 Review
Review

Affiliation:
1Institute of Biomedical Research of Barcelona, Spanish National Research Council (IIBB-CSIC), 08036 Barcelona Spain
†These authors contributed equally to this work.
ORCID: https://orcid.org/0009-0005-6616-3958
Affiliation:
1Institute of Biomedical Research of Barcelona, Spanish National Research Council (IIBB-CSIC), 08036 Barcelona Spain
2Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBERehd), Carlos III National Institute of Health, 28029 Madrid, Spain
3Faculty of Medicine, University of Barcelona, 08036 Barcelona, Spain
4Instituto de Investigaciones Biomédicas August Pi i Sunyer (IDIBAPS), 08036 Barcelona, Spain
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0001-9948-9720
Affiliation:
1Institute of Biomedical Research of Barcelona, Spanish National Research Council (IIBB-CSIC), 08036 Barcelona Spain
2Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBERehd), Carlos III National Institute of Health, 28029 Madrid, Spain
4Instituto de Investigaciones Biomédicas August Pi i Sunyer (IDIBAPS), 08036 Barcelona, Spain
Email: anna.moles.fernandez@gmail.com
ORCID: https://orcid.org/0000-0002-3915-0060
Explor Dig Dis. 2024;3:428–442 DOI: https://doi.org/10.37349/edd.2024.00059
Received: June 26, 2024 Accepted: August 06, 2024 Published: September 10, 2024
Academic Editor: Chunye Zhang, University of Missouri, United States
Metabolic-associated steatotic liver disease (MASLD) and its pathological version, metabolic dysfunction-associated steatohepatitis (MASH), are becoming the main leading causes of chronic liver disease almost worldwide and are the fastest growing aetiology of hepatocellular carcinoma (HCC), especially in the Western countries. The combination of high incidence and morbidity with limited treatment options for both MASH and HCC highlights an urgent need for the discovery of novel therapeutic candidates to inform drug development. The importance of lysosomes and cathepsins, their most abundant hydrolases, has been overlooked for decades. They were considered organelles only involved in the recycling of macromolecules, with cathepsins simply being their effectors. Contrary to this traditional view, recent findings have shed new light on the lysosome and its enzymes as drivers of essential cellular processes, such as apoptosis and autophagy. Bringing lysosomal activity and the regulation of cathepsins into the spotlight of MASH and HCC research can open new avenues for the development of novel drugs based on targeting cathepsin-driven lysosomal activity and its associated pathological processes. This review comprehensively summarises the current knowledge on the role and contribution of lysosomal cathepsins to MASLD/MASH and HCC progression.
Lysosomes are membranous organelles in charge of the recycling, degradation, and elimination of cellular waste, but they also take part in other signalling and metabolic pathways. These membrane-bound vesicles are known to participate both in the intracellular and extracellular trafficking pathways, via endocytosis, phagocytosis, and autophagy [1, 2]. For a long time, lysosomes have been studied as static and somehow ‘waste-bag’ organelles. However, latest research unveils a more dynamic and active role of these vesicles in the cellular homeostasis and metabolic regulation [3] and shows a clear cross-communication with other important cell organelles, such as the mitochondria [4].
Lysosomes are specialised on the processing of various macromolecules, including carbohydrates, lipids, nucleic acids, and proteins. The presence of degradative enzymes is key for this action. Lysosomal hydrolases are the most abundant and well-described group of proteolytic enzymes and are particularly active in the acidic pH of the endocytic pathway [5]. Different subtypes of lysosomal proteases finely tune cellular processes, like apoptosis, cell migration, immune activation, or extracellular matrix (ECM) remodelling. These networks are key for the physiological state of the cell (apoptosis or antigen presentation), but also play a key role on disease progression (cancer, fibrosis, or neurodegeneration) [6–9]. The malfunctioning of these proteolytic enzymes is responsible for a specific group of pathologies, commonly referred to as lysosomal storage diseases (LSDs) [10].
Different groups of lysosomal proteases have been studied according to their pH range of activation, cellular location, tissue of action, or type of substrate. Three classifications are currently in use, depending on their catalytic aminoacidic site (asparagine, glutamic, serine, threonine, aspartic, cysteine or metalloproteases) [11], their in-family homology and molecular structure, and their catalytic reaction. Most of the described proteolytic enzymes are integrated in the MEROPS database, which also catalogues their substrates and inhibitors [12]. This review focuses on cathepsins, which are the main group of lysosomal hydrolases and are commonly classified depending on their catalytic site.
Cathepsins are synthetized as inactive forms, which follow several modifications in the endoplasmic reticulum, the Golgi apparatus, and then are directed to the lysosomal compartment, where they become active through the cleavage of their pro-region, until now thought to be an auto-inhibitor. This specific and complex processing pathway ensures that the proteases are only active once they reach the endo-lysosomal compartments. Besides this, cathepsins can be directed by alternative routes to other cellular locations (cytoplasm, nuclei, and the extracellular space) where they can also exert extra-lysosomal functions, despite the neutral pH of these locations [13, 14], usually for a short period of time. Cathepsin activity is regulated not only by pH but also by exogenous and endogenous inhibitors, such as cystatins and stefins [15].
Most cathepsins are ubiquitously distributed across tissues but there are some exceptions such as cathepsin S (CtsS), CtsE, and CtsW, which are mainly expressed in immune cells, and CtsK, which is preferentially expressed in epithelial cells and osteoclasts. Within the liver there is also a differential cellular expression of some cathepsins. For example, CtsB is highly expressed in the periportal sinusoids and Kupffer cells, while CtsL shows an opposite expression pattern, with a higher expression in the centrilobular hepatocytes [16]. These cell-specific distributions suggest a differential functional role of cathepsins among the different liver cell populations. Cathepsins are involved in various processes for the maintenance of liver homeostasis and participate in essential signalling pathways, such as apoptosis and autophagy [17]. For example, lysosomal cysteine cathepsins are known to contribute to the activation of caspase-3 [18] and caspase-8 [19], participating in the canonical apoptosis signalling pathway in hepatocytes. In addition, lysosomal function driven by cathepsins has been reported to be an important factor for the progression of several liver disorders, including metabolic dysfunction-associated steatohepatitis [MASH, formerly known as non-alcoholic steatohepatitis (NASH)], fibrosis/cirrhosis, and hepatocellular carcinoma (HCC) [20–22]. This review aims to summarise the role of cathepsins in the development and progression from MASH to HCC and to provide new insights of lysosomal proteases as potential therapeutic targets for these diseases.
MASH is mainly caused by a subjacent state of metabolic syndrome and is becoming the main cause of chronic liver disease almost worldwide. Nowadays, up to 20% of the global population suffers from some degree of MASH and only in Europe, more than 10 million people already have developed MASH. In addition, current predictions estimate a raise in its prevalence up to 55% by 2040 [23].
The involvement of cathepsins in fatty liver disease was first reported back in 1958 [24]. One of the main biological features of MASH is cellular lipotoxicity, which is caused by the vast amount of lipids stored in hepatocytes and other liver cells. Some cathepsins, like CtsB and CtsD, regulate the flux of free fatty acids (FFA) between the mitochondria and the cytoplasmic compartment, via c-Jun N-terminal kinase (JNK) [25] and LDL receptor-related protein-1 (LRP1), respectively [26]. Besides, metabolic-associated steatotic liver disease (MASLD) patients show abnormal autophagy [27], that seems to be the result of both a defective lysosomal acidification and an impairment in lysosomal protease activation [28–30]. Since the transcription factor EB (TFEB) is a master regulator of lysosomal function and autophagy, it is not surprising that contributes to MASH progression [31]. Evidence of a link between TFEB and cathepsins, as downstream targets of TFEB, are slowly emerging in the context of MASH [32, 33]. MASLD is not considered a pathological condition, but a pre-pathological state for MASH. For this reason, this review will mainly focus on the role of cathepsins in MASH, as the implication and association of these lysosomal proteases with obesity-induced ectopic accumulation of lipids in hepatic tissue has already been reported [34].
Cathepsins are responsible for the exacerbation of MASH by stimulating the hepatic immune system, which is dramatically dysregulated towards a pro-inflammatory pathological state [35]. In this context, CtsB has recently been related with the progression of MASH, demonstrating that CtsB-deficient mice treated with a fructose-palmitate-cholesterol diet display improved liver function. According to this report, CtsB-deficient mice were able to suppress de novo lipogenesis and liver inflammation. The same researchers pointed to a possible shift towards an M2 polarization state of hepatic arginase 1-positive macrophages due to CtsB depletion, together with a reduction in TGF-β1 signalling [36]. These results suggest that CtsB could induce a pro-inflammatory M1 state in macrophages. In agreement, other reports indicate that CtsB pharmacological inhibition using CA-074-methyl-ester limits NF-κB-dependent hepatic inflammation when performing in vitro experiments with primary parenchymal and non-parenchymal hepatic cell lines [37]. Furthermore, CtsB inhibition using CA-074 stimulates the anti-inflammatory state of Kupffer cells, limiting the NLRP3 inflammasome-dependent MASH formation, in which caspase-1 is involved [38]. Additionally, LDL receptor-deficient mice treated with a high-fat, high-cholesterol (HFC) diet for three weeks showed lower levels of caspase-1 and pro-inflammatory markers (TNF-α or CCL2) when other lysosomal proteases, such as CtsD, were inhibited with Pepstatin A (PepA)—an exogenous inhibitor of aspartyl proteases [39]. Moreover, NLRP3 inflammasome is known to be activated by some cathepsins, such as CtsS and CtsL [40] and CtsB [41].
CtsD has shown potential to become a MASH biomarker, as serum CtsD correlates with MASH progression in humans when compared to a control population [42]. Moreover, plasma CtsD levels also correlate with insulin resistance, which is one of the main features contributing to MASH progression from MASLD [43]. Since these findings, several publications have been published deciphering the role of CtsD in MASH. In vitro studies demonstrated that CtsD inhibition with PepA in wild-type bone marrow-derived macrophages reduced inflammation and improved cholesterol metabolism in this cultured cell type [39]. Additionally, treating LDL receptor-deficient mice with an extracellular CtsD inhibitor, known as CTD-002, reduced hepatic lipid accumulation and increased faecal bile acid excretion. Moreover, extracellular CtsD inhibition promoted a switch towards an anti-inflammatory immune profile in the livers of these mice. Surprisingly, intracellular CtsD inhibitors did not affect lipid metabolism in this same murine model [44].
Since fibrosis contributes to both MASH and HCC, it is important to note that cathepsins actively contribute to fibrosis development by modulating hepatocyte apoptosis after injury [45], controlling hepatic stellate cell transdifferentiation into activated fibroblasts [21], influencing the inflammatory and autophagic responses [37] and participating in ECM remodelling [46]. Recent reviews have nicely covered this topic and more information can be found there [20, 47].
As far as we are concerned, only one drug has been approved by the Food and Drug Administration (FDA) to treat MASH and stop the disease from progressing: Resmetirom (an oral thyroid hormone receptor beta-selective agonist) [48–50]. The lack of effective and liver-targeted drugs to cure MASH urges further investigation into the cellular and molecular mechanisms driving this liver pathology. Lysosomal proteases, including cathepsins, are called to play a key role in several steps of MASH progression (Figure 1 and Table 1). Current research should focus on the discovery of experimental therapies to improve or revert MASH by restoring correct lysosomal activity, diminishing lipotoxicity, favouring an anti-inflammatory switch, and balancing cathepsin activity. For all of this, more research is needed to find out the key points of these recently reported anti-inflammatory pathways and how cathepsins influence MASH pathological state.
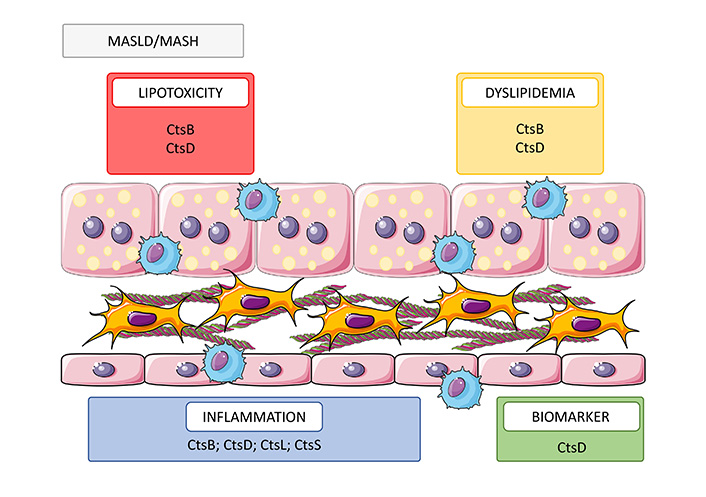
Role of cathepsins in MASLD/MASH. Cathepsins participate in multiple cellular processes contributing to MASLD/MASH progression through increasing lipotoxicity (CtsB and D) and dyslipidemia (CtsB and D), promoting inflammation (CtsB, D, L, and S) and biomarker (CtsD). MASLD: metabolic-associated steatotic liver disease; MASH: metabolic dysfunction-associated steatohepatitis; Cts: cathepsin
Note. This figure contains (modified) images from Servier Medical Art, licensed under CC BY 4.0.
Biological roles of cathepsins during MASLD/MASH and HCC development and progression
| Type | Name and MEROPS ID | Biomarker role | MASLD/MASH | HCC |
|---|---|---|---|---|
| Aspartate | Cathepsin D (A01.009) | |||
| Cysteine | Cathepsin B (C01.060) | Expression in tumour as a marker of HCC and bad prognosis [55] | ||
| Cathepsin C (C01.070) | - | - |
| |
| Cathepsin H (C01.040) | - | - |
| |
| Cathepsin K (C01.036) | Expression in tumour as a marker of HCC [61] | - |
| |
| Cathepsin L (C01.032) | Expression in tumour as a marker of HCC and bad prognosis [60] |
| ||
| Cathepsin S (C01.034) | Expression in tumour as a marker of HCC [59] |
| ||
| Cathepsin L2/V/U (C01.009) | Expression in tumour as a marker of HCC and bad prognosis [63] | - |
| |
| Cathepsin X/Z (C01.013) | Expression in tumour as a marker of HCC [64] | - |
| |
| Serine | Cathepsin A (S10.002) | Tumour expression and serum levels may be diagnostic and prognostic markers for HCC [57, 58, 65] | - | - |
| Cathepsin G (S01.133) | Expression in tumour as a marker of HCC [62] | - | - |
MASLD: metabolic-associated steatotic liver disease; MASH: metabolic dysfunction-associated steatohepatitis; HCC: hepatocellular carcinoma; FFA: free fatty acids; IGF-1: insulin-like growth factor 1; TAM: tumour-associated macrophage
Liver cancer is the third leading cause of cancer-related deaths worldwide, and despite research efforts, its incidence and mortality rates are steadily increasing each year [51]. Most primary liver cancers are HCCs, which have a very poor prognosis and are highly resistant to systemic therapies [52]. Around 20% of MASH patients will develop cirrhosis, and 1–2% of them will progress to HCC yearly [53]. Thus, MASH is becoming the fastest growing aetiology of HCC, especially in the Western countries [54].
Cathepsins play a key role in tumour progression, as they regulate many cellular processes related to tumour development, such as tumour growth, ECM remodelling, migration, and metastatic dissemination. Overexpression of CtsB [55], CtsD [56], CtsA [57, 58], CtsS [59], CtsL [60], CtsK [61], CtsG [62], CtsV [63], and CtsX/Z [64] have been reported in HCC, mainly as an indicator of poor prognosis and shorter overall survival of the patients. Moreover, serum levels of both CtsA [65] and CtsD [56] have been proposed as biomarkers to diagnose HCC and to distinguish it from other liver diseases such as cirrhosis.
Cathepsins regulate signalling pathways that contribute to cell proliferation and tumour growth. Both CtsB [66] and CtsK [61] have been described to activate the PI3K/AKT signalling pathway in human HCC cell lines, promoting cell proliferation and survival. Another mitogenic signalling pathway controlled by cathepsins is the insulin-like growth factor 1 (IGF-1), which activates both PI3K and MAPK pathways. CtsD has been proposed to cleave IGF-binding proteins (IGFBPs), thereby increasing the activity of IGF-1 in hepatoma cell lines [67, 68]. Furthermore, CtsB has been described to be the effector protein of IGF-1 pathway [69], as CtsB genetic depletion protects against IGF-1-driven tumour growth and metastasis in HCC cells, both in vitro and in vivo. In addition, CtsL has also been reported to stimulate HCC tumour growth, since injection of human HCC cells overexpressing CtsL resulted in increased tumour growth compared to the control and similar results were obtained in vitro [60]. Finally, small interfering RNA (siRNA) silencing of CtsS [70] and CtsV [63] has been shown to strongly suppress HCC cell proliferation in vitro and in vivo, respectively.
Apoptotic cell death is also widely known to be controlled by several cathepsins during HCC. Lysosomal membrane permeabilization (LMP) allows cathepsins to translocate into the cytoplasm, where they cleave several proteins from the Bcl-2 family, contributing to cytochrome C release from the mitochondria. Briefly, LMP in HCC cell lines can be mediated by either the TNF-related apoptosis-inducing ligand (TRAIL) pathway [71], TNF-α combined with cycloheximide [72] or JNK inhibition [73], resulting in the release of both CtsB and CtsD into the cytosol. There, they cleave the pro-apoptotic protein Bid into truncated Bid (tBid), which binds to the mitochondrial membrane, allowing Bax and Bak oligomerization and subsequent cytochrome C release [71, 74, 75]. On the contrary, some cathepsins have been described to play an anti-apoptotic role in HCC. Knockdown of CtsS induces apoptosis in human HCC cell lines and increases their chemosensitivity by regulating NF-κB nuclear translocation [76]. Similar results have been obtained when CtsS is pharmacologically inhibited in other types of cancer cell lines, such as glioblastoma [77] or renal cell carcinoma [78, 79] using molecules such as ZFL or CS-PEP1. CtsH silencing also sensitises HCC cell lines to apoptosis, reversing radioresistance via metabolic switch and subsequent release of pro-apoptotic factors [80].
Several studies have robustly linked CtsB expression with key steps of tumour progression, such as ECM remodelling, invasion, migration, and active contribution to the metastatic dissemination of tumour cells. First, CtsB expression mediated by CD147 facilitates migration and invasion of hepatoma cells in vitro [81]. In addition, in vitro and in vivo experiments show that CtsB overexpression promotes cell invasion through upregulation of MMP-9, whereas CtsB downregulation has the opposite effect [55]. Similarly, interaction of hepatitis B spliced protein with CtsB increases secretion and activation of proteolytic enzymes and activation of MAPK/AKT signalling pathway, resulting in hepatoma cell motility and invasion [82]. Recently, CtsB secretion by HCC tumour-initiating cells has been described to play a key role in mediating their migration and invasion capabilities [83]. Although there are fewer studies on other proteases, many cathepsins also play a role in tumour invasion. CtsD secretion to the extracellular space, together with CtsB, also promotes migration and invasiveness of the HCC cell line HepG2 after tetrabromobisphenol A exposure [84]. Furthermore, CtsC and CtsS are associated with metastasis development via activation of TNF-α/p38 MAPK [85] and VEGF-A/VEGFR-2/MEK1/ERK1/2 [86] signalling pathways, respectively. Other proteases, like CtsX/Z are overexpressed in HCC cells and are reported to upregulate the epithelial-to-mesenchymal transition and the metastatic potential of cancer cells [64]. On the contrary, CtsL downregulation has been described to increase cancer cell migration through stabilization of integrin beta 1 in HCC cells expressing HBV X protein [87].
In addition to cancer cells, tumour microenvironment (TME), which consists of immune cells, stromal cells, blood vessels and ECM, may also play an important role in tumour development, generating both pro- and anti-tumoral responses. Angiogenesis is a critical process in TME generation, as tumours rely on oxygen and nutrient supply in order to grow. To that respect, it has been described that CtsS overexpression in HUVEC results in increased angiogenesis in an endothelial cell tube formation assay via activation of VEGF-A/VEGFR-2/MEK1/ERK1/2 pathway [86]. In line with these results, medium from CtsS and CtsB knockdown hepatoma cells leads to decreased endothelial cell tube formation [70, 82]. Another key feature of the TME is immune evasion: tumours reshape both innate and adaptative immune responses towards immunosuppression, allowing tumour cells to escape from immunosurveillance. Tumour-associated macrophages (TAMs) play a significant role in creating an immunotolerant environment, by secreting cytokines, chemokines and inhibiting T cell-mediated anti-tumour response [88]. Single-cell RNA sequencing analyses of human HCC have shown that SPP1+ and FOLR2+ TAM subclusters overexpress several cathepsins, mainly CtsD, CtsB, and CtsL. High number of SPP1+ TAMs are associated with bad prognosis and overall survival in HCC patients [89–91]. In line with these observations, genetic inhibition of CtsB and CtsS [92] in TAMs leads to decreased tumour volume in vivo, probably due to a polarization shift towards an M1 phenotype. Pharmacological inhibition of CtsB, CtsS, and CtsL [93] using a novel inhibitor, GB111-NH2, has been shown to induce this phenotypic switch in vitro. Similarly, it has been observed that CtsD deletion in cancer cell lines induces TAM reprogramming from M2 to M1 subtype through TGFBI-CCL20 signalling [94].
In conclusion, cathepsins play an important role in the regulation of many processes associated with tumour growth and development (Table 1 and Figure 2). Therefore, they have been proposed as therapeutic targets in HCC and their pharmacological and genetic ablation is being tested both in vitro and in vivo for the treatment of many cancers besides liver cancer [95, 96]. However, current cathepsin inhibition strategies present some limitations that need to be addressed before being translated into the clinic. Firstly, cathepsins involvement in multiple signalling pathways both in physiology and pathology must be carefully considered before developing therapies based on modulating their activity. For example, both CtsB and CtsD are pro-apoptotic proteins and their inhibition could lead to immortalisation of HCC cancer cells under certain circumstances [74, 75]. Secondly, some cathepsins display a high degree of structural homology, which might limit the development of specific inhibitors. Indeed, many of the cathepsin inhibitors currently under investigation have a broad spectrum of activity. Therefore, non-targeted cathepsin inhibition could potentially lead to undesirable side effects and adverse patient outcomes. Fortunately, advances in drug design have opened new avenues for the development of specific inhibitors for individual cathepsins, such as CA-074 for CtsB or CLIK-148 for CtsL [97]. Small-molecule inhibitors targeting only secreted cathepsins have also been developed, such as CTD-002 for CtsD [44]. Moreover, other strategies such as antibody inhibitors are also being tested to improve the efficacy and specificity of cathepsin inhibition [98].
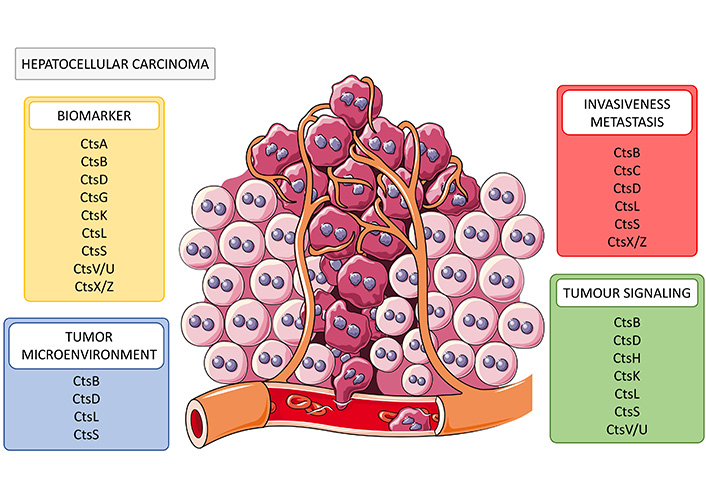
Role of cathepsins in HCC. Cathepsins have been described as potential biomarkers (CtsA, B, D, G, K, L, S, V/U, X/Z) and as participants in several cellular processes contributing to HCC development and progression through increasing tumour invasiveness and metastasis (CtsB, C, D, L, S, and X/Z), enhancing tumour microenvironment immunosuppression (CtsB, D, L, and S) and contributing to several tumour-promoting signalling pathways (CtsB, D, H, K, L, S and CtsV/U). Cts: cathepsin
Note. This figure contains (modified) images from Servier Medical Art, licensed under CC BY 4.0.
These limitations may have hampered the success of cathepsin modulation as a therapeutic strategy for cancer, as no cathepsin-related therapy has been tested in clinical trials for primary tumours or metastases other than bone metastases. In this regard, the CtsK-selective inhibitor Odanacatib was evaluated in phase III clinical trials for breast-derived bone metastases and showed some positive results in inhibiting bone reabsorption before being discontinued [99]. Of note, cathepsins inhibitors are also being evaluated in clinical trials for other pathologies, such as osteoporosis, osteoarthritis or psoriasis, however, none of them have been approved so far [100].
To overcome all these limitations, further research is needed to elucidate the cell-specific roles of cathepsins and their downstream effectors. Furthermore, cathepsin levels in both tumour and serum seem promising as diagnostic and prognostic markers for HCC; therefore, research efforts are needed to translate them into the clinical practice.
Mounting evidence points towards lysosomal cathepsins as important drivers of essential cellular processes for the development, progression and perpetuation of the pathological state associated to MASH and HCC. While in MASH they mainly contribute to MASH-associated lipotoxicity and inflammation, in HCC they can participate in tumour proliferation and growth, invasiveness, epithelial-to-mesenchymal transition, immunosuppression, metastasis, and chemoresistance. Uncontrolled protease activity is potentially very harmful; so it is not surprising that cathepsins functions are finely tuned and cell or time-restricted in several biological scenarios. Because of this, targeting cathepsins is not an easy task, but this should not be seen as a downside, but rather as an opportunity to bring into the clinical practice novel cell-specific and time-dependent treatments with less side-effects. To get closer to the development of potential therapeutic approaches targeting cathepsins for MASH or HCC, we need to increase our biological understanding of their partners and associated signalling pathways to precisely select the most appropriate candidate for drug development.
Since lysosomal function and cathepsin activity are intimately intertwined, cathepsin/lysosomal equilibrium and how its dysregulation affects important biological processes such as autophagy, apoptosis, and lipotoxicity, which are key in the progression of MASH and HCC, should be further studied. Over the next years, the combination of classical preclinical and functional studies using cell-specific knock-out mouse strains with new technologies such as spatial transcriptomics, single-cell proteomics, and AI data mining will surely increase our biological understanding about cathepsin-associated signalling pathways in MASLD/MASH and HCC, identifying novel therapeutic candidates and inhibitors around them.
Cathepsins play important biological functions and actively contribute to the development and progression of MASH and HCC. Despite that our understanding about cathepsin biology has increased over the last decades, many questions about their biology still remain, so further research is needed in the forthcoming years to unlock the full potential of cathepsins as therapeutic candidates for MASH and HCC.
Cts: cathepsin
ECM: extracellular matrix
FFA: free fatty acids
HCC: hepatocellular carcinoma
IGF-1: insulin-like growth factor 1
JNK: c-Jun N-terminal kinase
MASH: metabolic dysfunction-associated steatohepatitis
MASLD: metabolic-associated steatotic liver disease
PepA: Pepstatin A
TAMs: tumour-associated macrophages
TFEB: transcription factor EB
TME: tumour microenvironment
AdCC and MFF: Writing—original draft. AM: Conceptualization, Visualization, Writing—original draft, Writing—review & editing.
The authors have no conflict of interest to disclose.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was funded by MCIN/AEI/10.13039/501100011033/FEDER, UE through the project grants [PID2021-123652OB-I00] and [RTI2018-097475-A-100] (AM); by MCIN/AEI/10.13039/501100011033 and El FSE invest in your future through the contract [RYC-2016-19731] (AM); by Pfizer grant [#77131383] (AM); by CSIC contract [JAEINT_23_01245] (AdCC); and by Ministerio de Universidades, Spain contract [FPU20/01367] (MFF). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

