [71].
In the neutral pathway, two mechanisms have been proposed for the feed-back regulation of CYP7A1 gene transcription by primary bile acids. First, primary bile acids activate directly FXR in the liver to induce small heterodimer partner (SHP), which represses trans-activation of the CYP7A1 and CYP8B1 genes. Moreover, primary bile acids entering the enterocytes through the ASBT transporter, activate FXR in the intestine to induce fibroblast growth factor 15 (FGF15, or human orthologue FGF19), which reaches the liver activating hepatic FGF receptor 4/Klotho signaling to inhibit CYP7A1 gene transcription via extracellular regulated protein kinases 1/2 (ERK1/2)/c-Jun of the mitogen-activated protein kinase (MAPK) pathway [72]. The inhibition of ASBT reduces FGF19/15 secretion from the ileum, promoting bile acid synthesis and decreasing levels of hepatic cholesterol. However, the alternative mitochondrial pathway is not controlled by these mechanisms responding directly to bile acids or cholesterol. However, given that the limiting step of this pathway is the transport of cholesterol to mitochondria mediated by STAR, the regulation of expression and activity of this transporter is pivotal for the availability of cholesterol for the CYP27A1. However, little is known about regulation of STAR in the liver. STAR has been reported to be regulated by LXR activation [65] and induced by ER stress [73], and both features play important roles in MASLD and likely to the initiation and progression of HCC.
Alterations of cholesterol metabolism in HCC
As mentioned above, there are evidences that show that cholesterol is deregulated in the transition from simple steatosis to MASH. In line with this, cholesterol has also been identified as a tumor promoter in the MASH-to-HCC progression [74–80]. Basically, the pathological excess of free cholesterol and its derivatives in several cellular membranes may increase the likelihood of the initiation of cancer stem cells, their progression to the fully differentiated HCC cells and their survival, as will be discussed in the next sections.
Role of cholesterol in ER stress in MASH-derived HCC
The accumulation of lipid species in hepatocytes is initiated in MASLD by de novo lipogenesis, whose rate is up to 3-fold higher in MASLD patients [81]. Hence, lipogenesis was speculated to contribute to MASH progression by the generation of lipotoxic free fatty acids (FFA) and the induction of liver damage; fibrosis and inflammation in the transition from bland MASLD to MASH are believed to be a consequence of lipotoxicity. However, some studies attribute the main lipotoxicity in the MASH progression to nonesterified cholesterol accumulation [82–84]. Importantly, given that de novo lipogenesis and cholesterol synthesis share regulatory pathways, increased hepatic triglyceride levels are associated with increased cholesterol synthesis in the liver [85, 86]. Therefore, the lipotoxicity attributed to triglyceride accumulation in steatosis in MASLD may be, at least in part, due to increased non-sterified cholesterol in different cell compartments. In line with this, mitochondrial cholesterol loading cause mitochondrial dysfunction in hepatocytes [15] rendering them more susceptible to a second insult exerted by TNF-alpha leading to liver damage, which is a characteristic feature of MASH. Of note, TNF-alpha is also a critical mediator of MASH and HCC progression [13].
An important part of the lipotoxicity found in MASH is manifested with the persistent hepatocellular ER stress. Compared to normal liver tissues, MASLD tissues have been detected to have higher levels of C/EBP homologous protein (CHOP), cleaved ATF6 and spliced X-box binding protein 1 (XBP1) indicating the activation of the unfolded protein response to cope with ER stress in MASLD [87]. Moreover, the presence of chronic ER stress in livers from obese individuals is not simply by protein overloading but also driven by compromised folding capacity, in which lipid metabolism may have a function given the known ability of the incorporation of palmitate and cholesterol to the ER membrane to induce ER stress [88]. The implication of ER stress in MASH and the development of HCC were demonstrated in experimental animals where by means of chemical chaperones or liver overexpression of the protein chaperone BiP/Grp78 completely abrogated the MASH signs [13]. In line with this, a SREBP2 activation mechanism was described whereby ER stress drives continued cholesterol synthesis through the activation of Casp2-mediated activation of Sp1 [75]. This mechanism overrides the inhibition of de novo cholesterol synthesis where ER is sensitive to cholesterol levels, creating a cholesterol overload as found in states of chronic liver disease preceding HCC development such as MASH [82, 85]. Moreover, cholesterol accumulation triggers ER stress by altering the critical cholesterol-to-phospholipid ratio of the ER membrane, necessary to maintain its fluidity. The resulting stiffening of the ER membrane inhibits enzyme conformational changes and impairs ER activity and eventually triggering cell apoptosis. Even minimal increases of cholesterol loading on ER are able to impair the activity of SERCA, a sarco/ER calcium ATPase Ca2+ pump, decreasing the intra-ER Ca2+ concentrations, resulting in an impairment of protein-folding capacity and ultimately triggering ER stress [88]. This axis would create a feed forward mechanism of exacerbated cholesterol synthesis and ER stress elicited by ER membrane cholesterol overload [88], both situations found in MASH and HCC.
Lastly, ER stress is also associated with long-term inflammation, as a result of an overproduction of ROS and the activation of nuclear factor kappa-light-chain-enhancer of activated B (NF-κB) and JNK pathways. This creates a reciprocal and bilateral relationship between ROS production and ER stress, which drives the progression from MASLD to MASH. Oxidative stress and inflammation feeds on persistent ER stress in liver with MASH which in turn induces a deregulated increase of cholesterol synthesis and accumulation of free cholesterol [75]. This accumulation of cholesterol and persistent ER stress in HCC precursor cells might perpetuate the ER stress and increase its transport to other organelles. It is plausible that these occurrences contribute to lipotoxicity, promoting necrotic and apoptotic cell death, tissue damage and compensatory proliferation [13], potentially leading to a heightened mutagenesis rate and thereby promoting HCC initiation.
Role of mitochondrial cholesterol trafficking in HCC
Cancer cells are characterized by an array of metabolic transformations, which promote their unrestricted growth and ability to escape apoptosis by counteracting cell death pathways and evasion of immune surveillance [30, 89]. Cholesterol metabolism is deregulated in tumors [47] and it has been previously described that cholesterol trafficking to mitochondria in tumor cells, due to overexpression of the mitochondrial cholesterol transporter STAR, increase the resistance of HCC cells to cell death induction [90, 91], in addition to the role of STAR-mediated trafficking of cholesterol to mitochondria in MASLD pathogenesis [40, 70]. Moreover, this cholesterol transporter has been found upregulated in HCC cells [82, 85] and in patients with MASH-derived HCC [74]. Consequently, the combined dysregulation of cholesterol metabolism and its increased trafficking to mitochondria may be impacting in the initiation, survival, cell death evasion and immune surveillance of HCC cells by different mechanisms, as will be discussed below. A diagram illustrating the possible ways in which specifically the buildup of cholesterol in mitochondria could play a role in the formation of HCC is shown in Figure 1.
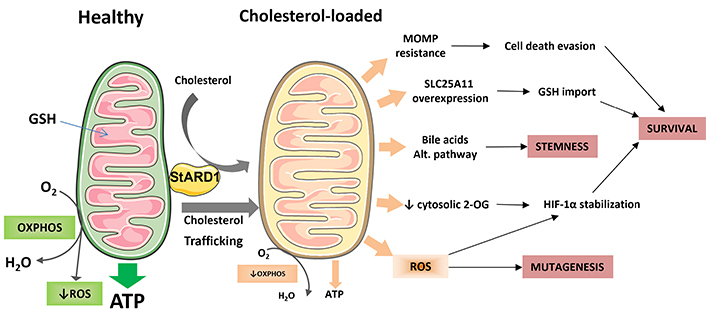
Impact of mitochondrial cholesterol loading on hepatocellular carcinoma (HCC). This diagram provides an overview of the potential mechanisms by which the accumulation of cholesterol in mitochondria leads to the onset and progression of HCC. Healthy mitochondria produce ATP through oxidative phosphorylation (OXPHOS) and imports glutathione (GSH) from cytosol to counteract the production of mitochondrial reactive oxygen species (ROS). However, when mitochondrial membranes become overloaded with cholesterol, it may lead to resistance to mitochondrial outer membrane permeabilization (MOMP), impaired OXPHOS activity and ATP production, increased ROS production, and enhanced synthesis of bile acids through the alternative bile acid pathway. The diminished activity of the solute carrier family 25 member 11 (SLC25A11) may impede the export of 2-oxoglutarate (2-OG), resulting in the stabilization of hypoxia-inducible factor 1 alpha (HIF-1α) due to increased ROS production. To survive, HCC cells can overcome this limitation by overexpressing the SLC25A11 transporter to maintain mitochondrial GSH levels. Finally, these mechanisms may have an impact on the mutagenesis rate; stemness of HCC initiated cells and overall survival of HCC cells. Parts of the figure were used from or adapted from pictures provided by Servier Medical Art, licensed under CC BY 4.0
Mitochondrial cholesterol in cell death evasion in HCC
One characteristic feature of cancer cells is the development of mechanisms that lead to resistance against apoptosis [30, 89]. This resistance can also result in chemotherapy refractoriness, particularly when the mitochondria, which play a central role in apoptotic signaling, are targeted [92]. The increased accumulation of cholesterol in mitochondrial membranes resulting in a decrease in membrane fluidity, has been implicated in various human pathologies including Alzheimer’s disease, lysosomal storage disorders, and cancer [73, 93, 94] in addition to chronic liver disease [95, 96] and importantly, in HCC cancer cells [90, 91].
The accumulation of mitochondrial cholesterol has been found to contribute to resistance to mitochondria-targeted chemotherapy in cancer cells, due to the crucial role of mitochondria in cell death regulation and mediating chemotherapy [97–100]. The resistance to antineoplastic drugs such as arsenic trioxide, lonidamine, thapsigargin, and doxorubicin, which induce cell death through various mechanisms involving mitochondria, has been observed in HCC cells [99–100]. These chemotherapeutic agents trigger apoptotic cell death but their effectiveness is diminished in the presence of accumulated cholesterol in mitochondrial membranes. The accumulation of cholesterol in mitochondria of cancer cells has been shown to impair the release of cytochrome c, an essential step in apoptosis, demonstrating the significant role of mitochondrial cholesterol in regulating cell death [90, 101–103]. Indeed, removal of cholesterol from mitochondrial membranes and fluidization of the membranes can restore the ability of Bax to induce apoptosis, further highlighting the importance of membrane dynamics in apoptosome activation [103, 104]. Interestingly, lovastatin was found to reverse the resistance of H35 or HepG2 hepatoma cells to chemotherapeutic agents by inhibiting cholesterol synthesis and thereby preventing the accumulation of cholesterol in mitochondrial membranes. Furthermore, the inhibitor the squalene synthase inhibitor YM-53601 sensitized the cells to the chemotherapeutic agents without affecting isoprenoid metabolism [90], suggesting that reducing cholesterol accumulation in mitochondria may enhance the efficacy of cancer treatment. Some studies have proposed the combination of statins as coadjuvants to enhance the effectiveness of systemic chemotherapies [105, 106].
However, the antiapoptotic role of cholesterol by increasing the membrane order of mitochondria is masked by the depletion of the mitochondrial glutathione pool, which in normal conditions would render cells more sensitive to cell death induction [15]. In primary hepatocytes, the decrease in membrane fluidity provoked by cholesterol loading, affects the function of specific mitochondrial carriers, such as the 2-oxoglutarate carrier SLC25A11, leading to mitochondrial glutathione depletion and increased sensitivity to oxidative stress-induced cell death [15]. Interestingly, the inhibitory effect of mitochondrial cholesterol on the 2-oxoglutarate carrier is target-specific, as other membrane carriers are not affected by cholesterol accumulation. Conversely, mitochondria in HCC cells accumulate cholesterol, but this accumulation does not deplete mitochondrial glutathione. This apparent paradox occurring in HCC cells respect to the primary hepatocytes is explained by the overexpression of the SLC25A11 carrier in cancer cells, which allows overcoming the glutathione transport blockade and consequently increase their resistance to oxidative stress. This adaptation permits HCC cells to protect themselves from apoptosis and promotes tumor growth by maintaining enough oxidative phosphorylation [91, 107], while mitochondrial cholesterol accumulation in primary hepatocytes lead to mitochondrial respiratory defects and ROS production [70].
Overall, the augmented cholesterol load in mitochondria of HCC cells could potentially explain the acknowledged dysfunction of mitochondria and their involvement in cancer chemotherapy resistance. However, the advantageous overexpression of SLC25A11 in HCC cells might contribute to the promotion of cancer growth, ultimately leading to resistance to mitochondrial outer membrane permeabilization, and insensitivity to chemotherapy. In line with this, results in animals with diethylnitrosamine-induced HCC fed with high-fat diet supplemented with cholesterol (DEN-HFHC), demonstrated that the overexpression of STAR in mouse liver increased the incidence of HCC tumors concomitant with an increased expression of tumor markers, while its ablation specifically in hepatocytes decreased tumor burden. Analogous results were obtained using an animal model with displaying spontaneous MASH and HCC when fed HFHC diet (MuP-UPA-HFHC) [74]. These findings further support the notion that STAR-mediated mitochondrial cholesterol trafficking is playing a relevant for HCC onset in MASH [47].
In addition to cholesterol, cardiolipin, which is an acidic phospholipid exclusively found in the mitochondrial inner membrane (MIM), has been identified as a molecule that can modulate mitochondrial outer membrane permeabilization [101]. It is worth noting that cardiolipin contains a carbon backbone with four unsaturated fatty acyl chains, making it vulnerable to peroxidation. In fact, studies have shown that peroxidized cardiolipin has the ability to counteract the inhibitory effect of cholesterol on liposome permeabilization [91]. Thus, in the early stages of cancer development, the accumulation of cholesterol in mitochondria favors the formation of peroxidized cardiolipin through the depletion of mitochondrial glutathione. In this complex scenario, both cholesterol and cardiolipin peroxidation exert opposing effects on mitochondrial outer membrane permeabilization, and the final outcome may depend on the severity of mitochondrial glutathione depletion as well as the relative levels of cardiolipin and cardiolipin peroxidation generated. Interestingly, in established cancer cells such as human hepatoblastoma cells, the observed accumulation of cholesterol does not lead to mitochondrial glutathione depletion due to the overexpression of SLC25A11. This overexpression of SLC25A11 enables the preservation of cardiolipin in its reduced state, thereby tilting the balance towards resistance to apoptosis [91].
Mitochondrial cholesterol in hypoxia-inducible factor 1 alpha activation
Hypoxia is a well-recognized characteristic of cancer, particularly solid tumors, and it plays a significant role in tumor development. The disorganized structure and architecture of tumor vasculature leads to inefficient oxygen delivery, resulting in hypoxia, which is considered a negative prognostic factor for cancer patients, affecting their response to treatment and survival [108–110]. Activation of hypoxia-inducible factor 1 alpha (HIF-1α) enhances the production of pyruvate and lactate from glucose by increasing the transcription of key enzymes and transporters involved in glycolysis [111]. HIF-1α also inhibits the conversion of pyruvate to acetyl-CoA, compromising mitochondrial function and promoting aerobic glycolysis, therefore diverting glycolysis from mitochondrial oxidation towards to the less energy-efficient lactate production [112]. Additionally, HIF-1α induces the degradation of a subunit of cytochrome c oxidase, decreasing oxidative metabolism, and triggers mitochondrial autophagy, reducing the metabolism of fatty acids and glucose [113].
The HIF-1 is a central pathway involved in oxygen sensing and adaptation to hypoxic conditions, activating genetic programs and regulating key pathways, thereby contributing to cancer cell survival in low oxygen environments. HIF-1 is composed of stable β subunit (HIF-1β/ARNT) and labile α subunit (HIF-1α), which encompass three family members. In normoxia, HIF-1α is degraded by the proteasome after hydroxylation by prolylhydroxylases (PHDs) and ubiquitination by Von Hippel Landau (VHL) tumor suppressor. In low oxygen conditions, PHDs are inactivated, leading to HIF-1α stabilization and translocation to the nucleus for gene regulation [114, 115]. Importantly, PHDs are also dependent on iron (Fe2+), ascorbate and on the tricarboxylic acid cycle (TCA) intermediate 2-oxoglutarate as cofactors. Besides oxygen, PHDs are also regulated by oxidative stress and ROS originating from mitochondria during hypoxia [116]. Therefore, the activity of PHDs can modulate HIF-1α regulation during normoxia in the context of ROS generation.
HIF-1α is overexpressed in human cancers due to intratumoral hypoxia and genetic alterations in oncogenes and tumor suppressor genes. Therefore, inhibiting HIF-1α has become an area of interest for cancer treatment [108, 117]. In this regard, various small molecules have been developed to target different levels of HIF-1α inhibition [118]. Additionally, an increase in cholesterol has been linked to HIF-1α activation through a mechanism involving ROS generation from mitochondria. Interestingly, cholesterol-mediated HIF-1α stabilization has been observed in normoxia and requires elevated nitric oxide levels and mitochondrial ROS production [119, 120]. Moreover, the depletion of mitochondrial DNA abolishes cholesterol-induced HIF-1α, supporting the mitochondrial origin of the ROS.
Mitochondrial cholesterol accumulation in normoxia can lead to decreased 2-oxoglutarate in cytosol, due to the inhibition of SLC25A11 which transports 2-oxoglutarate from TCA cycle out of the mitochondria. Given that 2-oxoglutarate is a PHD cofactor, this cytosolic decrease results in impaired PHDs activity and promotes subsequent HIF-1α stabilization. The disruption of the 2-oxoglutarate dehydrogenase complex controls HIF-1α stability in aerobic conditions [121]. Other intermediates of the TCA cycle, such as succinate, may also inhibit PHDs. The 2-oxoglutarate analog 3-oxoglutarate has been shown to decrease normoxic HIF-1α in cancer cells, inducing cell death and reducing tumor growth [122].
In line with these mechanisms, it is conceivable that STAR-mediated cholesterol trafficking to mitochondria results in HIF-1α stabilization through mitochondrial ROS generation and changes in TCA metabolites affecting PHD-induced HIF-1α hydroxylation. STAR expression is regulated by HIF-1α, consistent with the stimulating effect of hypoxia in steroidogenesis, suggesting a mutual upregulation that promotes mitochondrial cholesterol accumulation and sustained HIF-1α stabilization [123–126]. Consequently, STAR and HIF-1α interplay may be playing a potential role in HCC survival and resistance to chemotherapy.
Activation of alternative bile acid synthesis pathway
As stated before, there is evidence indicating that the rate of cholesterol transport to the mitochondrial mediated by STAR, rather than CYP27A1, is the rate-limiting step of the alternative pathway of bile acid synthesis [68, 69]. In the context of MASH-driven HCC development, modulating STAR expression resulted in alterations in the generation of bile acid species and the total bile acid pool [74]. These effects were independent of the expression levels of Cyp27a1/Cyp7b1, highlighting the pivotal role of STAR in the biotransformation of cholesterol into bile acids. Moreover, the trafficking of cholesterol to mitochondria would increase the synthesis of bile acids by the alternative pathway, which is not controlled by the feed-back inhibition as in the classical pathway [66]. These findings suggest that in diseased states where both cholesterol and STAR are induced, such as MASH-driven HCC, the STAR-dependent alternative pathway may surpass the classical pathway regulated by CYP7A1 as the predominant route of cholesterol-mediated bile acid synthesis. Additionally, ER stress, a significant factor in MASH-HCC development, has been identified as a mechanism that regulates bile acid synthesis by reducing the expression of CYP7A1. Cholic acid is the predominant bile acid synthesized through the classical pathway while chenodeoxycholic acid is predominantly synthesized through the alternative pathway via CYP27A1 and CYP7B1. In humans, chenodeoxycholic acid is further metabolized by intestinal bacteria to LCA and in this context, it has been found that liver tumor-inducer cells treated in vitro with the secondary bile acids lithocholic and deoxycholic acid induced the expression of genes related to self-renewal, stemness, and inflammation [74], which are involved in HCC development [127, 128]. Moreover, these observations are consistent with the recognized role of bile acids in promoting MASH progression and HCC development [64] associated with an increase in circulating bile acids in human and in mouse models [129–133].
Role on mutagenesis and tumor initiation
The threshold at which apoptosis becomes irreversible is widely accepted to be the point at which mitochondrial outer membrane permeabilization occurs. Nevertheless, cancer cells can evade this step and inhibit caspase activation, ensuring their survival under certain conditions. This mechanism, observed in post-mitotic cells and certain cancer cells, allows them to recover and evade cell death once the stimuli triggering mitochondrial outer membrane permeabilization are removed [134, 135]. For example, the release of cytochrome c through mitochondrial outer membrane permeabilization can be targeted for degradation by the proteasome, preventing the assembly of the apoptosome complex [136]. In addition to caspase inhibition, the survival of cancer cells following mitochondrial outer membrane permeabilization requires the presence of a pool of intact mitochondria [135]. Moreover, it has been shown that sub-lethal apoptotic stimuli, inducing limited mitochondrial permeabilization, can promote DNA damage, genomic instability, and ultimately, carcinogenesis, through the partial activation of apoptosis nucleases [137–139]. This mechanism has two important implications for cancer initiation and progression. Firstly, low-level limited apoptosis can drive mutagenesis in surviving cancer cells, serving as a driving force towards malignancy. In other words, this mechanism partially explains the association between genetic instability in liver tissue when toxic and/or metabolic insults that lead to liver injury, ultimately result in an inflammatory environment characterized by cycles of hepatocellular cell death and compensatory proliferation, as observed in MASH and ASH. Secondly, sub-lethal apoptotic anticancer therapies have the potential to increase the tumorigenic potential of surviving cancer cells by promoting the occurrence of new mutations that favor relapse and confer resistance to chemotherapy.
Besides, it is plausible that cholesterol loading in mitochondria, which renders HCC precursor cells more resistant to apoptosis, induces a greater number of incomplete mitochondrial outer membrane permeabilization and heightened apoptotic stimuli, thereby creating a driving force towards mutagenesis and cell transformation, which ultimately lead to carcinogenesis. In fact, studies have shown that treating mice with ezetimibe, a cholesterol absorption inhibitor that effectively reduces liver cholesterol levels, decreases the incidence of tumors in both induced [74] and in a spontaneous model of HCC [140]. Therefore, it is worth noting the significant effect of reducing liver cholesterol levels on decreasing the occurrence of liver tumors in various models, suggesting that decreased cholesterol burden may reduce mutagenesis in cancer precursor hepatocytes, although there are likely additional mechanisms at play.
Given the central role of hepatocytes in biotransformation and metabolism of xenobiotics, ROS production constitutes an important burden in liver physiology [141]. In addition, STAR-mediated trafficking of cholesterol to mitochondria in MASLD setting compromises mitochondrial respiration [40, 70] and hinders import of glutathione to mitochondria, subsequently leading to excess ROS production from mitochondria. Oxidative stress originated from mitochondria serves as a catalyst for the elevation of the mutagenic rate [142] while inducing stabilization the pro-survival HIF-1α [120], further supporting the progression towards malignancy of HCC precursor cells.
Immunomodulatory effects of cholesterol HCC
Another critical survival characteristic of tumors is their ability to evade immune surveillance, which is the process by which transformed cells are usually detected and eliminated by the immune system. As transformed cells usually display neoantigens in their surface, this ability allows that only cells with the capacity to escape immune surveillance are able to establish a tumor during the microevolution process towards malignancy. Definitely, the immune system in the liver plays a vital role in tumor surveillance and the failure of this system can lead to the development of liver cancer [143]. Additionally, the interaction between immune cells and cancer cells is influenced by the metabolic changes that occur in immune cells [144]. The adaptive immune system, particularly CD8+ T cells and lymphotoxin β, has been identified as a key mediator in liver injury and the development of HCC [145]. In this regard, the antitumor function of natural killer cells is known to increase with high serum cholesterol levels, leading to a reduction in the growth of liver tumors [146, 147]. While cholesterol in some immune cells activate their activity, the accumulation of cholesterol in liver tissue and/or the presence of its metabolites, such as oxysterols or bile acids, may create an immunosuppressive microenvironment in this tissue. Indeed, there is an observed immunosuppressive effect of a cholesterol-enriched tumor microenvironment, which contributes to unrestricted tumor growth [148, 149]. Consistent with this, an increase in liver cholesterol levels was found to enhance the expression of lymphocyte exhaustion genes such as Pdl1 and Ctl4 among others, in mice with DEN-HFHC-induced HCC and in the spontaneous MuP-UPA transgenic mice model of HCC. Importantly, pharmacological treatment with ezetimibe, which reduces liver cholesterol levels, repressed the expression of these genes and the incidence and growth of tumors, suggesting that this treatment could potentially restore the cytolytic activity of CD8+ cells [140] in both the induced and spontaneous model. Furthermore, in another study using the MuP-UPA transgenic spontaneous HCC model, treatment with checkpoint inhibitors was able to induce regression of HCC tumors [150]. These results, although very promising, showed a partial response as it has been found in humans in the clinical setting, indicating that additional factors are blunting the cytolitic response against tumors in HCC that cannot be completely corrected with checkpoint inhibitors. These results prompt to the use of pharmacological interventions for reducing liver cholesterol burden as a coadjuvant to immunotherapies.
Potential of cholesterol modulation in MASH-driven HCC
Statins, also known as HMG-CoA reductase inhibitors, are the primary class of drugs prescribed for lowering serum cholesterol levels by reducing the endogenous synthesis of cholesterol. A recent meta-analysis has presented evidence supporting the significant therapeutic benefits of statins, as they have been found to significantly decrease liver biochemical indicators in individuals with MASLD [151]. Importantly, meta-analyses have found consistent associations with a reduced HCC risk among patients with chronic liver disease for statins [37], and therefore, it has been proposed that these drugs may also reduce the risk of HCC in patients with MASLD [152]. Notably, lipophilic statins appear to have a greater potential for benefit compared to hydrophilic statins, as seen in a study of viral hepatitis HCC patients in Sweden [153]. However, the mechanism of action of statins extends beyond their cholesterol-lowering effect alone; they also possess anti-inflammatory, anti-proliferative, and anti-angiogenic properties [154], which complement their well-established cholesterol-lowering effect. Nonetheless, it remains unclear whether the cholesterol-lowering effect of statins is limited to the circulation or if it also reduces non-esterified cholesterol in liver tissue, which is responsible for the lipotoxic and pro-carcinogenic effects. Interestingly, ezetimibe treatment has been shown to reduce tumor burden in mice with excess dietary cholesterol, and this effect is associated with decreased hepatic cholesterol levels [74, 140]. Therefore, cholesterol-targeted therapies for HCC should consider mechanisms for specifically reducing cholesterol in the liver, while avoiding depletion of cholesterol in immune cells where it is required for activation and proper function. It is also important to determine whether dietary cholesterol or endogenous synthesis is the main source of cholesterol driving cholesterol accumulation in hepatocytes.
Conclusions
In the course of the transformation from healthy hepatocytes to fully differentiated HCC a microevolution selection process occurs, where cells with specific metabolic features, resistance to cell death, stemness, proliferative potential, genetic instability, mutagenesis, and immune evasion are chosen for survival. As previously discussed, there is evidence proposing that dysregulated cholesterol metabolism plays a significant and often underestimated role in the driving forces behind the transition from MASLD to MASH to the onset hepatocellular carcinogenesis. Initially, free cholesterol accumulates in hepatocytes, either as a result of increased synthesis and lipogenesis in MASLD or from dietary sources. This accumulation of cholesterol in the ER leads to ER stress, which in turn drives further cholesterol synthesis, lipotoxicity, and ultimately inflammation. Besides, the trafficking of cholesterol to mitochondria impairs mitochondrial respiration and blocks the import of glutathione into mitochondria, resulting in additional toxicity to hepatocytes. This creates a cycle of cell death and regeneration, leading to a fibrotic and inflammatory environment characteristic of MASH. Subsequently, the increased expression of STAR in response to ER stress induces mitochondrial cholesterol trafficking, rendering initiated hepatocytes resistant to apoptosis and increasing the likelihood of incomplete apoptosis. Consequently, incomplete apoptosis, along with oxidative stress, an inflammatory milieu and heightened compensatory proliferation, leads to genetic instability, mutagenesis and epigenetic changes that are transmitted to the daughter cells. Once genetic and epigenetic instability is established, survival mechanisms such as overexpression SLC25A11 are selected in initiated hepatocytes. The increased cholesterol and STAR overexpression in these cells activate alternative bile acid synthesis and its metabolites, which promote self-renewal, stemness, and inflammation programs. Furthermore, stabilization of HIF-1α induced by ROS and a decrease in cytosolic 2-oxoglutarate promote survival and a shift towards glycolytic metabolism, which is characteristic of proliferating and cancer cells. These mechanisms work in conjunction with immune evasion mechanisms such as overexpression of PDL1 and CTL4 in cancer cells which is induced by cholesterol accumulation by an undefined mechanism, which drive CD8+ T cell exhaustion and failure of cytolytic intratumoral response. Other mechanisms that result in immune dysfunction, such as microenvironment acidification due to highly glycolytic metabolism or oxidative stress also play a role in immune supression. The processes involved in the transition from healthy to MASLD to MASH and the onset and progression of HCC are briefly summarized in Figure 2.
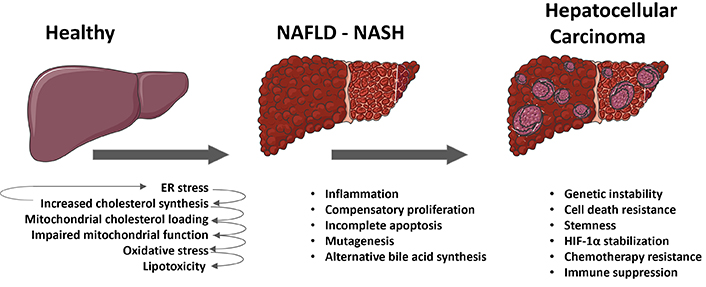
Role of cholesterol in the transition from metabolic dysfunction-associated fatty liver disease (MASLD) to metabolic dysfunction-associated steatohepatitis (MASH) to hepatocellular carcinoma (HCC). The schematic summary delineates the various processes and mechanisms involved in the transition from a healthy liver to MASLD, then progressing to steatohepatitis (MASH), ultimately resulting in the onset of HCC in MASH. External metabolic or toxic insults give rise to endoplasmic reticulum (ER) stress in hepatocytes, consequently leading to an upregulation in cholesterol synthesis. This endogenous elevation in cholesterol levels, coupled with the dietary intake of cholesterol, perpetuates ER stress and induces the expression of steroidogenic acute regulatory protein (STAR), thereby promoting the trafficking of cholesterol to mitochondria. The augmented mitochondrial cholesterol load impairs mitochondrial function and impedes the import of glutathione, ultimately causing oxidative stress and lipotoxicity in hepatocytes. As MASH ensues, cycles of cell death and regeneration stimulate compensatory proliferation within an inflammatory milieu, while the accumulation of mitochondrial cholesterol contributes to an increased frequency of incomplete apoptosis, alternative bile acid synthesis, oxidative stress, genetic instability, stemness, and further mutagenesis in initiated hepatocytes. Upon the onset of HCC, in addition to these mechanisms, cholesterol accumulation may also drive additional survival strategies such as stabilization of hypoxia-inducible factor 1 alpha (HIF-1α), resistance to chemotherapy, and immune exhaustion and suppression. Parts of the figure were used from or adapted from pictures provided by Servier Medical Art, licensed under CC BY 4.0
While there is evidence supporting these mechanisms, there are still many areas that remain obscure, and our understanding of how cholesterol accumulation in hepatocytes and cancer cells exerts these effects is still incomplete. Additionally, HCC and other tumors display a wide range of heterogeneity in terms of metabolism, mutation burden, resistance to chemotherapeutics, and invasiveness. Therefore, the specific contribution of each proposed mechanism is likely to be highly heterogeneous as well. However, empirical and epidemiological evidence supports the therapeutic potential of cholesterol modulation.
Overall, cholesterol metabolism plays a multifaceted role in various aspects of HCC genesis, progression, survival, and immune suppression. Given the existing pharmacological strategies to control cholesterol metabolism, modulating it could potentially lead to exciting new approaches for liver cancer prevention and treatment. These strategies may also be applicable to other tumors where cholesterol plays a pivotal role, such as breast, prostate, or ovarian cancer. Despite the cholesterol classification as a neutral lipid in terms of its physicochemical properties, there is strong evidence pointing to that cholesterol is not a neutral player in the pathophysiology of the transition from MASLD to MASH to HCC.
Abbreviations
| ABC: | ATP-binding cassette |
| ACAT: | acyl-CoA cholesterol acyltransferase |
| ASH: | alcoholic steatohepatitis |
| DEN-HFHC: | diethylnitrosamine-induced hepatocellular carcinoma fed with high-fat diet supplemented with cholesterol |
| ER: | endoplasmic reticulum |
| FGF15: | fibroblast growth factor 15 |
| FPP: | farnesyl pyrophosphate |
| FXR: | farnesoid X receptor |
| HCC: | hepatocellular carcinoma |
| HDL: | high density lipoproteins |
| HIF-1α: | hypoxia-inducible factor 1 alpha |
| HMG-CoA: | 3-hydroxy-3-methylglutaryl-CoA |
| HMGCR: | 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase |
| LDL: | low-density lipoprotein |
| MASH: | metabolic dysfunction-associated steatohepatitis |
| MASLD: | metabolic dysfunction-associated fatty liver disease |
| NPC1: | Niemann-Pick type C protein 1 |
| PHDs: | prolylhydroxylases |
| ROS: | reactive oxygen species |
| SLC25A11: | solute carrier family 25 member 11 |
| SREBP2: | sterol regulatory element protein 2 |
| STAR: | steroidogenic acute regulatory protein |
| TCA: | tricarboxylic acid cycle |
| VLDL: | very low-density lipoprotein |
Declarations
Acknowledgments
The elements in the figures have been obtained from Servier slides (https://smart.servier.com/smart_image/slide/).
Author contributions
VR: Conceptualization, Investigation, Writing—original draft, Writing—review & editing.
Conflicts of interest
The author declare that he has no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2024.