
Abstract
Patients with advanced chronic liver disease can develop specific pulmonary complications related or unrelated to pre-existing lung disease. The three major pulmonary complications in this patient population include hepatopulmonary syndrome (HPS), portopulmonary hypertension (PoPH), and hepatic hydrothorax (HH). These entities are most often revealed by increasing dyspnea together with signs of portal hypertension. The prevalence of these complications remains underestimated due to the lack of routine screening of the cirrhotic population. The pathophysiology of HH is better understood than that of HPS and PoPH. The clinical features, diagnosis, and therapeutic options of these pulmonary complications are extensively discussed in this chapter. Liver transplantation may offer a curative therapy in highly-selected cases and MELD exception points allow priority access to liver transplantation, thus avoiding potential deterioration while awaiting transplant and providing a better post liver transplant survival. The complexity of managing these pulmonary complications requires a multidisciplinary team approach, especially when liver transplantation is indicated.
Keywords
Portopulmonary hypertension, hepatopulmonary syndrome, hepatic hydrothoraxIntroduction
Patients with advanced chronic liver disease are prone to pulmonary complications independently of any pre-existing lung disease. When dyspnea occurs, practitioners should investigate their patients for common pulmonary disorders as well as those related to the liver, including hepatopulmonary syndrome (HPS), portopulmonary hypertension (PoPH), and hepatic hydrothorax (HH). In recent years, knowledge and understanding of these liver-related pulmonary complications have improved considerably. In this review, we summarize the pathogenesis, clinical features, and treatment of PoPH, HPS, and HH.
Portopulmonary hypertension
Epidemiology
Pulmonary arterial hypertension (PAH) associated with portal hypertension was first described in 1951 by Mantz and Craige [1], but the term “portopulmonary hypertension (PoPH)” was first used by Yoshida et al. [2] in 1993 in association with the first successful liver transplant in a 39-year-old man with decompensated autoimmune cirrhosis and pulmonary hypertension (PH). PoPH has a prevalence of 5% to 6% among patients with advanced liver disease, while PAH occurs in 5% to 15% of patients in PH registries [3–7]. The prevalence of PoPH is surely underestimated due to the lack of routine screening of the cirrhotic population who are not being considered for liver transplantation (LT).
Pathogenesis
The underlying mechanisms for the development of PoPH remain poorly understood. PoPH may develop in the presence of either cirrhotic or non-cirrhotic portal hypertension, such as Budd-Chiari syndrome, portal vein thrombosis, schistosomiasis, and Abernethy malformation [4, 8, 9]. One current theory refers to the increase in venous return from portosystemic shunts leading to an increase in shear stress on pulmonary arteries thus prompting remodeling of these vessels over time [10]. The hyperkinetic syndrome (characterized by an increased cardiac output and a decreased peripheral vascular resistance) and renal sodium and water retention are the hallmarks of portal hypertension and increase the shear stress on the small pulmonary arteries contributing to endothelial dysfunction with a deficient nitric oxide (NO) availability. A strong association was also demonstrated between large portosystemic shunts and PoPH, suggesting a substantial contribution of the hyperkinetic syndrome to the occurrence of PoPH [11]. Another mechanism for the development of PoPH involves excessive production of vasoconstrictors (norepinephrine, endothelin-1, angiotensin II, leukotrienes, and thromboxane A2) that are not eliminated by the liver in the presence of portosystemic shunts thus reaching the pulmonary vascular bed to produce their vasoconstrictor effects.
There is no clear correlation between the severity of liver disease and the prevalence and severity of PoPH. Hadengue et al. [12] did not observe any correlation between hepatic venous pressure gradient and pulmonary vascular resistance (PVR) level. However, all patients with portal hypertension will not develop PoPH, suggesting that genetic or other factors are involved. Roberts et al. [13] identified genetic polymorphisms of oestrogen-related genes and other pathways involving cell growth/apoptosis and oxidative stress that may influence emergence of PoPH. A retrospective cohort study of 190 adult candidates (87 women) for LT who had PoPH showed that women were more likely to have autoimmune liver disease, had lower MELD score and higher baseline PVR, as compared with men [14]. Those observations support the involvement of genetic polymorphisms in the pathogenesis of PoPH.
Diagnosis
The clinical classification of PH distinguishes five groups, including PAH which belongs to group 1 [15] (Table 1). The 2018 World Symposium on PH defined PH as mean pulmonary arterial pressure (mPAP) > 20 mmHg [11], a threshold having a prognostic relevance in US veterans (portal hypertension was rare in this cohort) [16, 17]. However, an isolated increase in mPAP does not constitute a clinical entity per se, because it could be due to increased cardiac output and/or excessive fluid retention, two conditions frequently observed in portal hypertension. Based on recent literature, the upper limit of normal PVR and the lowest prognostic threshold of PVR, was recognized to be around 2 Wood units (WU) rather than PVR ≥ 3 WU as previously recommended [18, 19]. The 2022 European Society of Cardiology and European Respiratory Society (ESC/ERS) PH guidelines adopted this new PVR threshold for an earlier diagnosis of PAH (and by default PoPH, Table 1) [20]. Thus, PoPH is currently defined by right heart catheterization as pre-capillary PH by the concomitant presence of mPAP > 20 mmHg, pulmonary arterial wedge pressure (PAWP) ≤ 15 mmHg and PVR > 2 WU in the absence of other causes of PH, such as chronic thromboembolic PH or intrinsic lung disease [20]. These new PAH diagnostic criteria have recently demonstrated that they were associated with poor global and transplant-free survivals in cirrhotic patients with portal hypertension [21]. PAWP measured by right heart catheterization discriminates pre-capillary PH associated with pulmonary arterial disease (high PVR) from PH related to hyperkinetic syndrome or volume overload. Depending on PAWP, PH is subdivided into pre-capillary PH (PAWP ≤ 15 mmHg), and post-capillary PH (PAWP > 15 mmHg) associated with left heart disease (Table 1).
Hemodynamic definitions of pulmonary hypertension
| Definition | Hemodynamic criteria | Clinical groups |
|---|---|---|
| PH | mPAP > 20 mmHg | |
| Pre-capillary PH | mPAP > 20 mmHgPAWP ≤ 15 mmHgPVR > 2 WU | Group 1: PAH: idiopathic, heritable, associated with drugs/toxins, with concomitant diseases (including portal hypertension), with features of venous/capillary involvementGroup 3: PH associated with lung diseases and/or hypoxiaGroup 4: PH due to pulmonary artery obstructionsGroup 5: PH with unclear and/or multifactorial mechanisms |
| Isolated post-capillary PH | mPAP > 20 mmHgPAWP > 15 mmHgPVR ≤ 2 WU | Group 2: PH associated with left heart diseaseGroup 5: PH with unclear and/or multifactorial mechanisms |
| Combined pre- and post-capillary PH | mPAP > 20 mmHgPAWP > 15 mmHgPVR > 2 WU |
Portopulmonary hypertension belongs to PAH (group 1 PH) characterized by precapillary PH in the absence of other causes of pre-capillary PH, such as chronic thrombo-embolic PH and PH associated with lung diseases. More details are reported in Humbert M, et al. Eur Heart J 2022 [20]. mPAP: mean pulmonary arterial pressure; PAH: pulmonary arterial hypertension; PAWP: pulmonary arterial wedge pressure; PH: pulmonary hypertension; PVR: pulmonary vascular resistance; WU: Wood units (1 WU = 80 dyn.s.cm-5)
Note. Adapted with permission from “Portopulmonary hypertension: An unfolding story” by Thévenot T, Savale L, Sitbon O. Clin Res Hepatol Gastroenterol. 2020;45:101492 (https://www.sciencedirect.com/science/article/abs/pii/S221074012030187X?via%3Dihub). © 2020 Elsevier Masson SAS.
Clinical presentation
Symptoms of PH are nonspecific and mainly linked to right ventricle dysfunction, and typically associated with exercise in the earlier course of the disease [20]. Dyspnea upon exertion and fatigue are the most common symptoms, but lack of specificity leads to potentially harmful delayed diagnosis of PH. The median time between diagnosis of liver disease and PoPH was 5 years in a large U.S. registry of patients ineligible for LT [22]. Other symptoms may occur, such as coughing, palpitations, and syncope during or shortly after exertion. At the late stage, weight gain with edema of the legs secondary to right-sided heart failure may be observed. Other symptoms are less frequently reported, such as hoarseness, wheeze, and angina. These symptoms must draw the attention of practitioners to the possible existence of PH in order to ensure a prompt referral to expert centres.
Screening
Since the symptoms of PoPH are nonspecific and its diagnosis may compromise LT eligibility [or transjugular intrahepatic portosystemic shunt (TIPS) placement], it is recommended to screen all LT candidates for PAH using echocardiography and to annually repeat this examination as long as the patient remains on the waiting list, although the optimal interval between two examinations remains unclear [9]. Doppler echocardiography allows calculation of systolic pulmonary arterial pressure (sPAP) using the modified Bernoulli equation [sPAP = 4 × tricuspid regurgitation velocity (TRV)² + RAP; TVR is the peak tricuspid regurgitation velocity, RAP is the right atrial pressure], which aids decisions to perform more invasive heart catheterization. There is no consensus regarding the best threshold of sPAP to trigger heart catheterization. By combining a sPAP > 38 mmHg and a dilatation of the right ventricle > 3.3 cm, the specificity and the positive predictive value were respectively of 93% and 41% [23]. Considering the inaccuracies in estimating RAP, guidelines recommend using a peak TRV > 2.8 m/s (and not the estimated sPAP) together with additional echocardiographic signs of right ventricle pressure overload (details are reported in [20]) for assigning the probability of PH. Echocardiogram is a useful tool for routine screening of PoPH, but it cannot reliably differentiate patients with PoPH from patients with elevated right ventricle systolic pressure but a normal PVR due to an increased cardiac output and/or volume overload [24]. So, right heart catheterization remains essential to confirm or rule out PoPH and to evaluate its severity.
Electrocardiogram abnormalities associated with PH include right axis deviation, right ventricular hypertrophy, right bundle branch block, and a nonspecific prolonged QTc interval. Enlargement of the pulmonary arteries, right atrium, and right ventricle may be observed on chest-X ray in patients with PAH. Pulmonary function tests and arterial blood gas are also of value to detect concomitant lung disease and the need for supplementary oxygen.
Impact of PoPH on survival
The survival of untreated patients with PoPH is poor. The 5-year survival and the median survival of 19 patients who received no specific therapy for PH and no LT were respectively of 14% and 15 months, and 54% had died within 1 year of diagnosis [25]. In the U.S. REVEAL registry, patients with PoPH (n = 174) had worse outcomes compared with patients suffering from idiopathic or heritable PAH (5-year survival of 40% vs. 64%, respectively), even though pulmonary haemodynamics appeared more favorable in PoPH patients [5]. Globally, the reported one-year survival of patients with PoPH ranges approximately between 35% to 46% without treatment [26]. The survival observed in the French registry (69% at 3 years) is higher because these patients mainly received specific treatment for PAH [27]. The survival of these patients is significantly influenced by the severity of both the liver disease and pulmonary hemodynamics [4].
Management of PoPH
General measures
Hypoxic conditions (such as long-stay at high altitudes) may aggravate PH and should be avoided. Diuretics and low sodium diet are useful for reducing right ventricular volume overload. Anticoagulation is considered in patients with idiopathic PAH [20] but is not indicated in PoPH, due to an increased risk of bleeding in patients with portal hypertension [28]. Nonselective beta-blockers, used for the primary and secondary prevention of variceal bleeding in cirrhotic patients, should be avoided in the setting of PoPH, as they may worsen cardiac output and exercise capacity [20, 29]; consequently, endoscopic band ligation represents a safer alternative therapy and should be considered.
Specific-PAH therapies
Epoprostenol IV was the first molecule approved by the FDA for PAH in the early 1990s. Currently, there are three major classes of PAH-targeted drugs approved for PAH, the endothelin receptor antagonists, prostacyclin analogs, and drugs targeting the NO-soluble guanylate cyclase-cyclic guanosine monophosphate (cGMP) pathway, but none of them are FDA-approved for PoPH because almost all randomized controlled trials have excluded patients with PoPH. A recent systematic review with meta-analysis confirmed that PAH therapies, with or without LT, significantly improve pulmonary hemodynamics (mPAP and PVR) and overall survival in patients with PoPH (1-year and 3-year survival of 86% and 69%, respectively); more importantly, 44% of patients became eligible for LT [30]. The largest series of patients with PAH confirms the effectiveness of PAH treatments on pulmonary hemodynamics and exercise capacity with a post-LT survival of 81% at 5 years [27]. A retrospective study of 160 patients with PoPH ineligible for LT (mostly due to uncontrolled PH) confirmed the poor prognosis of non-transplanted patients having a 3-year survival of 51%; the causes of death were mainly attributed to liver disease (38%) and right-sided heart failure (23%) [22].
Endothelin receptor antagonists
This therapeutic class includes bosentan, ambrisentan, and macitentan. Bosentan, the first-in-class, has been associated with elevated aminotransferases in 3% to 18% of cases, and thus has been used with great caution in patients having both end-stage liver disease and PoPH [30, 31]. The more recent pharmacotherapies, ambrisentan and macitentan, have not been associated with significant hepatotoxicity. The PORTICO study is the first randomized controlled trial of PAH therapy specifically dedicated to symptomatic patients with PoPH, but excluded patients with severe liver disease (i.e., Child-Pugh C and MELD ≥ 19) [32]. In this trial, 43 and 42 patients respectively received macitentan 10 mg/day orally or placebo for a 12-week double-blind period, followed by a 12-week open-label period, during which all patients received macitentan. The primary endpoint was reached and showed a 35% decrease in PVR at week 12 with macitentan, together with improvements in cardiac index and mPAP. However, no significant improvement was observed in the 6-minute walk distance [32]. Overall, macitentan was well tolerated with side effects typical for endothelin receptor antagonists, including anemia, headache, and peripheral edema. There was no worsening of either hepatic function or portal hypertension, as shown by the absence of any increase in hepatic venous pressure gradient with macitentan, despite the increase in cardiac output.
Drugs targeting the NO-guanylate cGMP pathway
The pulmonary vascular bed is rich in phosphodiesterases-5, enzymes involved in the breakdown of cGMP. By blocking the breakdown of cGMP, phosphodiesterases-5 inhibitors cause vasorelaxation. Two phosphodiesterase-5 inhibitors, sildenafil, and tadalafil, and the soluble guanylate cyclase stimulator riociguat are currently approved as PAH therapies. Riociguat acts independently of endogenous NO to promote vascular remodeling and vasorelaxation. Studies using these agents have reported improvement in pulmonary hemodynamics, exercise capacity, right ventricular function, and eligibility for LT [30, 33, 34]. The PATENT-1 study, a multicenter randomized controlled trial of riociguat vs. placebo for the treatment of PAH, included 13 patients with PoPH (11 and 2 randomized to the riociguat and placebo arms, respectively). Riociguat was found to be well tolerated and associated with an improvement in the 6-minute walk test and PVR [34].
Prostacyclin analogs
Endothelial dysfunction leads to a decrease in the production of the vasodilators prostacyclin (prostaglandin I2) and NO. Epoprostenol has been the first synthetic prostaglandin I2 approved for PAH therapy; because of the short biological half-life (3–5 min) of this drug requiring a continuous intravenous administration via a tunnelled catheter, new stable analogues were developed, such as iloprost, beraprost, and treprostinil or more recently the non-prostanoid prostacyclin receptor agonist, selexipag. The most common adverse events related to prostacyclin analogs are due to systemic vasodilation and include headache, flushing, jaw pain, and diarrhea. At this time, we did not identify any published case reports or studies describing the safety or efficacy of oral treprostinil or selexipag in patients with PoPH.
Treatment approach
No data are available for the efficacy of PAH drugs in patients whose mPAP is < 25 mmHg and whose PVR is < 3 WU. Only one retrospective study suggests that “early PoPH” using recent PoPH diagnostic criteria had a poor prognosis, comparable to that of patients with PoPH who receive PAH therapy [21]. The best management and optimal drug approaches remain unclear for patients with PoPH mainly due to the exclusion of PoPH from almost all randomized controlled trials of PAH [3]. The 2022 ESC/ERS guidelines recommend that initial monotherapy should be considered in PoPH, followed by sequential combination if necessary, taking into consideration the underlying liver disease and indication for LT. The French PH Registry (2007–2017) confirmed that oral monotherapy was the most frequently used strategy (74%) while upfront dual and triple combination therapy were used in only 15% and 0.8%, respectively [27]. However, upfront monotherapy or combined therapy may differ greatly according to the geographical origin of healthcare practitioners and their personal preference and experiences with PAH therapy [35].
Liver transplantation
In a large single-center study of patients with PoPH prospectively enrolled between 1988 to 2019 demonstrated that these patients who did not receive LT had a poor prognosis despite the use of PAH-targeted therapies and improvements in hemodynamics [22]. The impact of LT on PoPH (and vice versa) is complex, and to date, it has not been possible to predict outcomes of PoPH following LT. In a series from the Mayo Clinic before the era of modern management of PAH, cardiopulmonary-related mortality after LT was 100% in patients with mPAP ≥ 50 mmHg, and 50% if preoperative mPAP was 35–50 mmHg with PVR ≥ 3.1 WU [36]. Access to LT is improved by early use of PAH-targeted treatments, while MELD exception points offer the possibility of priority access to LT, thus avoiding potential deterioration of PAH while awaiting transplant [9, 27]. In 2021, the MELD exception criteria for PoPH have been revised and indicated that patients with mPAP between 35 and 45 mmHg together with a PVR < 3 WU could receive LT [37, 38]. For other patients, LT should be deferred until improvement of hemodynamic parameters on heart catheterization (Figure 1). The collaboration between surgeons and anesthesiologists is crucial for the successful management of PoPH. PAH therapy should not be discontinued abruptly after LT because there is a high risk of PoPH worsening during the 6-month post-LT period; however, beyond this period, stabilization, improvement, and normalization of pulmonary hemodynamics have been reported, with or without PAH-targeted therapy [39]. A single-center study and a recent meta-analysis reported better survival in patients who were transplanted after being medically treated than in those who were treated with PAH therapies only [30, 40]. Analysis of data from the French PH registry (2007–2017), including 637 PoPH patients (63 of whom had LT), showed that survival of patients receiving LT was better, regardless of Child-Pugh and MELD scores, compared to non-transplanted patients [27]. The survival benefit of LT in PoPH patients raises the question of whether LT should be considered in selected PoPH patients who do not have a liver transplant indication because of mild liver disease. At this point, this question remains unanswered. Nevertheless, the improved survival of transplanted patients may be driven by the selection of patients without severe comorbidities. No well-established predictors of hemodynamic response to LT are available, and nearly half of PoPH patients need to continue PAH therapy after LT. In a large retrospective cohort of patients enrolled in 3 US transplant centers, 42% of patients who had undergone LT could discontinue PAH-specific therapies after LT [41]. Consequently, LT should not be considered as a “cure therapy” for PoPH. Overall, post-LT survival was satisfactory in most series, with 3-year survivals ranging from 63% to 87% [3]. Slightly worse results were reported in a retrospective English series (1992–2012), with 3- and 5-year post-LT survival of 59% and 54%, respectively; however, only 8/28 transplant patients had received PAH treatment before LT [42].
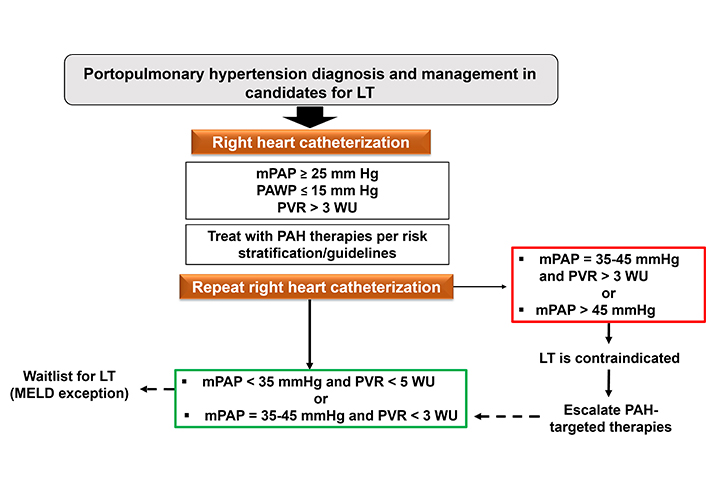
Algorithm for the management of liver transplant candidates in the presence of portopulmonary hypertension. LT: liver transplantation; mPAP: mean pulmonary arterial pressure; PAWP: pulmonary arterial wedge pressure; PVR: pulmonary vascular resistance; PAH: pulmonary arterial hypertension; WU: Wood units
Given the challenging management of PoPH, a multidisciplinary team approach is required. Despite significant breakthroughs in the understanding of PoPH, important unresolved questions remain regarding the optimal noninvasive evaluation of perioperative risk, the best medical PAH-therapy, and the predictors of hemodynamic response to LT. The answers to these research questions will require strengthened collaboration among scientists.
Hepatopulmonary syndrome
Epidemiology and definitions
Although the association between pulmonary functional alterations and liver diseases has been recognized for more than 100 years, the term “hepatopulmonary syndrome” appeared only in 1977 after the recognition of the involvement of intrapulmonary vasodilatations (IPVDs) in the alteration of gas exchanges [43, 44]. Diffuse or localized dilations of pre-capillary and capillary pulmonary vessels and intrapulmonary arteriovenous communications are common in patients with advanced chronic liver disease, with an estimated prevalence between 13% and 56% [45]. HPS affects 10% to 30% of patients with advanced liver disease requiring LT [46]. The diagnostic criteria for HPS, defined in 2004, are reported in Figure 2 [47]. The severity of HPS is classified by the degree of hypoxemia while breathing room air (Figure 2). Beside liver cirrhosis, HPS may also occur in patients with non-cirrhotic portal hypertension, such as Budd-Chiari syndrome, portal vein thrombosis or congenital extra-hepatic portosystemic shunts (Abernethy malformations) [8, 48]. Of note, the presence or severity of HPS does not reliably correlate with the severity of the underlying liver disease [46, 49].
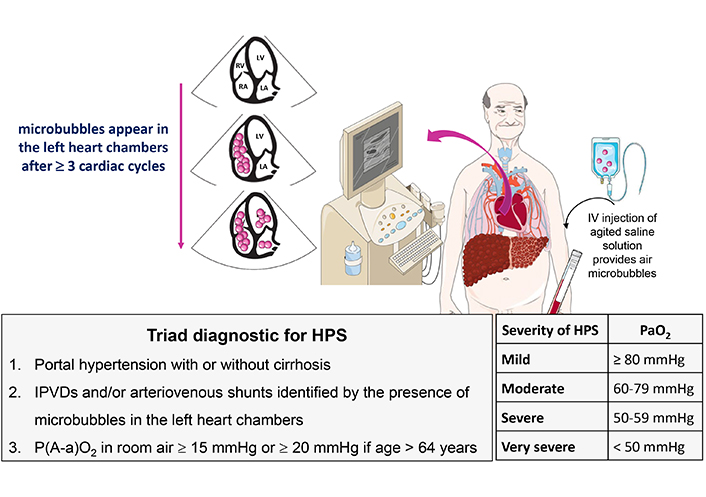
Diagnostic criteria and severity of HPS. Microbubbles are intravenously injected and appear in left heart chambers in 3 to 6 cardiac cycles after opacification of right atrium, indicating the presence of IPVDs and/or shunts. HPS: hepatopulmonary syndrome; IPVDs: intrapulmonary vascular dilatations
Note. Adapted with permission from “Syndrome hépato-pulmonaire” by Weil D, Sayadi A, Di Martino V, Coilly A, Raevens S, Thévenot T. Hépato-Gastro & Oncologie Digestive. 2023;30:902–10 (https://www.jle.com/fr/revues/hpg/e-docs/syndrome_hepato_pulmonaire_333482/article.phtml). French. © John Libbey Eurotext Plc.
Pathophysiology
The pathophysiology of HPS contributing to gas exchange abnormalities remains poorly understood, but three types of mechanisms characterized HPS (Figure 3): i) a shunt effect (especially in the lower pulmonary zones), ii) an incomplete diffusion of oxygen between the alveoli and the red blood cells and, iii) presence of direct arteriovenous communications causing blood bypassing alveoli [48, 50]. The mechanisms involved in the development of IPVDs mainly result from an imbalance between vasodilators and vasoconstrictors, with a predominant role of vasodilation. The main mediators involved in vasodilatation and angiogenesis are NO, carbon monoxide (CO), enthodelin-1, tumor necrosis factor-α and vascular endothelial growth factor-A. Bacterial translocation play a key role in the excessive production of vasoactive mediators, such as NO, as evidenced by 1) the increase in exhaled NO in patients with HPS and normalization of NO levels following LT, and 2) the normalization of NO production after the administration of L-NAME (NO-synthase inhibitor) in experimental models [51, 52]. The improvement of hypoxemia or prevention of HPS has been observed following administration of anti-TNF- antibodies, pentoxifylline (inhibitor of TNF- production) and norfloxacin to decrease bacterial translocation [53–55].
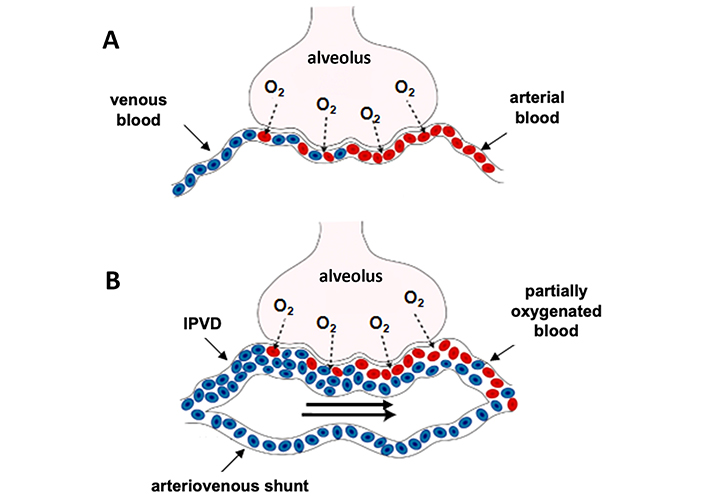
Pathophysiology of gas exchange impairment in HPS. Figure A (healthy subject) illustrates the oxygen exchange between alveolar space and a normal capillary. Figure B illustrates an impaired gas exchange in HPS attributed to a ventilation/perfusion mismatch (mainly due to hyperkinetic syndrome), a diffusion restriction (oxygen must travel a greater distance to bind haemoglobin due to dilated pulmonary capillary) and the presence of arteriovenous shunts that allow mixing of venous and arterial blood. IPVD: intra-pulmonary vascular dilation
Note. Adapted with permission from “Hepatopulmonary syndrome: Update on recent advances in pathophysiology, investigation, and treatment” by Grace JA, Angus PW. J Gastroenterol Hepatol. 2013;28:213–9 (https://onlinelibrary.wiley.com/doi/10.1111/jgh.12061). © 2012 Journal of Gastroenterology and Hepatology Foundation and Wiley Publishing Asia Pty Ltd.
In experimental rat models of HPS with common bile duct ligation, endothelin-1 produced by cholangiocytes stimulates pulmonary vascular endothelin-B receptor leading to eNOS activation. Moreover, iNOS and heme oxygenase-1 are overstimulated in pulmonary monocytes resulting in overproduction of NO and CO which contribute to IPVD in experimental HPS. Finally, pulmonary vascular development is also regulated by several angiogenic growth factors (the most studied being VEGF-A) produced by diverse cell types including pulmonary activated monocytes and macrophages [10]. Other pro-angiogenic biomarkers (such as angiopoietin-2, vascular cell adhesion molecule-1 and platelet-derived growth factor) and abnormalities in von Willebrand factor metabolism are increased in HPS and could participate in the pathological angiogenesis of HPS [56]. More recently, Baweja et al. [57] showed that fingolimod, a sphingosine-1-phosphate receptor modulator, improved arterial blood gas exchange, reduced systemic inflammation, portal pressures and attenuated hepatic fibrogenesis in animal models; however, the potential hepatotoxicity of this molecule needs further evaluation in cirrhotic patients [58]. Pathological pulmonary angiogenesis associated with reduced bone morphogenetic protein 9/10 and elevated endoglin may promote HPS [59]. A deficit in surfactant production and an increased apoptosis in alveolar epithelial cells could also aggravate HPS may also aggravate HPS [60].
Clinical presentation
HPS is most often asymptomatic and respiratory symptoms are nonspecific. Dyspnea is the most common symptom but other causes of dyspnea must be considered (Table 2). Platypnea (i.e., dyspnea that worsens from lying to standing or sitting positions) and orthodeoxia (i.e., a fall in arterial oxygen saturation ≥ 5% or PaO2 ≥ 4 mmHg in the upright position) are suggestive of HPS, but are present in a minority of cases (approximately 20% of cases) [48]. Platypnea and orthodeoxia result from the preferential basal lung localization of IPVDs and the gravitational effect of upright position that increases perfusion to areas affected by IPVDs/shunts, thereby increasing ventilation/perfusion mismatch. Other clinical signs are mainly represented by digital clubbing and spider nevi.
Main causes of dyspnea in cirrhotic patients
| Pulmonary vascular complications associated with portal hypertension | Hepatopulmonary syndrome and portopulmonary hypertension |
|---|---|
| Pleural effusion associated with portal hypertension | Hepatic hydrothorax |
| Extra-pulmonary conditions associated with cirrhosis | Cirrhotic cardiomyopathyLarge volume ascitesSaropeniaAnemia |
| Lung diseases linked to the cause of cirrhosis | Alpha-1-antitrypsin deficiencyDiffuse interstitial lung disease associated with scleroderma, primary biliary cholangitis or sarcoidosisVenous thromboembolism |
| Cardio-pulmonary manifestations associated with other disorders | ObesityLung diseases associated with cigarette smokingPleuropulmonary infectionsIschemic/hypertensive/alcoholic cardiomyopathy |
HPS screening and diagnostic tools
All patients awaiting LT should be screened for HPS since HPS is usually asymptomatic but may affect survival after LT. Assessment of oxygen saturation (SpO2) in an upright position by pulse oximetry is recommended by practices guidelines [9]. A SpO2 ≤ 97% had a sensitivity of 96% and a specificity of 75% for the detection of arterial hypoxemic (PaO2 < 70 mmHg) and a SpO2 ≤ 94% detected all patients with a PaO2 < 60 mmHg [61]. However, a recent US prospective study showed that a SpO2 cutoff < 96% to detect HPS had a suboptimal sensitivity (only 28%) and negative predictive value (82%, meaning that 18% of patients with SpO2 ≥ 96% had HPS) [62].
Due to a rightward shift in the oxyhemoglobin dissociation curve in cirrhotic patients, small changes in SpO2 are paralleled by large changes in PaO2 and consequently, SpO2 is not sufficiently accurate for identifying hypoxaemia in HPS [63]. Given the limited performance of SpO2, it seems preferable to screen for HPS by contrast echocardiography and/or arterial blood gas in all LT candidates for [62]. HPS is also frequently associated with a reduced diffusing capacity for CO [48].
The diagnosis of HPS is made by the demonstration of IPVDs and/or shunts in combination with impaired gas exchange. IPVDs are documented by microbubble transthoracic echocardiography, which is the gold standard, or 99m-technetium-labeled macroaggregated albumin scan. During echocardiography, hand-shaken saline, resulting in microbubbles of > 10 μm diameter, is injected intravenously. In healthy condition, the bubbles are trapped in the lung capillaries (< 8–15 μm) and absorbed by the alveoli. ‘Delayed’ opacification of the left atrium, i.e., 3 to 6 cardiac cycles after appearance of the microbubbles in the right atrium, indicates the presence of IPVDs and shunts (15–100 μm) (Figure 2). Macroaggregated albumin scan relies on the same principle, but with the limitation that it cannot distinguish between intracardiac and intrapulmonary shunts. In case of intracardiac or intrapulmonary shunts, the aggregates are retained in the brain and kidneys. macroaggregated albumin scan allows quantification of pulmonary and systemic radionuclide uptake, whereas contrast echocardiography cannot [64].
The characteristic finding of HPS on chest computed tomography (CT) are dilated peripheral vasculature and bibasilar reticulonodular opacities, but CT scan is especially useful for excluding other coexistent chronic respiratory conditions. Pulmonary angiography may be used in patients with persistent hypoxemia despite inhaled pure oxygen to identify potential large arteriovenous malformations amenable for embolization [48].
Natural history
Patients with HPS have a reduced quality of life and a risk of mortality twice as high as those without HPS, regardless of age and the severity of the underlying liver disease [46]. In a retrospective study comparing 37 cirrhotic patients with HPS to 47 control patients without HPS, the median survival and 5-year survival without LT of patients with HPS and controls were respectively of 24 and 87 months, and 23% and 76%. In this study, PaO2 decreased over time in 85% of HPS patients, with an average decline of approximately 5 mmHg per year [65]. In 102 Canadian patients with moderate to very severe HPS, the decline in PaO2 was less severe (3.7 mmHg/year) [49]. The prognosis is particularly poor when PaO2 is less than 60 mmHg with a median survival of less than 3 months [66]. Alongside progressive forms of HPS, there are slowly or non-progressive disease and even spontaneously resolving forms of HPS [67].
Pharmacological treatment
Despite recent advances in our understanding of the pathophysiology of HPS [68], there are currently no effective medical treatment to cure HPS. Oxygen therapy is useful when PaO2 is below 60 mmHg. Case series have evaluated methylene blue or N-nitro-L-arginine methyl ester (inhibitors of NO synthase), pentoxifylline, aspirin, somatostatin, indomethacin, or mycophenolate mofetil with null or even contradictory results [48]. A small randomized controlled study suggest that garlic supplementation may have some beneficial effects on hypoxemia in patients with HPS [69].
The prophylactic use of antibiotics can improve severity of HPS in experimental rat models by inhibiting Gram-negative bacterial translocation, and thus by decreasing NO production by pulmonary intravascular macrophages [51]. A small pilot randomized crossover study of norfloxacin (9 patients; 400 mg BID for one month) failed to show any improvement in the alveolar-arterial oxygen gradient over the placebo [70]. Sorafenib, a tyrosine kinase inhibitor, reversed pulmonary vascular proliferation and improved gas exchange in animal model of HPS [71]. In a randomized, double-blind, placebo-controlled trial, sorafenib 400 mg/day (16 patients) or placebo (12 patients) for 12 weeks had no effects on AaPO2, on exercise capacity or symptoms of dyspnea, and worsened quality of life [72].
Liver transplantation
HPS was considered a contraindication for LT for a long period of time. In the early 1990s, a series of case reports in adults and children with varying severity of HPS demonstrated that arterial hypoxemia normalized following LT. The increasing success of LT for patients with HPS and the poor outcome in the absence of LT (5-year survival rate of 23%) led to consideration of HPS as an established indication for LT [73]. To date, LT is the only curative treatment for HPS with similar post-LT survival rates between patients with and without HPS [74]. However, analysis of more than 700 patients with HPS who underwent liver transplant in the US between 2002 and 2012 demonstrated a worse post-LT survival in those with a pre-LT PaO2 ≤ 44 mmHg when compared with those with a PaO2 of 44.1–54.0 mmHg. In this study, the 3-year post-LT survivals were of 68%, 84%, and 86% respectively in patients with pre-LT PaO2 ≤ 44 mmHg, between 44.1 and 54 mmHg, and between 54.1 and 61 mmHg [75].
Given that LT remains the unique and definitive treatment of HPS, the Organ Procurement Transplant Network and United Network for Organ Sharing recommend allocation of MELD exception points to patients with severe HPS (PaO2 < 60 mmHg) in order to prioritize LT [76, 77]. US and European data, as well as a recent systematic review, show that since the implementation of policies for prioritization for LT, patients with pre-LT HPS demonstrate improved 5-year post-LT survival (73% in the pre-MELD period vs. 87.3%) [48, 78]. The threshold of PaO2 considered “severe” and associated with an increased risk of post-LT mortality may differ in literature (44 mmHg for Goldberg et al. [75], 45 mmHg for Kadry et al. [79], and 54 mmHg for Jose et al. [80]) but experts agree on the significantly increased risk of post-LT mortality when pre-LT PaO2 is less than 60 mmHg [80].
Management of post-LT hypoxemia
Normalization of arterial hypoxemia generally occurs between 6 and 12 months following LT. A retrospective analyzes of HPS patients included in the Canadian HPS Program showed a faster improvement of PaO2 [49]. The one-year post-LT PaO2 (42 patients) improved with a slope of 6.5 mmHg per month and the median time to normalize PaO2 was 149 (116–184) days. In such a scenario, a pre-LT PaO2 of 54 mmHg normalizes at 4 months after LT (PaO2 ≥ 80%). Furthermore, there was no significant difference in the probability of post-LT normalization in PaO2 between HPS severity classes [49].
Hypoxemia may increase in the immediate post-LT period and may delay extubation. Severe post-LT hypoxemia, defined by the need for 100% FiO2 to maintain oxygen saturation ≥ 85%, is associated with heavy peri-LT mortality which may affect 45% of patients [48], while the overall early post-LT mortality for HPS patients is approximately 4% [81]. The management of post-LT hypoxemia may require a strict supine position or alternating lateral decubitus position, or even Trendelenburg, the use of epoprostenol or inhaled NO and/or intravenous methylene blue [48]. Veno-venous extracorporeal membrane oxygenation (ECMO) has been used successfully in getting through a difficult pre- or post-LT period [82].
Coexistence of hepatopumonary syndrome and portopumonary hypertension
Although HPS and PoPH have distinct pathophysiology and thought to be mutually exclusive, their coexistence is probably underestimated. In a retrospective study, IPVDs were detected in 59% of patients with PoPH by contrast echocardiography and may be associated with worse survival [83]. In most reported cases, HPS precedes the diagnosis of PoPH [84]. In the pretransplant period, HPS could potentially mask the presence of PoPH. With the resolution of the HPS following LT, PoPH can raise its ugly head months after transplant [85]. Consequently, a follow-up echocardiography post-LT in patients with pre-LT HPS is important.
Hepatic hydrothorax
Epidemiology
Pleural effusion associated with liver disease was initially described by Laennec in the 19th century and the term “hepatic hydrothorax (HH)” was introduced in 1958 [86]. HH refers to the accumulation of fluid in the pleural space (classically greater than 500 mL) secondary to portal hypertension, in the absence of any other cause of cardiopulmonary or malignant origin [87].
The prevalence of HH varies considerably (from 16% to 70%) depending on the definitions used [88]. A retrospective analysis of 495 cirrhotic patients from US tertiary centers found 77 cases of HH, representing a prevalence of 16% after excluding a large number of incomplete files. The main characteristics were a predominance of male (64%), a preferential right-sided location of HH (73% of cases, 17% left-sided and 10% bilateral), the clinical absence of detectable ascites in 9% of cases, and a poor outcome (nearly 60% of death over the next year) [89]. Overall, the exact prevalence of HH remains poorly determined because large prospective studies are lacking, but HH affects approximatively 5% to 15% of patients with decompensated cirrhosis [88].
Pathophysiology
HH is the consequence of a unidirectional transfer of ascites into the pleural cavity through diaphragmatic defects and facilitated by negative intrathoracic pressure and intra-abdominal pressure caused by ascites. HH develops when the accumulated volume of pleural fluid exceeds the resorptive capacity of the pleural lymphatic system. Huang et al. [90] differentiated 4 types of diaphragmatic defects during thoracoscopy: Type 1—no macroscopically visible defects, Type 2—blebs lying on the diaphragm, Type 3—broken defects (fenestrations) in the diaphragm originating from ruptured bullae, and Type 4—multiple gaps in the diaphragm. The preferential location of HH is on the right side because the right hemidiaphragm is less thick and muscular than the left side and has more areas of tendon weakness. The diagnosis of HH may be difficult in the absence of clinical ascites, but can be confirmed by a peritoneal technetium-99m scintigraphy demonstrating the unidirectional flow of ascites towards the pleura [91]. In practice, scintigraphy is rarely done and diagnosis of HH is typically based on clinical presentation/context and laboratory analysis as discussed in the following sections.
Diagnosis
In the absence of cardiac or pulmonary disease, HH is typically diagnosed during follow-up of decompensated liver disease or, less frequently, portal hypertension without cirrhosis. Low-volume pleural effusions are generally asymptomatic and incidentally discovered, whereas large-volume HH may manifest as restrictive respiratory failure or even acute tamponade. Acute respiratory failure requiring hospitalization is common, accounting for 70% of patients in the retrospective series by Osman et al. [92]. Symptoms in patients with HH are similar to those observed in other causes of pleural effusion, namely dyspnoea, cough, fatigue and chest pain [93]. The onset of symptoms associated with HH will depend on the volume of the effusion, the rapidity of its constitution and the existence of other cardiopulmonary disease. HH is generally accompanied by ascites and/or edema of the lower limbs.
Spontaneous bacterial empyema
The clinical presentation of spontaneous bacterial empyema (SBE), defined as spontaneous infection of pre-existing HH, is non-specific and may include dyspnea, fever, abdominal pain, encephalopathy and worsening of renal function. In the absence of systemic infection, malignancy or pancreatic disease, the diagnosis of SBE is based on pleural fluid showing a neutrophil count of over 250/mm3 associated with a positive culture, or a neutrophil count of over 500/mm3 even if the culture is negative [94]. This more complex definition, as compared to that used for spontaneous bacterial peritonitis (SBP), probably accounts for the low percentage of correct answers (17.6% of hepatogastroenterologists and 5.6% of pulmonologists) when asked to define SBE in an international practice survey [95]. The most common bacteria found are enterobacteria (mainly Klebsiella spp, Escherichia coli) suggesting that the pathogenic mechanism precipitating SBE is intestinal bacterial translocation; bacteria originating from the skin are mainly observed when an invasive procedure is performed. In 40% to 50% of cases, SBE can occur in the absence of SBP [94, 96]. Recurrence of SBE is frequent; in a remarkable series of 16 patients with SBE, three had recurrence, including one patient who had SBE four times [94]. The prognosis of SBE is poor, with in-hospital mortality ranging from 20% to 38% [94, 96]. An exploratory pleural puncture should be performed whenever SBP (and, a fortiori, SBE) is suspected.
Evaluation of suspected HH
HH is a diagnosis of exclusion, requiring exploration of other causes of pleural effusion. Chest X-ray is the first examination to confirm the presence of pleural fluid, but a significant volume (250 mL to 600 mL) of fluid is required to be visible in the upright position [97]. A chest CT scan can be used to assess the underlying lung parenchyma for infiltration or tissue damage, and to diagnose loculated pleural fluid collections. Hepatic Doppler ultrasonography is used to identify subclinical ascites, evaluate portal flow, and rule out an intrahepatic tumor.
An exploratory pleural puncture (thoracentesis) will best indicate the transudative or exudative nature of pleural fluid, according to Light’s criteria; the negative predictive value and sensitivity of these criteria were 96% and 98% respectively (Table 3) [98]. In patients on long-term diuretic therapy, transudates may be misclassified as exudates due to contraction of extracellular volumes, increasing protein and LDH concentrations in pleural fluid. The main characteristics of pleural fluid in HH are shown in Table 3.
Main characteristics of pleural fluid allowing to diagnose or exclude hepatic hydrothorax
| Light’s criteria for exudative effusion if at least one criterion is present |
|---|
| Pleural fluid protein/serum protein ratio > 0.5 |
| Pleural fluid LDH > 200 IU/L or > 2/3 serum LDH |
| Pleural fluid LDH/serum LDH > 0.6 |
| Main characteristics of hepatic hydrothorax |
| Serum protein < 25 g/L |
| Pleural fluid protein/serum protein ratio < 0.5 |
| Pleural fluid LDH < 200 IU/L |
| Pleural fluid LDH/serum LDH < 0.6 |
| Albumin gradient (serum-pleural) > 11 g/L |
| Pleural fluid pH > 7.3 |
| Neutrophils in pleural fluid < 500/mm3 with negative culture |
LDH: lactate dehydrogenase. Hepatic hydrothorax is a transudative pleura effusion
When the etiology of HH remains unclear, other pleural fluid analyses can be performed, such as mycobacterial gene amplification, amylase for pancreatic pleurisy, cytology to exclude a tumoral origin, and triglyceride to identify a chylothorax. Peritoneal Tc-99m scintigraphy can exclude HH when the radioisotope does not migrate into the pleural cavity. Videothoracoscopy is an invasive procedure that can be used to visualize, and sometimes repair, diaphragmatic defects when the diagnosis of HH remains unclear [90].
Natural course
The prognosis of patients with HH is poor. In a retrospective study of 763 cirrhotic patients with ascites, the survival of 97 patients with HH was compared with that of 97 patients without HH and matched for age and severity of cirrhosis. The 5-year survivals were 15.4% and 30.9% respectively in patients with and without HH [99]. This study also showed a good prognostic performance of the MELD-Na score, unlike other studies [89, 100]. Refractory HH (RHH) has a very poor prognosis [92, 100, 101]. The study by Osman et al. [92] observed that patients with RHH (n = 47) had a decreased 1-year survival compared with patients with refractory ascites without HH and matched for age, gender, and MELD-Na score (survival of 19% and 51%, respectively). The most common causes of death were hypoxemic respiratory failure or septic shock. The unfavorable prognosis of RHH even persisted in the subgroup of patients with a MELD-Na score ≤ 20 [92]. A retrospective single-center study carried out on cirrhotic patients evaluated for LT compared the prognosis of patients with (n = 82 HH including 34 RHH) and without HH (n = 703). In this study, the refractory nature of HH was significantly associated with the risk of death without LT, after adjusting for the MELD-Na score, in the entire cohort or in the subgroup of 82 HH [101]. Finally, a recent study of cirrhotic American veterans compared the survival of 177 patients with RHH with that of 422 patients with refractory ascites. The two groups of patients were comparable in age and severity of cirrhosis (according to Child-Pugh and MELD-Na scores). RHH was associated with a greater risk of liver-related death than refractory ascites (adjusted OR: 4.63, CI 95%: 3.31–6.48), irrespective of the strata level of the baseline MELD-Na score (strata < 10, 10–15, 15–25 and ≥ 25). At the time of liver-related death, patients with RHH had a lower MELD-Na than those in the refractory ascites group (16.50 vs. 21.82, p = 0.002), suggesting that MELD-Na underestimates the risk of liver-related mortality in the presence of RHH. More remarkably, the MELD 3.0 score was similar in the two groups but significantly higher at the time of death in the RHH group, indicating that MELD 3.0 may better assess the prognosis of patients with RHH [100]. The increased risk of death in patients with RHH is at least partly related to the morbidity of repeated thoracentesis [102], more frequent nosocomial infections, and increased risk of acute-on-chronic liver failure [103].
Pharmacological treatment
Diuretics and low-salt diets
Since HH probably involves passage of ascites fluid from the peritoneal cavity into the pleural cavity, first-line treatment includes a low-salt diet and diuretics. Sodium restriction of 2 g (i.e., around 5 g of salt) is initially sufficient to reduce the volume of isolated ascites, provided that natriuresis exceeds intake. Sodium restriction should not be too rigorous because it may adversely result in nutritional deficiencies. Because of the secondary hyperaldosteronism associated with decompensated cirrhosis, anti-mineralocorticoids (spironolactone) are the diuretics of choice. Adding a loop diuretic (furosemide) to spironolactone may improve respiratory symptoms more rapidly. Despite this, around 20–30% of patients develop RHH. HH is considered refractory when it is “diuretic-resistant” (recurrent HH despite low-salt diet and intensive diuretic use) or “intractable” (HH cannot be treated or recurs as a result of inability to use an adequate diuretic regime due to diuretic-induced complications) [88]. These definitions require more widespread dissemination, as our recent practice survey found that 45% of hepatogastroenterologists thought there was no definition of RHH [95].
Vasoconstrictors
All the drugs limiting ascites formation are potentially useful for HH. However, no large studies or randomized trials have evaluated these agents (octreotide, midodrine, and terlipressin) in HH, and the evidence is largely limited to case reports or case series [104–106]. The combination of octreotide, midodrine, and albumin may improve survival in patients with hepatorenal syndrome but has not been evaluated in RHH. Anecdotal cases of resolution of RHH have been reported with the following combinations, octreotide-albumin [104], terlipressin-albumin [105] and octreotide-midodrine [106].
Interventional treatments
Approximately 20% to 30% of cirrhotic patients with HH fail to respond adequately to a low-salt diet combined with optimal doses of diuretics [88]. For those patients, LT or TIPS should be considered as a first-line treatment. However, many patients are ineligible for TIPS and LT, and other procedures should be considered.
Thoracentesis
Thoracentesis can provide rapid symptom relief. If ascites is present, it should be removed before pleural fluid. Thoracentesis should not exceed 1.5 liters to avoid re-expansion pulmonary edema. The use of albumin, the standard of care for the management of large-volume ascites, remains largely debated for thoracentesis and needs further investigation in this area [107]. Despite this lack of consensus, our survey found that 68% of hepatogastroenterologists used albumin after therapeutic thoracentesis [95]. The main pitfall of repeated thoracentesis is the risk of complications (pneumothorax, hemothorax, empyema). A retrospective case-control study observed 7.3% of complications after the 2nd pleural puncture (55 patients) and around 10% after the 4th puncture (28 patients) [108]; in this study, a high MELD score and low platelets predicted the risk of developing hemothorax, and patients with platelet count below 50 G/L were 10 times more likely to develop hemothorax. Consequently, thoracentesis remains an effective treatment in the very short term but, whenever possible, should be replaced by other therapeutic alternatives.
Chest tube
Chest tube placement is associated with high morbidity and mortality. Reported complications include worsening of renal function, electrolyte imbalances, pneumothorax, and empyema. A series of 17 patients with HH treated by chest tube showed a 3-month death rate of 35% (only one patient had no complication). In this series, 7 patients received a TIPS following the chest tube placement and 6 of them were alive at 3 months versus 5 of the 10 patients who did not receive a TIPS [109]. This study suggests that TIPS should be considered as soon as a chest tube is placed in order to facilitate its removal and prevent recurrence of HH. Thus, chest tubes have no role in the therapeutic arsenal for HH, and should be reserved for the treatment of empyema provided a small-calibre drain (< 20 F) is used [88].
Indwelling tunnelled pleural catheter
The indwelling tunnelled pleural catheter (ITPC) is a fenestrated catheter inserted percutaneously into the pleural space for intermittent drainage. This technique has proved to be helpful in the palliative treatment of pleural effusions caused by malignancy and cardiac or renal diseases. In a prospective study, 24 cirrhotic patients with HH received ITPC combined with 10 TIPS placed before (n = 5) or after (n = 5) ITPC. Spontaneous pleurodesis was observed exclusively when TIPS was placed before ITPC, allowing definitive removal of the catheter. Infection of the pleural fluid with cutaneous germs (4 cases/24, i.e., 16.7%) was the main pitfall of this technique [110]. Another retrospective monocentric series of 62 patients who received ITPC demonstrated the feasibility of this procedure pending LT. The most common side effect was empyema (16.1%), mainly associated with Gram-negative bacteria requiring removal of the catheter [111]. Pleural effusions are common after LT and may develop with or without HH. Majority of pleural effusions are exudative [112] and are associated with increased morbidity [113]. Thus, a PleurX ITPC may be required for several weeks with usual resolution.
Surgical repair of diaphragmatic defects
Video-assisted thoracoscopic surgery (VATS) for pleurodesis with or without diaphragmatic repair resolves HH in 53% to 80% of cases reported in small series [114]. This attractive technique remains difficult to implement in patients with advanced liver failure. In a series of 63 RHH, survival after VATS was respectively 53% and 80–85% in Child-Pugh C (n = 15) and Child-Pugh A/B patients (n = 48) after a median follow-up of 20 months, and the recurrence rate of HH was 6.3% [115]. Another difficulty is the exhaustive identification of diaphragmatic defects, which may be undetectable to the naked eye because of their small size and the large surface area of the diaphragm.
Pleurodesis
Chemical pleurodesis using talc or other sclerosing agents (such as vibramycin, bleomycin or minocycline), through a chest tube or under thoracoscopy, induces an inflammatory pleural reaction resulting in pleural symphysis, which reduces fluid accumulation in the reduced pleural space [88]. However, this procedure carries a risk of encysted pleurisy because of the dilution of the sclerosing agent and the difficulty to maintain in contact the visceral and parietal pleura due to the constant ascites fluid accumulation into the pleural space. A meta-analysis of 180 patients (13 case series) treated by pleurodesis for HH showed a complete response in 72% (CI 95%: 65–79%) of cases after a follow-up ranging from 1 to 36 months, and a complication rate of 82% (CI 95%: 66–94%). However, it is difficult to draw robust conclusions from this meta-analysis, which mixed pleurodesis methods (chemical in 90% of cases) and abrasive agents, and analyzed results without specifying the role of diaphragmatic defects repair under VATS; moreover, the follow-up period was variable and the definition of “complete response” was not homogeneous between case series [116].
Transjugular intrahepatic portosystemic shunt
TIPS placement for HH remains a marginal indication compared with other indications; in a Chinese retrospective study of 1,292 decompensated cirrhotic patients who received TIPS between 2015 and 2021, RHH as the indication for TIPS was noted only in 0.2% of patients, while esophageal variceal bleeding and refractory ascites represented the main indications (91% and 7.5%, respectively) [117]. A meta-analysis of 198 patients (6 studies) treated with TIPS (mostly uncovered) for RHH showed that the efficacy of TIPS was complete in 56% of cases and partial in 18% after a mean follow-up of 10 months. In this meta-analysis, 45-day post-TIPS mortality was 17.7%, and overall post-TIPS mortality over a maximum follow-up of 5 years was 50.2% [118]. Recent US recommendations propose that in patients with RHH or disabling clinical symptoms (e.g., dyspnea at rest), TIPS should be considered [119]. Elective TIPS for RHH should be considered in appropriately selected patients. Before TIPS creation, a contrast-enhanced multiphasic cross-sectional imaging (CT or MRI) of the liver together with echocardiography is useful to ensure the absence of contraindications, such as severe valvular heart disease or PH, hepatic lesions. Recurrent overt HE or uncontrolled systemic infection are also usual contraindications [119]. The persistence of HH despite TIPS combined with appropriate low-salt diet and diuretics should prompt a doppler ultrasound to assess TIPS dysfunction and the potential need for revision.
Liver transplantation
LT remains the most definitive treatment of HH. Post-LT survival of patients with HH is similar to that of other cirrhotic patients, with survival rates of 70% at 8 years [120, 121]. Using Taiwan’s national health insurance database, Hung et al. [122] identified 3,487 cirrhotic patients with pleural effusion requiring pleural drainage between 2007 and 2010. The 90-day, 1-year, and 3-year mortality rates in patients who received LT were respectively 13.4%, 20.4%, and 21.7%, and 41%, 61.2%, and 77.5% in non-transplanted patients [122]. These data highlight the beneficial effect of LT in patients with RHH. In the retrospective study by Osman et al. [101] assessing cirrhotic patients for LT, the highest 1-year survival benefit with LT was for patients with RHH (0.48 years compared to non-transplant patients), followed by patients with non-refractory HH (0.28 years), and then patients with other complications of cirrhosis (0.19 years) [101]. The same authors also observed that the incidence of post-LT complications (duration of intubation, tracheotomy, hemothorax, empyema, and other infections) was similar between patients with and without HH, apart from an increased proportion of patients with HH getting chest tubes [123].
The poor prognosis of HH, especially when refractory, and the significant survival benefit provided by LT, support the referral of patients more rapidly for LT evaluation. MELD exception points may be awarded on an individual basis in patients with chronic, recurrent, confirmed HH, providing that infectious and malignant causes have been ruled out, TIPS is contraindicated and diuretics are ineffective [124]. We propose an algorithm for the management of HH (Figure 4).
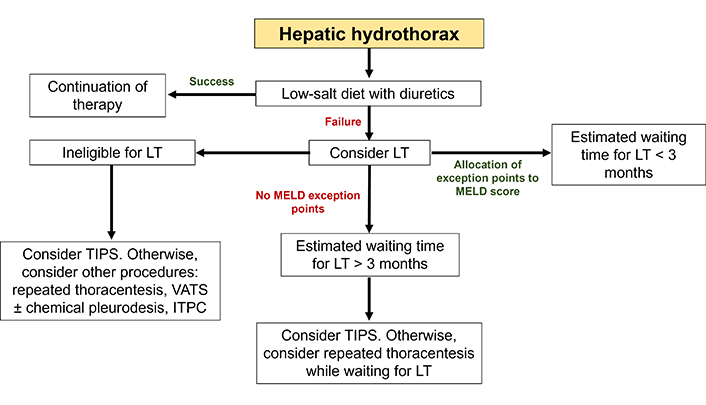
Algorithm for the management of hepatic hydrothorax. Initial treatment is based on a low-salt diet with diuretics. In cases of recurrent or hypoxemic HH, a history of empyema or asymptomatic HH associated with a MELD score > 15, LT should be systematically considered. In the absence of contraindications, a pretransplant TIPS may be relevant if the estimated waiting time for transplantation is long (arbitrarily greater than 3 months). When LT is not indicated, TIPS remains a satisfactory solution. ITPC: indwelling tunnelled pleural catheter; LT: liver transplantation; TIPS: transjugular intrahepatic portosystemic shunt; VATS: video-assisted thoracoscopic surgery
Conclusions
The challenging management of pulmonary vasculopathy including HPS and PoPH requires a multidisciplinary team approach. Despite significant breakthroughs in the understanding of HPS and PoPH, important unresolved questions remain, including the best medical therapy and the ideal time for LT. Given the significant prognostic impact of these vasculopathies, their prompt detection is advantageous. Finally, HH also displays a poor prognosis with limited therapeutic options, management requires a multidisciplinary approach with prompt referral to LT when indicated.
Abbreviations
| cGMP: | cyclase-cyclic guanosine monophosphate |
| CO: | carbon monoxide |
| CT: | computed tomography |
| HH: | hepatic hydrothorax |
| HPS: | hepatopulmonary syndrome |
| IPVD: | intrapulmonary vasodilatations |
| ITPC: | indwelling tunnelled pleural catheter |
| LT: | liver transplantation |
| mPAP: | mean pulmonary arterial pressure |
| NO: | nitric oxide |
| PAH: | pulmonary arterial hypertension |
| PAWP: | pulmonary arterial wedge pressure |
| PH: | pulmonary hypertension |
| PoPH: | portopulmonary hypertension |
| PVR: | pulmonary vascular resistance |
| RHH: | refractory hepatic hydrothorax |
| SBE: | spontaneous bacterial empyema |
| sPAP: | systolic pulmonary arterial pressure |
| TIPS: | transjugular intrahepatic portosystemic shunt |
| TRV: | tricuspid regurgitation velocity |
| VATS: | video-assisted thoracoscopic surgery |
| WU: | Wood units |
Declarations
Author contributions
TT: Writing—original draft, Validation, Writing—review & editing, Supervision. SR, AA, BAB, and HMD: Writing—original draft. JFDC: Visualization. All authors read and approved the submitted version.
Conflicts of interest
Prof. Jean François D. Cadranel is the Editorial Board Member of Exploration of Digestive Diseases, but he had no involvement in the journal review process of this manuscript. The other authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2024.