 Review
Review

Affiliation:
1Victorian Infectious Diseases Reference Laboratory, Royal Melbourne Hospital at the Peter Doherty Institute for Infection and Immunity, Melbourne 3000, Australia
2Department of Microbiology and Immunology, The University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Melbourne 3000, Australia
Email: laura.mccoullough@mh.org.au
ORCID: https://orcid.org/0009-0002-6770-3889
Affiliation:
1Victorian Infectious Diseases Reference Laboratory, Royal Melbourne Hospital at the Peter Doherty Institute for Infection and Immunity, Melbourne 3000, Australia
3Department of Infectious Diseases, The University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Melbourne 3000, Australia
ORCID: https://orcid.org/0000-0002-8206-2664
Affiliation:
1Victorian Infectious Diseases Reference Laboratory, Royal Melbourne Hospital at the Peter Doherty Institute for Infection and Immunity, Melbourne 3000, Australia
3Department of Infectious Diseases, The University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Melbourne 3000, Australia
Email: peter.revill@vidrl.org.au
ORCID: https://orcid.org/0000-0003-2715-2541
Explor Dig Dis. 2024;3:326–343 DOI: https://doi.org/10.37349/edd.2024.00054
Received: February 26, 2024 Accepted: June 21, 2024 Published: August 07, 2024
Academic Editor: Youhua Xie, Fudan University, China
The article belongs to the special issue Viral Hepatitis
Chronic hepatitis B (CHB) disease caused by persistent infection with hepatitis B virus (HBV) is a global health problem affecting almost 300 million people worldwide, resulting in up to 1 million deaths each year. The factors contributing to HBV mediated liver disease are yet to be fully resolved, however, multiple studies have suggested that HBV splice variants may be a contributing factor. Recent studies have indicated that novel fusion proteins encoded by splice variants, or the splice-derived RNA itself, may impact replication of wild-type HBV, although the direct mechanisms for these interactions are largely unknown. This review explores the latest knowledge regarding the contribution of splice variants to liver disease and their impact on HBV replication.
Persistent hepatitis B infection leads to cirrhosis and hepatocellular carcinoma (HCC), which causes up to 1 million deaths annually. Prior to nuclear export, the hepatitis B pregenomic RNA (pgRNA) may be spliced by the host cell spliceosome to form shorter RNA sequences, which are reverse transcribed and secreted from the cell as defective hepatitis B virus (HBV) genomes. The role of splice variants in the HBV replication cycle is still largely unknown, however a recent study has shown that the major splice variant Sp1 impacts the structure of the HBV nucleocapsid, as a splicing-deficient mutant impaired HBV infectivity in human hepatocyte chimeric mice, as well as viral entry in HepG2-NTCP cell line [1]. In addition, splice variants are associated with liver disease progression [2–5] and liver cancer [6], are variably expressed throughout chronic hepatitis B (CHB) natural history [7, 8] and the sequences of splice donor and acceptor sites are highly conserved across HBV genotypes [9]. Together, this suggests they are more than mere artefacts of HBV processing that may play a key but still to be defined role in the HBV “life-cycle” and disease pathogenesis. This review focuses on the reported impact of HBV splice variants on HBV replication and pathogenesis.
HBV encodes a 3.2 kb partially double-stranded relaxed circular DNA (rcDNA) genome [10] of 4 open reading frames (ORFs) that encode the surface/envelope proteins, hepatitis B X protein (HBx), polymerase (pol) protein and the precore/core protein. These are transcribed to form five non-spliced mRNA transcripts. The 3.5 kb precore mRNA encodes the precore protein which is processed to form the secreted hepatitis B e antigen (HBeAg) [11]. A slightly smaller 3.5 kb pgRNA, encodes the core and pol proteins and is also the template for reverse transcription to HBV DNA. The 2.4 kb mRNA transcript encodes the large hepatitis B surface protein (LHBs) (preS1, preS2, and S) and the 2.1 kb mRNA transcript encodes the middle (MHBs) (preS2 and S), and small (SHBs) surface proteins, which together form the viral envelope protein. Finally, a 0.7 kb mRNA transcript encodes HBx [12–15].
Following attachment of the antigenic loop within the S protein to cell-associated heparan sulphate proteoglycans [16], viral entry is triggered by binding of the N-terminal sequence of the pre-S1 region to the sodium taurocholate co-transporting polypeptide (NTCP) receptor (Figure 1) [17]. The rcDNA genome is released into the nucleus and the pol protein is released from the rcDNA, perhaps by the host DNA-repair enzyme TDP2 [18, 19] although this mechanism is yet to be fully resolved. The plus strand is repaired by the host cell DNA pol and DNA topoisomerases and both strands are ligated to form a covalently closed circular DNA (cccDNA) [20–24]. The cccDNA undergoes chromatinization to form the cccDNA minichromosome, which is the HBV transcriptional template and the reservoir of HBV DNA that is a major contributor to HBV persistence [25, 26]. cccDNA is very stable in non-dividing human hepatocytes and can survive for the whole lifespan of the cell [27, 28]. The cccDNA is transcribed by the host DNA pol to produce the pgRNA, precore mRNA, preS/S mRNAs and HBx mRNA, which are exported from the nucleus where they are translated into the viral proteins [29]. The core proteins form a nucleocapsid which packages the pgRNA and the pol protein [30] and pgRNA is then reverse transcribed by the pol protein to form the double-stranded DNA HBV genome [31–33].
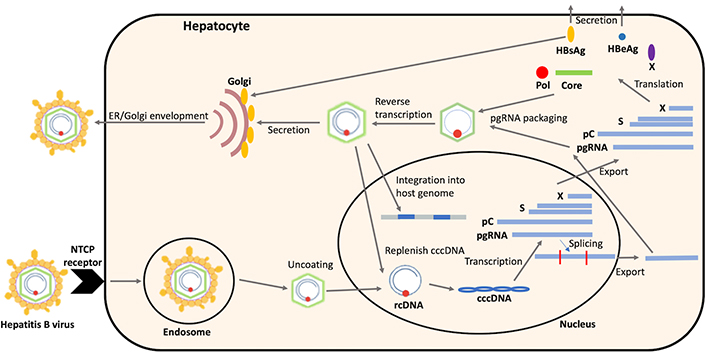
The hepatitis B virus (HBV) replication cycle. NTCP: sodium taurocholate co-transporting polypeptide; ER: endoplasmic reticulum; rcDNA: relaxed circular DNA; cccDNA: covalently closed circular DNA; pC: precore mRNA; pgRNA: pregenomic RNA; Pol: polymerase; HBsAg: hepatitis B surface antigen; HBeAg: hepatitis B e antigen
In eukaryotes, DNA is initially transcribed into pre-mRNA, which must be processed into mature mRNA for protein synthesis. This process includes splicing of the RNA to remove the introns, which are the non-coding sequences. The exons, the coding sequences, are then ligated to produce a mature mRNA strand. Splicing can either be constitutive—one pre-mRNA strand makes one mature mRNA strand, or alternative, where one pre-mRNA strand has different splice sites and so more than one mature mRNA strand is produced. RNA splicing is mediated by the spliceosome, a ribonucleoprotein (RNP) complex comprised of several small nuclear RNAs (snRNAs) [34–36]. The spliceosome binds to the intron in the pre-mRNA strand which is defined by a 5’ donor splice site, a 3’ acceptor splice site, and a branch site. The branch site is usually 18–40 nucleotides upstream of the 3’ acceptor splice site and in higher eukaryotes, the branch site is followed by a polypyrimidine tract [37, 38]. The 5’ donor splice site sequence consists of nine nucleotides of the sequence MAG/GTRAGT (A: adenine, G: guanine, T: thymine, M: adenine or cytosine, R: guanine or adenine) and the 3’ acceptor splice site sequence consists of four nucleotides of the sequence YAG/G (Y: cytosine or thymine, A: adenine, G: guanine) [39], which are conserved in humans [40]. A number of cellular factors regulate this process, including serine and arginine-rich proteins such as SF2/ASF, SC35, 9G8, and SRSF1 which bind to exonic splicing enhancers on the pre-mRNA strand to facilitate spliceosome assembly [41]. Alternatively, heterogenous nuclear RNA (hnRNP) proteins, such as hnRNP A1 and polypyrimidine tract-binding protein 1 (PTB1), bind to exonic splicing silencer regions on the pre-mRNA strand to repress splicing, usually by preventing spliceosome assembly [42]. Viruses may hijack this process to splice their own RNA to increase the diversity of RNA transcripts and proteins produced. This is particularly important for human immunodeficiency virus (HIV) replication, which is completely dependent on the host cell spliceosome to produce an additional seven regulatory proteins that are required for infectivity [43].
Both the HBV pgRNA and preS/S mRNAs may be spliced by the host cell spliceosome. Studies in occult HBV patients have suggested that spliced preS/S mRNAs are important for hepatitis B surface antigen (HBsAg) secretion, as a mutation that prevents preS/S mRNA splicing reduced HBsAg expression [44, 45]. The HBV pgRNA can be alternatively spliced prior to nuclear export to form shorter spliced RNAs (Figure 1). Spliced pgRNA is the major focus of this review.
Spliced pgRNAs are reverse transcribed using HBV pol supplied in trans [46] to form shorter double-stranded linear DNA (dsDNA) genomes, which are enveloped and secreted as defective viral particles [47]. Unlike viruses such as HIV which require splicing to express key viral proteins that support viral replication, splice variants are not essential for HBV replication, as mutation or deletion of HBV splice sites has no discernible effect on replication [48, 49]. However, their role in the HBV replication cycle and/or pathogenesis largely remains unknown.
The pgRNA contains at least five 5’ donor splice sites and five 3’ acceptor splice sites that are used interchangeably to produce over 20 splice variants that have been described thus far [8, 9, 50, 51] (Table 1, Figure 2).
Location of splice sites and sequences responsible for generating various spliced HBV pgRNA
| 5’ donor splice site nucleotide position* | Splice site sequence (based on HBV genotype D) | Splice variants produced by splicing at this splice site |
|---|---|---|
| 2067/8 | CAG/GCAAGC | Sp2, Sp3, Sp8, Sp10, Sp12, Sp15, Sp16, Sp17 |
| 2087/8 | GGG/GTGAGT (genotype B and C only) | Sp4, Sp5 |
| 2447/8 | AAT/GTTAGT | Sp1, Sp2, Sp4, Sp7, Sp8, Sp9, Sp10, Sp12, Sp13, Sp15 |
| 2471/2 | AAG/GTGGGR | Sp6, Sp11, Sp16, Sp18 |
| 2985/6 | AAG/GTAGGA | Sp7, Sp8, Sp14, Sp18, Sp19, Sp20 |
| 3’ acceptor splice site nucleotide position* | Splice site sequence (based on HBV genotype D) | Splice variants produced by splicing at this splice site |
| 2234/5 | GAA/G | Sp12, Sp15 |
| 2349/50 | TAG/A | Sp2, Sp4, Sp8, Sp10, Sp16 |
| 2901/2 | CAG/T | Sp7, Sp8, Sp13, Sp18 |
| 281/2 | TAG/G | Sp9, Sp10, Sp11, Sp12, Sp17, Sp20 |
| 488/9 | CAG/G | Sp1, Sp2, Sp3, Sp4, Sp5, Sp6, Sp7, Sp8, Sp14, Sp15, Sp16, Sp18 |
* Numbering is based on EcoR1 start site. HBV: hepatitis B virus; C: cytosine; A: adenine; G: guanine; T: thymine; R: adenine or guanine
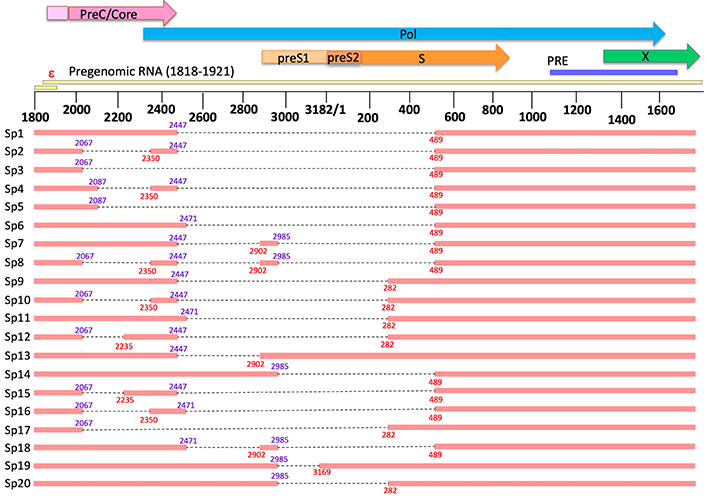
Linear map of all detected hepatitis B virus (HBV) splice variants. The top of the figure shows the open reading frames of wild-type HBV and proximity of the epsilon sequence on the pregenomic RNA (pgRNA). Numbering is based on the EcoR1 site (genotype D). Parts of the genome missing due to splicing are indicated by the black dotted lines. Locations of donor splice sites are indicated in purple and locations of acceptor splice sites are indicated in red. PreC: precore; Pol: polymerase; PRE: post-transcriptional regulatory element; ε: epsilon encapsidation signal
Note. Reprinted from “The in vitro replication phenotype of hepatitis B virus (HBV) splice variants Sp3 and Sp9 and their impact on wild-type HBV replication” by McCoullough LC, Sadauskas T, Sozzi V, Mak KY, Mason H, Littlejohn M, et al. J Virol. 2024;98:e0153823 (https://journals.asm.org/doi/10.1128/jvi.01538-23). CC BY.
All splice variants retain the encapsidation signal in epsilon critical for packaging of pregenomic (and spliced) RNA, and the X gene, but have deletions in the pol as well as core and/or surface ORFs. As they do not produce a full-length pol protein, splice variants are unable to replicate autonomously and require rescue by wild-type HBV pol for replication [46]. Spliced and unspliced HBV RNA have also been identified in the sera of HBV-infected patients [52, 53], suggesting these RNAs may be packaged into nucleocapsids but fail to be reverse transcribed by the viral pol protein. Several splice variants encode precore RNA, which encodes the secreted HBeAg, a key immunomodulatory protein that is critical for establishment of chronic HBV infection [54–56]. The role of splice variants as a regulator of HBeAg expression is unknown.
Sp1 was the first splice variant to be detected in the hepatocytes of patients living with CHB [57]. Sp1 is spliced between the 5’ donor splice site at nucleotide 2447 (within the penultimate amino acid of the core gene) and the 3’ acceptor splice site at nucleotide 489, mid-way through the pol gene and S genes [9]. It is the most common splice variant detected in patient sera and accounts for up to 30% of total splice variants [7, 49, 57, 58]. There are conflicting reports on which is the second most common splice variant. Some studies in cell culture report Sp3, spliced between nucleotides 2067 and 489, is the second most common splice variant [46, 58, 59], and was more common than Sp1 in one patient sample [60]. Other cell culture studies have suggested that Sp9, spliced between nucleotides 2447 and 282, is the second most common splice variant [8, 61], which was also observed in HBV patient samples [8, 62]. In turn, Sp13 has been reported to be the second most common splice variant in patient samples [7]. Sp6 has also been identified as a common splice variant in cell culture [63]. These differences may be due to differences between HBV genotypes and/or phase of natural history analysed as well as differences between cell culture and human studies.
HBV splicing depends on the sequences of the 5’ donor and 3’ acceptor splice sites as mutations at these splice sites abolish splicing [48, 50]. However, as HBV relies on the full-length pgRNA to replicate, HBV splicing must be strongly regulated to facilitate export of the unspliced full-length pgRNA. The mechanisms regulating production of HBV splice variants remain largely unknown. HBV encodes a post-transcriptional regulatory element (PRE; nucleotides 1151–1684) that facilitates the nuclear export of unspliced pgRNA [64, 65]. PRE is required for production of splice variants, as deletion of this region results in a ~60% reduction of splicing [66, 67]. The PRE contains both splicing enhancer and silencing elements, and so both positively and negatively regulate splicing [67, 68]. Splicing regulator element 1 (SRE-1), a splicing enhancer element, is located between nucleotides 1252 and 1288. Deletion of this region reduces splicing, however, deleting the whole PRE resulted in a greater reduction, suggesting that other unidentified splicing enhancer elements are located in the PRE [67]. Alternatively, a splicing silencer was identified at the 3’ end of the PRE, located between nucleotides 1481 and 1585 [68]. This element folds into a double hairpin structure, and both sense and antisense strands were found to strongly suppress splicing. However, this element did not suppress the production of Sp1, the most common splice variant, and therefore its ability to reduce splicing may be co-dependent on its location.
Cellular splicing factors may also play a role in regulation of HBV splicing. The PTB binds to the PRE and represses splicing, facilitating export of the full-length genome [69]. In contrast, the PTB-associated splicing factor (PSF) enhances splicing, perhaps by interacting with an element in the pgRNA, as knocking out PSF led to a decrease in splicing even in the absence of the PRE [67]. PSF binds to the polypyrimidine tract near the 3’ acceptor splice site and has been suggested to replace one of the 3’ splice site binding factors to promote splicing [70]. The increase in splicing by PSF was confirmed in vivo which showed that overexpression of PSF increased Sp1 RNA [71]. Furthermore, overexpression of the splicing factor La increased Sp1 production whereas overexpression of SRSF1 decreased Sp1 production [71]. Cells expressing LUC7 like 3 pre-mRNA splicing factor (LUC7L3) had a higher proportion of spliced RNA than unspliced RNA, suggesting that LUC7L3 also promotes splicing [72]. Additional host proteins may also be involved in regulating HBV splicing. Overexpression of histone deacetylase 5, an enzyme that removes acetyl groups from histone and non-histone proteins, upregulated expression of spliced HBV RNA in cell culture co-transfection experiments, suggesting that this host factor may be involved in regulating HBV splicing [73], whereas knocking out expression of trans-active response DNA binding protein (TARDBP) increased pgRNA splicing, suggesting that this host factor may inhibit pgRNA splicing [74]. However, as both of these studies were performed in cell culture, it remains unknown whether these host factors influence pgRNA splicing in vivo.
Taken together, these studies suggest that cellular splicing factors and host proteins contribute to tightly controlled regulation of HBV splicing.
Due to splicing and the overlapping reading frames, spliced HBV RNAs may encode novel fusion or truncated proteins, very few of which have been characterised [9]. The functions of four novel fusion proteins have been partially characterised—a 10 kDa pol fusion protein and 21 kDa C-terminal truncated core protein produced by Sp1, a 15 kDa pol-surface fusion protein produced by Sp7 and a 43 kDa pol-surface fusion protein produced by Sp13 [1, 75–77]. A Sp10 novel core-surface fusion protein has also been detected via western blotting, although its function is currently unknown [78] (Table 2, Figure 3).
Selected novel fusion proteins derived from hepatitis B virus (HBV) splice variants and their proposed role and/or effect on HBV replication
| Splice variant | Novel protein | Size (kDa) | Proposed role/effect on HBV replication | References |
|---|---|---|---|---|
| Sp1 | HBSP | 10 |
| [75, 79, 80] |
| [81] | |||
| [5] | |||
| [82] | |||
| [83–85] | |||
| [86] | |||
| p21.5 | 21.5 |
| [88] | |
| C-terminal truncated core protein | 21 |
| [87] | |
| [1] | |||
| N-terminal truncated pol protein | 39 |
| [48] | |
| Sp3 | Precore-pol | 48 |
| [9, 89] |
| Core-pol | 47 |
| [9, 89] | |
| Sp7 | HBDSP | 15 |
| [76] |
| Sp9 | Precore-surface | 45 |
| [89] |
| Core-surface | 44 |
| [89] | |
| C-terminal truncated pol with new open reading frame | 12 |
| [89] | |
| N-terminal truncated pol | 50 |
| [89] | |
| Sp10 | Core-surface | 35 |
| [78] |
| Sp13 | Pol-surface | 43 |
| [90] |
| [77] | |||
| [77] |
HBSP: hepatitis B spliced protein; pol: polymerase; HBDSP: hepatitis B doubly spliced protein; HBsAg: hepatitis B surface antigen
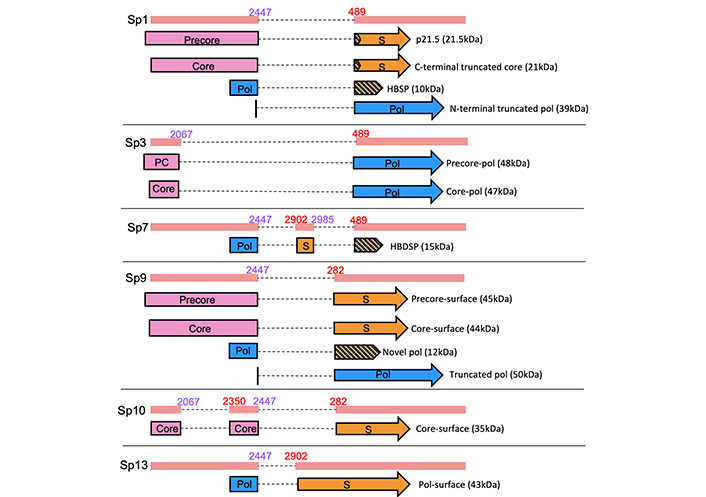
Selected novel fusion proteins derived from hepatitis B virus (HBV) splice variants. S: surface; Pol: polymerase; HBSP: hepatitis B spliced protein; PC: precore; HBDSP: hepatitis B doubly spliced protein
Sp1 RNA encodes four novel proteins: a 10 kDa pol fusion protein, a 21 kDa C-terminal truncated core protein, a 21.5 kDa precore protein derivative and a 39 kDa N-terminal truncated pol protein.
The 10 kDa small pol fusion protein, known as the hepatitis B spliced protein (HBSP), was one of the first novel fusion proteins to be detected in chronic HBV patients [75]. It consists of the first 46 amino acids of the pol sequence fused to an additional 47 amino acids which correspond to a new ORF. Initial studies of this protein revealed that it had no impact on HBV replication, however, cell viability assays showed that HBSP ectopic expression induced cell death, suggesting a role in apoptosis [75]. This was confirmed by Lu et al. [79] who found a BH3 domain in the N-terminus of HBSP, a known pro-apoptotic motif and in vitro studies showed that cells expressing higher levels of HBSP had higher caspase-3 protease activity. This was further confirmed by fluorescence-activated cell sorting (FACS) analysis which showed that cells expressing HBSP were more apoptotic than cells not expressing HBSP [80]. However, other studies have found that HBSP suppresses apoptosis, perhaps to promote cell proliferation and therefore promote HBV chronicity. Cells expressing HBSP increased cell viability when exposed to the agonistic anti-Fas antibody CH11 which is known to cause apoptosis. In addition, HBSP-expressing cells were found to reduce the amount of active caspase-8 and caspase-3 and therefore reduce Fas-induced apoptosis [81]. These seemingly conflicting findings may be due to the different genotypes used or different experimental conditions. Studies in patient samples found that HBSP is associated with higher levels of viral replication and severe liver fibrosis [5], whereas studies in murine models showed that HBSP limits the extent of liver inflammation by modulating the TNF-α-stimulated signalling pathways [82]. HBSP interacts with fibrinogen γ chain to inhibit fibrin polymerisation [83], cathepsin B to promote cell migration and invasion [84] and microsomal epoxide hydrolase to enhance the metabolism of benzo [alpha] pyrene, causing DNA mutations which increases the risk of cancer [85]. HBSP has also been shown to induce T-cell responses in mice models and HBV patients, which may assist with immune-mediated liver damage and HBV pathogenesis [86].
The 21 kDa C-terminal truncated core protein can replace the wild-type core protein to rescue wild-type HBV replication [87]. This protein is only truncated by one amino acid at the C-terminus of the core protein. More recently, this protein has also been shown to contribute to nucleocapsid formation and is required for infectivity in vitro and in vivo [1].
The 21.5 kDa novel precore protein is also only truncated by one amino acid at its C-terminus, however, it inhibits nucleocapsid formation by forming homodimers which interact with the core protein, preventing its assembly. This protein may therefore play a role in viral persistence by suppressing HBV replication [88]. The role of the 39 kDa N-terminal truncated pol protein, if any, is unknown [48].
Sp3 RNA, spliced between nucleotides 2067 and 489, is predicted to produce a novel 48 kDa precore-pol and a novel 47 kDa core-pol fusion protein [9, 89]. These proteins are yet to be characterised, and their role if any, is unknown.
Sp7 RNA, doubly spliced between nucleotides 2447 and 2902, 2985 and 489, produces a pol-surface fusion protein known as hepatitis B doubly spliced protein (HBDSP). This protein was first detected via western blotting using rabbit anti-HBDSP polyclonal antibodies after transfection of Sp7 DNA. It acts as a Gal4 activation domain, and the amino acid residues 48 to 75 were essential for this activity. Further analysis showed that HBDSP had transactivation activity on viral genes [76].
Sp9 RNA, spliced between nucleotides 2447 and 282, is predicted to produce a novel 45 kDa precore-surface fusion protein, a novel 44 kDa core-surface fusion protein, a novel 12 kDa C-terminal truncated pol protein joined to a new ORF and a novel 50 kDa N-terminal truncated pol protein. The roles of these proteins are largely unknown, however recent in vitro studies showed that Sp9 novel precore-surface and core-surface fusion proteins are involved in Sp9-mediated suppression of wild-type HBV replication [89].
Sp10 RNA, spliced between nucleotides 2067 and 2350, 2447 and 282, produces a core-surface fusion protein which was first detected by western blotting using an HBV core antibody [78]. The function of this protein is unresolved.
Sp13 RNA, spliced between nucleotides 2447 and 2902, produces a larger 43 kDa pol-surface fusion protein than Sp7 HBDSP. Furthermore, due to the shorter splicing sequence, Sp13 DNA retains most of the S ORF. The pol-surface fusion protein was first detected by immunoprecipitation using pol antibodies. This protein can replace LHBs in replication, as Dane particles were still produced upon co-transfection of LHBs-deficient wild-type HBV and the pol-surface fusion protein [90]. These studies were performed using the HBV ayw subtype. However, Park et al. [77] examined this protein in a different HBV subtype (adw), showing that it was produced following splicing between nucleotides 2455/2943. They found that this protein partially colocalised with the ER, nuclear pore complex, microtubules and vimentin filaments and prevented the secretion of HBsAg [77].
Even though HBV replication does not depend on the production of splice variants, splice variants have been shown to differentially impact wild-type HBV replication. Sp1, the most common splice variant, decreased wild-type HBV replication in vitro [88]. Co-transfection of wild-type HBV with Sp1 RNA decreased the production of pgRNA, HBV DNA and nucleocapsid formation. In this study, the effect was believed to be due to the production of its novel p21.5 protein. Wang et al. [88] showed that p21.5 forms homodimers that interact with the wild-type core protein, therefore inhibiting nucleocapsid formation, which decreases replication. However, a drawback of this study is that a truncated Sp1 RNA was used, and furthermore, transcription was driven by a cytomegalovirus (CMV) promoter, which overexpressed Sp1 RNA and the p21.5 protein. This did not accurately reflect the natural HBV replication cycle, and production of splice variants and their novel fusion proteins. A more recent study co-transfected a full-length Sp1 construct with wild-type HBV, and saw a similar inhibiting effect on wild-type HBV replication. However, mutating the start codon of p21.5 and therefore preventing its production did not completely restore HBV replication, suggesting other mechanisms may be involved [91].
Sp10 also decreased wild-type HBV replication in vitro. Tsai et al. [78] showed that Sp10 acted as a non-coding RNA that interacted with the TATA-binding protein (TBP) in the cytoplasm. TBP is essential for activating transcription in the nucleus. The interaction between Sp10 and TBP in the cytoplasm reduced the amount of nuclear TBP available, which lowered the rate of transcription and therefore decreased HBV replication [78]. Sp13 also decreased HBV replication in vitro. This effect depended on the production of the Sp13 pol-surface fusion protein as knocking out this protein partially restored HBV replication. However, the exact mechanism behind this effect remains unclear [77].
In contrast, Sp7 expression increased HBV replication in cell culture studies. Sp7’s effect on replication depended on the production of HBx, as there was no impact on replication when Sp7’s X protein was mutated [92]. Although yet to be investigated, the Sp7 HBDSP may also play a role in increasing replication as HBDSP has been shown to behave as a transcriptional activator [76]. In contrast, another splice variant, known as ASN, enhanced wild-type HBV replication independently of the X protein, suggesting that splice variants may have different mechanisms to enhance wild-type HBV replication [93].
We have recently shown that Sp9 profoundly impacts HBV replication in cell culture, whereas Sp3 had no effect [89]. The impact of Sp9 was largely due to the novel precore-surface fusion protein, as knocking out expression of this protein almost completely restored intracellular core-associated DNA and completely restored intracellular RNA and core protein expression. The Sp9 novel core-surface fusion protein was also involved in suppressing wild-type HBV replication, although not to the same extent as the novel precore-surface fusion protein. The mechanism of how these novel fusion proteins impact wild-type HBV replication remains to be determined.
These studies are limited by predominantly being performed in cell culture, apart from Wang et al.’s Sp1 study [88] which was partly performed in vivo. Therefore, it remains unknown whether similar effects occur in patients, where splice variants are largely detected at low levels. In cell culture, up to 5% of plasmid expressing Sp1 RNA relative to wild-type HBV plasmid reduced HBV replication [88]. In patient samples, Sp1 RNA made up to ~20–30% of total HBV RNA, with one sample having 83% Sp1 RNA [8, 62]. Up to 33% of plasmid expressing Sp7 RNA increased HBV replication in cell culture [92], whereas Sp7 RNA made up ~1% of total HBV RNA in patient samples [8]. Similarly, up to 10% and 12% of plasmid expressing Sp9 and Sp13 relative to wild-type HBV plasmid reduced HBV replication in cell culture [77, 89], whereas these splice variants made up < 10% of total HBV RNA in patient samples, with one patient sample having up to 30% of Sp13 RNA [8, 62]. Sp10 RNA is not often detected in patient samples and therefore the proportion remains unknown, although only 2% of Sp10 RNA was required to reduce HBV replication in cell culture [78]. Despite these comparisons, the levels of splice variants required to impact HBV replication in cell culture cannot be directly correlated to whether splice variants may impact HBV replication in patients. In addition, splice variants may be present for several years in patients with CHB, and it remains unknown whether the length of time splice variants are present for may be more influential than their abundance. Due to difficulties detecting splice variant novel fusion proteins, their abundance in patients with CHB remains unknown.
Different HBV genotypes express different quantities and types of splice variants. For example, the proportion of splice variants in serum of patients with genotypes B and C, which are most prevalent in Asia, is 8% higher than in the serum of patients with genotypes A and D, which are most prevalent in Europe and North America [94, 95]. Overall, splice variants are most common in genotype C [50]. Interestingly, patients with genotype C are more likely to develop HCC, which may suggest an association between the levels of splice variants and HCC development [96]. Sp1 is the most common splice variant in all HBV genotypes examined to date [8, 97, 98]. Due to the sequence variation between genotypes, not all genotypes produce every splice variant. For example, the doubly-spliced Sp10 is produced by genotypes A and B, but not genotype D, due to a single nucleotide change in the polypyrimidine tract between the nucleotide 2067 donor and nucleotide 2350 acceptor splice site [59]. Sp4 and Sp5 are only produced by genotypes B and C, as the consensus donor splice site sequence (nucleotide 2087) is only present in genotypes B and C [7, 8]. To date, the prevalence, impact and characterisation of splice variants in genotypes E to H have not been studied.
The association between HBV splice variants and drug resistance in HBV patients remains unknown. One study found that patients who did not respond to interferon (IFN)-α treatment had a higher ratio of spliced to wild-type HBV DNA in their serum compared to HBV-infected patients who did respond to treatment [50]. Furthermore, in vitro studies showed that the Sp1 HBSP and the Sp1, Sp2, and Sp3 RT’-RNase H domain interfere with IFN-α treatment. Sensitivity to IFN-α treatment was recovered after mutation of the most common donor (2447/2448) and acceptor (488/489) site which prevented the production of splice variants Sp1–8 and 13–16, suggesting that splice variants and their novel fusion proteins play a role in IFN-α drug resistance. However, it remains unknown how Sp9–12 and Sp17–20 and their novel proteins affect IFN treatment [50].
Mutations associated with drug resistance to direct acting antiviral therapy result in a lower production of splice variants. In a study by Preiss et al. [99], a lower proportion of splice variants was observed in patients with lamivudine resistant HBV compared to patients infected with wild-type HBV. This may be due to the mutations preventing or slowing the rate of reverse transcription of the splice variant [99].
HBV splice variants are associated with liver disease progression. Soussan et al. [4] showed there was a higher proportion of defective HBV DNA, produced by reverse transcription of spliced HBV RNA, compared to wild-type DNA in patients with severe fibrosis and liver necrosis compared to patients with mild disease [4]. Subsequently Redelsperger et al. [3] also observed higher ratios of spliced HBV in patients with severe liver disease. This association may be due to the partial loss of the core and S genes in the spliced DNA as core and S mutants have been associated with progressive liver disease [100, 101]. Although a separate study found no association between HBV splice variants and liver disease severity [99], studies showing that spliced HBV DNA levels fluctuated during disease progression [3] or HBV natural history [7] suggest that such associations may be overlooked in the absence of longitudinal analysis.
It is unknown whether HBV spliced DNA may promote liver disease or whether the development of liver disease promotes HBV splicing. Murine studies suggest the latter, as the induction of liver fibrosis or inflammation in mice resulted in the overexpression of splicing factors PSF, SF1, SRSF1, and La, increasing the production of splice variants [71]. Alternatively, the additional HBx protein contributed by splice variants may promote the production of liver disease as HBx has been shown to upregulate inflammatory cytokines, contributing to liver inflammation [102]. HBSP may also promote liver disease as high anti-HBSP levels have been associated with liver fibrosis, and HBSP has been shown to activate a T-cell response, which may cause liver damage [5, 86]. However, as previously mentioned, a conflicting study suggests that HBSP limits liver inflammation by modulating the TNF-α-stimulated signalling pathways [82] and the role of Sp1 and HBSP in liver disease progression remains unclear.
An increased proportion of splice variants in patient sera has been associated with the development of HCC, as HCC patients had higher levels of spliced HBV DNA in their sera than non-HCC controls [6]. Furthermore, serum spliced HBV DNA levels increased approximately 0.1% every year prior to diagnosis, suggesting that spliced HBV DNA levels may indicate a higher risk of HCC development [6].
The reason as to why the presence of splice variants are associated with the development of HCC remains unclear. Sp7, isolated from an HCC patient, increased HBV replication, which may contribute to viral persistence in HCC patients [92]. Splice variants may also encode proteins that assist with HCC development. All characterised splice variants produce the HBx protein [9]. HBx is a cofactor in hepatocarcinogenesis [103] and interacts with a number of cellular proteins to disrupt the cell cycle [104–108] and signalling transduction pathways [109, 110], key factors in HCC development. HBSP has been associated with HCC metastasis and has been shown to increase hepatoma cell invasion and mobility, perhaps through the interaction of cathepsin B, a lysosomal protease [84]. Integration of splice-derived HBV DNA may also contribute to cancer development, as a splice-derived protein from integrated HBV DNA was found to have oncogenic properties [111]. In contrast, the development of HCC may lead to the increased production of splice variants as the splicing factor SRSF2 is upregulated in human HCC [112].
A major limitation is that most studies on HBV splice variants are exclusively cell culture-based which may not correspond to the impact of splice variants on HBV replication and pathogenesis in patients with CHB. Further studies examining the association between clinical outcomes and levels of spliced RNA are required to further elucidate the impact of splice variants in patients with CHB. In addition, a number of studies use expression plasmids to express spliced RNA which do not accurately reflect the true production of splice variants [77, 78, 88]. Ideally, the impact of splice variants on HBV replication should be determined using an infection model or in vivo models, although this proves difficult with current HBV infection models as splice variants require HBV proteins in trans to replicate. A number of studies have also used short-read sequencing methods to map HBV spliced RNAs [7, 62], which limits the detection of multiple-spliced transcripts such as Sp2, Sp4, Sp8, Sp10, Sp12, Sp15, Sp16, and Sp18. Long-read sequencing permits detection of these splice variants [8, 60, 63], and therefore should be the preferred approach to determine levels of these splice variants and any association with HBV replication and pathogenesis.
There is increasing evidence that HBV splicing, through the generation of novel variants and proteins, impacts HBV replication, protein expression, and infectivity more than previously credited. These studies should be further explored using in vivo models, noting that this is difficult as splice variants are incapable of autonomous replication in the absence of the HBV pol supplied in trans. Nonetheless, recent studies using “knockout” HBV clones that do not splice, by mutation of the two most common 3’ splice sites (A487C and A280C), compared to replication (and splice) competent wild-type HBV clones offers a way forward to study the impact of splice variants in cell culture and murine studies [1]. Indeed, findings from these studies have already suggested the importance of the Sp1 C-terminal truncated core protein in nucleocapsid formation and infectivity [1]. Further studies on the association of individual splice variants with liver disease progression and liver cancer would also prove enlightening.
cccDNA: covalently closed circular DNA
CHB: chronic hepatitis B
HBDSP: hepatitis B doubly spliced protein
HBeAg: hepatitis B e antigen
HBsAg: hepatitis B surface antigen
HBSP: hepatitis B spliced protein
HBV: hepatitis B virus
HBx: hepatitis B X protein
HCC: hepatocellular carcinoma
IFN: interferon
LHBs: large hepatitis B surface protein
ORF: open reading frame
pgRNA: pregenomic RNA
pol: polymerase
PRE: post-transcriptional regulatory element
PSF: polypyrimidine tract binding protein-associated splicing factor
PTB1: polypyrimidine tract binding protein 1
rcDNA: relaxed circular DNA
TBP: TATA-binding protein
LCM: Investigation, Writing—original draft, Writing—review & editing. ML: Writing—review & editing, Supervision. PAR: Conceptualization, Writing—review & editing, Supervision.
PAR has received investigator-initiated industry-funded grants from Gilead. None of this support is relevant to the work in this manuscript. The remaining authors declare no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
LCM, ML and PAR were funded by National Health and Medical Research Council (NHMRC) grant [1145977]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Hao Xiong, Jinsheng Guo
Chunzheng Li ... Xianguang Yang
Juntian Yao ... Youhua Xie
