
Abstract
Steatotic liver disease (SLD) has been known for a long time, but our understanding of this disease has remained poor until the past decade. Despite extensive research, our ability to comprehend the etiopathogenesis and natural course of SLD is far from the desired level of comprehension. This is required to develop a universally effective novel therapeutic agent. This review aims to concisely elaborate the conceptual approach and advancement in the understanding of global disease burden and etiopathogenic process, identifying the gaps and the pathophysiologic mechanism behind developing novel therapeutic agents. We searched two major databases, PubMed and Google Scholar, to identify publications related to the abovementioned topics. All publications, including original papers, reviews, and commentaries, were reviewed. Findings: Metabolic dysfunction-associated steatotic liver disease (MASLD) is not limited to obese individuals, rather, it may develop in any individual independent of weight. Visceral adiposity is strongly associated with MASLD and subsequent risks of cirrhosis, hepatocellular carcinoma, and cardiovascular disease. MASLD is associated with diabetes mellitus independent of underlying pathogenic mechanisms, and there is a bidirectional connection between MASLD and diabetes mellitus, making the situation quite challenging. Not all patients with MASLD exhibit atherogenic dyslipidemia and thus do not have a higher risk of cardiovascular disease. The overlap of these metabolic risk factors is not straightforward. There is a differential contribution of these risk factors based on age, gender, race, ethnicity, alcohol consumption, and microbiota composition. Poor dietary habits and lifestyle directly affect the microbiota, modulators, and mediators, thereby affecting the final biochemical processes leading to steatosis, steatohepatitis, fibrosis, and oncogenesis. In conclusion, MASLD is a complex and pathogenically heterogeneous disease with significant interpatient variation in the natural course and outcome. Understanding the precise mechanism of variability is the key gap and a limiting factor in the development of a novel therapeutic agent.
Keywords
Metabolic dysfunctions-associated steatotic liver disease (MASLD), visceral adiposity, obesity, pathogenesisIntroduction
Globally, the overconsumption of calories with and without physical inactivity is the most common cause of weight gain and obesity. Congenital or hereditary abnormalities are far fewer common causes of obesity, although weight gain is the result of uncontrolled polyphagia. However, not all obese people are at risk of life-threatening illnesses [1]. People with normal nutrient metabolism may develop obesity (peripheral adiposity) as defined by body mass index (BMI). Still, they may not be at risk for life-threatening illnesses such as cardiovascular disease (CVD), metabolic-dysfunction-associated steatohepatitis (MASH), or cancers. Instead, peripheral obesity may protect them against cardiometabolic risks [1, 2]. This draws attention to the importance of metabolic health in the pathogenesis of disease regardless of BMI.
Fat is deposited in subcutaneous tissue in healthy individuals, and deposition of fat in other locations in the body is termed ectopic fat (visceral fat), categorized as fat deposition in parenchymal or stromal cells (cellular steatosis) or surrounding the organ such as intestine (mesenteric fat) or heart (pericardial fat). Thus far, it is unclear if hepatic steatosis is an expansion of mesenteric fat or if it manifests concurrently, independent of mesenteric adipose tissue volume. A strong association between mesenteric fat volume and hepatic steatosis has been reported [3]. A higher cardiometabolic risk has been described with increased mesenteric fat mass and hepatic steatosis independent of each other and BMI [4, 5].
The focus of this review is to help understand the pathophysiological basis of metabolic dysfunction-associated steatotic liver disease (MASLD), which will help us to comprehend the evolving landscape of pharmacologic therapies. We will address the following topics:
Epidemiology of MASLD
Pathogenetic heterogeneity of the disease
Pathogenetic mechanism
Steatosis
Inflammation
Fibrogenesis
The natural course of MASLD
Epidemiology of MASLD and risk factors
Several risk factors associated with MASLD have been identified. A BMI greater than 25 is considered one of the key risk factors for MASLD, which is the most prevalent chronic liver disease worldwide. A meta-analysis of the studies between 1990 and 2019 revealed an estimated global prevalence of 30.05% [6]. Similar rates (30.69%) were reported when it was defined based on ultrasound imaging. Among overweight and obese patients who underwent liver biopsy, the prevalence of MASLD was 69.99% and 75.25% respectively [7]. Approximately one out of five overweight or obese MASLD patients had clinically significant (F2–F4) fibrosis [7]. Among overweight and obese MASLD patients, the prevalence of advanced (F3 and F4) fibrosis was 6.65% and 6.68%, respectively. In a nutshell, the risk of MASLD in patients with BMI > 25 is high and is similar regardless of the severity of obesity. Interestingly, it has been shown that lean and normal-weight healthy individuals are not even free of risk of MASLD. A prevalence of 10% to 16% has been documented in recent studies [8]. In light of the nondiscriminatory effect of weight or BMI on MASLD, researchers sought to explore other features associated with MASLD. As described above, visceral adiposity is more strongly associated with cardiometabolic health risk than BMI [4, 5].
Insulin resistance (IR) and diabetes mellitus (DM) are other important risk factors for MASLD. A bidirectional relationship between IR and DM has been discovered [9]. All types of diabetes including type II, type I, maturity-onset diabetes of young (MODY), and ketone-prone diabetes increase the risk of MASLD [10–15]. The prevalence of MASLD is highest (55% to 76%) in patients with DM type II and it is lowest in patients with ketone-prone diabetics and MODY. A prevalence of up to 20% has been reported in DM type I. Overall, approximately 20% to 25% of MASLD patients have been diagnosed with DM independent of type.
Both obesity and DM manifest with dyslipidemia, which is also strongly tied to MASLD. In MASLD patients, the prevalence of dyslipidemia ranges from 20% to 80%, depending upon the presence of risk factors [16]. The prevalence of dyslipidemia among diabetics and non-diabetics is 63.4% and 16.7%, respectively [15, 17, 18]. Dyslipidemia in obesity or DM could be the cause or effect of IR. Nonetheless, primary hyperlipidemia, either familial combined hyperlipidemia or familial hypertriglyceridemia, is associated with MASLD independent of other risk factors.
Finally, some patients with MASLD do not possess any known risk factors. Such patients are thought to have genetic abnormalities such as patatin-like phospholipase domain-containing 3 (PNPLA3) p.I148M and transmembrane 6 superfamily member 2 (TM6SF2) p.E167K [19].
Heterogeneity of MASLD
MASLD is a complex and heterogeneous disease associated with a variety of risk factors and manifests with a wide spectrum of clinical and histopathological features. The natural disease course is significantly variable from one patient to another and thus has diverse adverse outcomes. The complexity and heterogeneity of diverse clinical courses and outcomes stem from variable degrees of combination of key pathophysiological mechanisms: increased uptake of fatty acids released by enhanced lipolysis of adipose tissue, de novo lipogenesis (DNL), impaired excretion of triglycerides from hepatocytes and impaired mitochondrial or peroxisomal beta-oxidation. Each risk factor can lead to MASLD by affecting one or more pathogenic mechanisms. Whether any of these mechanisms is associated with enhanced severity of the MASLD compared to others remains unknown. Each of these pathogenic mechanisms can result from a combination of risk factors. Several risk factors have been identified that are significantly associated with MASLD independent of other factors. Three categories: individual characteristics, metabolic health, and genetics/epigenetics effectively convey the risk factors responsible for the manifestation of MASLD. The uniqueness of MASLD cases can be attributed to the influence of individual characteristics. Individual characteristics include age, gender, ethnicity, dietary habits, activity level, alcohol consumption, and microbiota composition. Obesity (BMI vs. visceral), DM, dyslipidemia, and hypertension are categorized as metabolic health. The actual magnitude of the effect of genetic and epigenetic risk factors in the development of MASLD and transitioning from simple hepatic steatosis to active and aggressive MASH remains unknown. There is speculation surrounding the role of genetic/epigenetic factors in combination with individual and metabolic health risk factors, though pure genetic MASLD has been described and is a well-known entity. Both PNPLA3 p.I148M and TM6SF2 p.E167K single nucleotide polymorphisms (SNPs) are independently associated with impaired excretion of VLDL and eventually lead to MASLD. Both of these SNPs are quite prevalent in all ethnic groups. However, the precise magnitude of the disease associated with these SNPs remains unknown. Like any other genetic disease, such as hemochromatosis, their penetrance is expected to be low, meaning that having a genetic mutation does not necessarily signify the presence of an aggressive disease.
Among patients with MASLD identified by the US, the prevalence of obesity, dyslipidemia, and DM is almost 80%, 70%, and 30%, respectively. Though each of these metabolic factors is independently associated with MASLD and CVD, patients with all three metabolic factors are more likely to have MASH, but not every individual with overlap will develop MASH (Figure 1) [7, 9, 18–20]. It is speculated that genetic/epigenetic factors play a role in addition to metabolic health risk factors in developing aggressive diseases, leading to increased risks of MASH, cirrhosis, hepatocellular carcinoma (HCC), CVD, and death. Such interplay of these factors creates tremendous heterogeneity and variability of the risk of MASLD and CVD [21–24]. Concerning genetic susceptibility, researchers have extensively explored genetic mapping, and several hundreds of mutations have been identified associated with the risk of MASLD. Pathogenic contributions of each of these point mutations largely remain unknown. An effort has been made to develop a polygenic risk score (PRS), which could help stratify the risk of genetic susceptibility in conjunction with individual and metabolic health risk factors [25, 26].
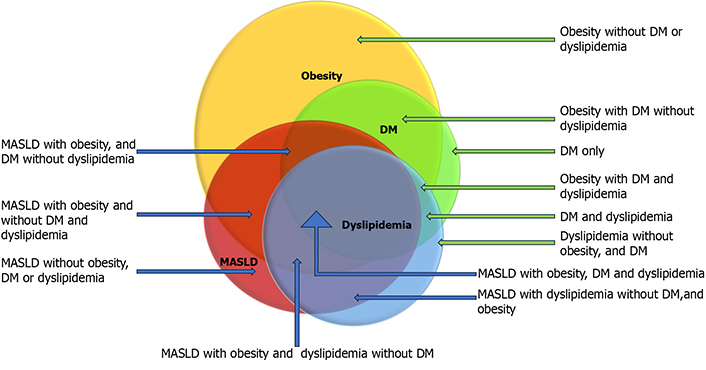
Overlap of metabolic comorbidities. This diagram highlights the heterogeneity of metabolic overlap by displaying the hypothetical overlap of metabolic risk factors. Data from epidemiological studies inferred the extent of overlaps. The blue arrow represents MASLD patients, and the green arrow represents non-MASLD patients with metabolic risk factors. DM: diabetes mellitus; MASLD: metabolic dysfunctions-associated steatotic liver disease. Reprinted from Habib [19]. © 2024 The Authors. CC BY-NC 4.0
Baratta et al. [22] from Italy have attempted to stratify the MASLD disease spectrum into three distinct categories: metabolic, genetic, and mixed. They reported variable degrees of risk of MASLD and CVD in these three subgroups. Patients with genetic MASLD have a lower risk of CVD compared to patients with acquired MASLD. However, genetic MASLD patients have the highest risk of progressive and aggressive MASH leading to cirrhosis and HCC (Figure 2). Other groups have also revealed that a combination of DM and IR adds to the risk of both liver disease and CVD [22]. Among 140 biopsy-proven MASH patients, the risk of MASH was evaluated based on the presence of genetic mutations compared to the metabolic risk factors. PNPLA3 rs738409 and TM6SF2 rs58542926 polymorphisms were independently related to advanced liver fibrosis. The presence of DM type II in conjunction with either SNP increased the risk of advanced fibrosis. Similarly, in non-diabetic patients, the IR homeostasis model assessment of insulin resistance (HOMA-IR ≥ 5.2) along with either SNP increased the risk of advanced fibrosis [23]. Moreover, in patients with a homozygous mutation (M variant and G allele), the risk of MASH, cirrhosis, and HCC is very high as opposed to in patients with the heterozygous mutation (M variant of G allele and I variant of C allele) [27].
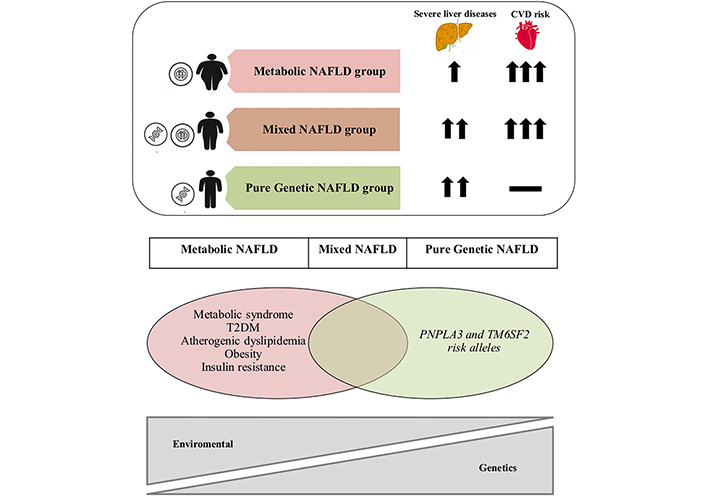
An overlap of metabolic and genetic factors in MASLD, known as NAFLD in the reprinted original article, and associated risk of advanced liver disease and CVD. CVD: cardiovascular disease; MASLD: metabolic dysfunction-associated steatotic liver disease; NAFLD: non-alcoholic fatty liver disease; PNPLA3: patatin-like phospholipase domain containing 3; T2DM: type 2 diabetes mellitus; TM6SF2: transmembrane 6 superfamily member 2. Reprinted with permission from Baratta et al. [22]. © 2022 Elsevier B.V.
To complicate the picture, a wide variety of risk factors and multiple pathogenic processes translate into a wide spectrum of clinical manifestations, and the different combinations of clinical manifestations may have disease behavior and outcomes. As of now, it has been reported that the presence of inflammation, fibrosis, or MASH based upon NAFLD/MASLD activity score (NAS) ≥ 4 is independently associated with disease progression. Ballooning degeneration is also considered a significant factor tied to progressive disease. Not all patients with MASLD have all the features of the definition of MASH at any given point because of the variability in the lifestyle of an individual and the control of metabolic health risk factors. Alteration in histological lesion severity is a rather dynamic process that is influenced by variables such as the time of exposure and the evolution (improvement or worsening) of the metabolic risk factors. This limits the utility of routine histopathologic evaluation to establish the diagnosis of MASH as it does not capture the dynamic nature of the disease. Nonetheless, immunohistochemical staining of α-SMA (alpha-smooth muscle actin), CD36, and peroxisome proliferator-activated receptor α (PPAR-α) might help characterize the behavior of the disease.
Furthermore, in addition to the factors discussed above, MASLD often overlaps with other chronic liver diseases such as alcohol liver disease, hereditary liver disease, autoimmune disease, and viral hepatitis. Because of the heterogeneity of MASLD, the natural history of MASLD is less predictable, and subtle variations in the associated risk factors can impact its manifestation and progression.
In conclusion, the combined contribution of genetic/epigenetic, individual characteristics, and metabolic factors creates a significant interpatient variation when considering the major driver of disease. Consequences of differential contribution of etiopathogenic factors include variability in the biological behavior of the disease, which translates into pathologic variability and might explain inconsistency among hepatic steatosis, inflammation, ballooning, apoptosis, and fibrosis. Eventually, such interpatient variation makes it difficult to standardize the cohorts in research settings. This may explain the lower response rates in MASH clinical trials than reported in clinical trials focused on other causes of chronic liver disease. Most of the phase II or III MASH clinical trials have elicited 20% to 30% response rates. Nonetheless, lifestyle intervention remains the focus of treatment. A general approach to lifestyle intervention may not apply to all subtypes of MASLD. For example, weight loss intervention in patients with genetic MASLD and lean diabetic MASLD is not required. In contrast, the restriction of high glycemic food and saturated fat would benefit patients with non-genetic lean and obese MASLD patients; however, the role of this strategy in genetic MASLD has yet to be investigated. Nevertheless, reducing polyunsaturated fatty acid consumption may be beneficial to genetic MASH patients. Based on the limited data, there are three distinct subtypes: insulin-resistant, diabetic, and genetic. The fourth category is the mixed type, with a variable degree of overlap of three distinct types. The natural course of disease subtypes remains unknown for the most part. However, the mortality and morbidity of risks related to cardiovascular events and progression to cirrhosis, and related complications, including HCC in metabolic subtypes, have not been studied extensively [22, 23, 28].
To characterize and predict the risk of MASH, cirrhosis, HCC, and CVD among patients with fatty liver disease, it would be beneficial to quantify hepatic fat, intra-abdominal visceral fat, dyslipidemia, and IR severity; determine the presence or absence of diabetes; conduct genetic testing for [PNPLA3 (adiponutrin gene) and TM6SF2] gene mutations; and obtain PRS. The risk is elevated in patients diagnosed with low adiponectin and high leptin levels in the presence of IR and/or diabetes. Routine adiponectin and leptin assessment is debatable. We are also not able to routinely assess genetic tests yet to evaluate genetic mutations in clinical practice.
Pathogenetic mechanisms
The role of the liver in metabolic dysfunction: a cause-and-effect relationship
The liver is a chemical factory and plays several metabolic functions. One of the key physiologic functions of the liver is to maintain metabolic homeostasis of both glucose and triglycerides as a source of energy during feeding and fasting states. It is essential to hormone synthesis via cholesterol metabolism and contributes to atherosclerosis prevention through excess cholesterol utilization for bile creation. The key metabolic processes related to nutrient metabolism are the intake of free fatty acids (FFA) released by lipolysis of adipose fat and transformation into triglycerides and storage into lipid droplets, creation of triglycerides from excess glucose and other monosaccharides, glycogenolysis, glycogenosis, gluconeogenesis, the production of lipoproteins, the secretion of triglycerides as VLDL, cholesterol synthesis and its excretion through bile acids. Moreover, hepatocytes produce a large variety of proteins that are used elsewhere in the body to maintain the vital functions of other tissues and organs. Up to now, 564 different proteins (hepatokines) have been identified by a mass spectrometry approach. Fat accumulation and liver damage affect many of these proteins, thus impacting the functions of other organs, which explains the occurrence of multiorgan failure in the setting of hepatic insufficiency. Vice versa, the dysregulated hepatokines are shown to negatively affect nutrient metabolism in the liver as well as in other peripheral tissues [29–33]. The first step in the regulation of glucose occurs in the liver. In contrast, adipose tissue has a preferential ability for the uptake of triglycerides from blood in prandial states after they are transported via lymphatics from the intestine. Namely, the liver stores excess nutrients in lipid droplets in hepatocytes and releases them back into circulation on demand. On the other hand, muscle and other end tissues store the excess nutrients and utilize them as a source of energy, but it does not have the potential to release them back into circulation on demand.
Hepatic steatosis as the consequence of metabolic dysfunction: an adaptation phenomenon
Metabolic stress, as a result of unhealthy dietary consumption and due to dysregulated adipose tissue, and pancreatic islet cells, affects the liver through several mechanisms, which leads to secondary effects on the liver, causing increased hepatic cellular uptake of glucose and triglycerides resulting in excess fat accumulation. The manifestation of hepatic steatosis can be attributed to multiple intracellular processes. These include increased fatty acid uptake, DNL, decreased excretion of VLDL, and decreased beta-oxidation of fatty acids. In a postprandial state, when glucose-stimulated insulin secretion occurs, the ingested glucose is partly stored as glycogen and partly oxidized. It is also responsible for the production of pyruvate, lactate, and amino acids like alanine (gluconeogenic substrates). Moreover, it also re-routed to DNL and glycerol synthesis. Fructose is the main driver of DNL, with very little glucose being utilized. Therefore, increased DNL can be triggered by excess sugar consumption, which is, in turn, exacerbated by a diet rich in saturated fat. In MASLD, DNL is increased more than threefold. DNL is tightly regulated via hormone signaling and transcription factors, such as sterol response element binding protein 1c (SREBP1c) and carbohydrate response element binding protein (ChREBP), which control the expression of the key lipogenic genes acetyl CoA carboxylase (ACC); fatty acid synthase (FAS) and ATP-citrate lyase (ACL). Insulin also promotes lipogenesis and adiposity. All these processes are regulated by several hormones, bile acids, nuclear receptors, and genes. IR develops as a consequence of cellular glucolipotoxicity. Excess FFA availability can contribute to inflammation, increased oxidative stress, mitochondrial dysfunction, and uncoupled oxidative phosphorylation, causing a subsequent hepatic stellate cell (HSC) fibrogenic response that can result in MASH progression and ultimately cirrhosis [34–38].
Simple intracellular hepatic fat deposition is an adaptation response for the maintenance of blood glucose and triglycerides homeostasis. Nutrient quantity, nutrient quality, number of meals, the timing of food intake, and fasting duration in 24 hours play significant roles in nutrient metabolism and the health of an individual. The consumption of fructose and saturated fats has been implicated in MASLD manifestation. Nutrients directly affect the modulators and mediators of fat and carbohydrate metabolism. Moreover, nutrients have an impact on the microbiota. Several factors influence the causation of dysbiosis with dietary factors being the key players. These include excessive sugar consumption, saturated fat consumption, calorie-dense foods, and alcohol consumption. Gastric acid suppression by proton pump inhibitors, or due to atrophic gastritis, has been implicated in the pathogenesis of dysbiosis, which plays a role in the transition of simple hepatic steatosis into MASH. Hepatic nuclear receptors play a pivotal role in the maintenance of cellular homeostasis while reinforcing crosstalk with adipose tissue and pancreatic islet β cells. Many factors affect these modulators and contribute to the progression of hepatic steatosis into steatohepatitis. IR has a cause-and-effect relationship with cellular glucolipotoxicity. Other players are insulin, PPAR, farnesoid X receptor (FXR), bile acids, adiponectin, and dysbiosis in the pathogenesis of MASLD and MASH (Figure 3).
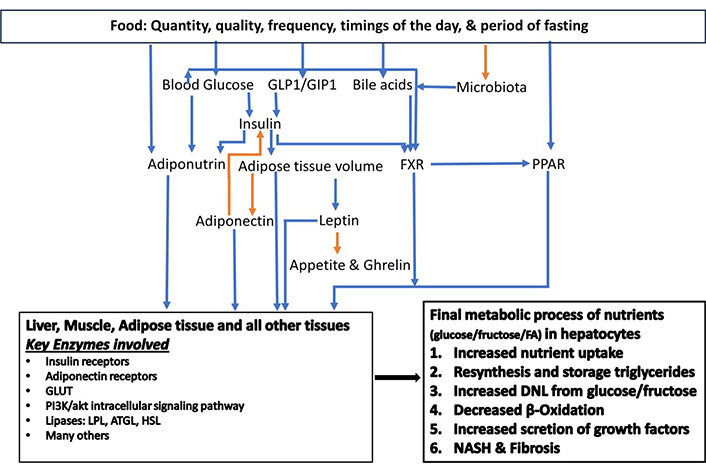
The key modulators of carbohydrate and fat metabolism. The orange arrow corresponds to a negative relationship, and the blue arrow corresponds to a positive relationship. ATGL: adipose triglyceride lipase; DNL: de novo lipogenesis; FA: fatty acid; FXR: farnesoid X receptor; GIP1: gastric inhibitory peptide 1; GLP1: glucagon-like peptide 1; GLUT: glucose transporter; HSL: hormone-sensitive lipase; LPL: lipoprotein lipase; NASH: non-alcoholic steatohepatitis; PPAR: peroxisome proliferator-activated receptor. Adapted from Habib [19]. © 2024 The Authors. CC BY-NC 4.0
In summary, these modulators (Figure 3) play an interconnected role in achieving the final effect of intrahepatic DNL through an enhanced uptake of triglycerides and glucose, an increased conversion of glucose and fructose into fat via tricarboxylic acid cycle in mitochondria, and a decrease in excretion of triglycerides (VLDL). Combustion via β-oxidation of excess nutrients is enhanced in all end organ tissues under the effect of PPAR-α. As the fat content of hepatocytes increases, it exerts negative feedback on insulin functions causing IR [30]. Hepatocyte excess fat deposition enhances adiponectin expression in adipocytes via PPAR-γ, which in turn enhances insulin sensitivity and lipogenesis in adipose tissue [30].
Evolution of hepatic steatosis into MASH: cellular metabolic exhaustion
With chronic ingestion of high-calorie food, hepatocytes continue to adapt to the exaggerated need to store energy in the form of lipid droplets under the influence of PPAR-α and FXR regulation. At some point, oxidative stress exceeds the enzymatic machinery, turning simple hepatic steatosis into steatohepatitis and progressive liver damage, which perpetuates the situation by further loss of functional hepatocytes. The intracellular consequences of ectopic lipid accumulation across most cell types are detrimental and include unfolded protein response (UPR)/ER stress activation, proinflammatory signaling, and oxidative stress. It also downregulates insulin signaling, resulting in IR. Overproduction and accumulation of lipid intermediates such as diacylglycerol and ceramide are potent inducers of cell cycle arrest, apoptosis, and mitochondrial dysfunction characterized by impaired oxidative phosphorylation. The metabolic stress of adipocytes can cause decreased adiponectin hormone secretion from adipose tissue. As a result, the decrease in adiponectin hormone secretion contributes to hepatocyte metabolic adaptation failure.
The fate of MASH: development of hepatic fibrosis or tissue repair and regeneration
It has been widely accepted that the liver is a regenerating organ. Studies have shown that hepatic fibrosis is a dynamic process and is influenced by a variety of factors. In select groups of patients, despite significant hepatic tissue injury, hepatic fibrosis may not develop. Among others, hepatic fibrosis ensues after exposure to a pathogen or poor lifestyle. Over time it can progress, regress, or remain stagnant [39, 40]. Despite mounting evidence, understanding of hepatic tissue repair, healing, and regeneration or progression of hepatic fibrosis remains elusive. In this section, the author summarizes the key concepts that have emerged thus far. A detailed review is beyond the scope of this paper and refer to dedicated review articles [41–52].
Several different cells and cellular pathways participate in the pathogenesis of hepatic fibrosis or tissue repair and regeneration [43]. Nonetheless, the HSC, previously known as Ito or fat-storing cell, plays a crucial part in tissue regeneration and in the pathogenesis of hepatic fibrosis, and malignancy. To augment the functions of HSC, hepatocytes, and cholangiocytes also differentiate into HSCs [41, 44]. The physiological functions of HSCs include vitamin A storage, fat storage, and maintenance of healthy liver architecture by regulating its extracellular matrix (ECM) replacement. Stellate cells play a key role in cell-to-cell interactions, cytokine networks, and sinusoidal blood flow modulation, and participate in hepatic immune tolerance through their immunomodulatory properties [53]. Upon exposure to an injury, HSCs undergo morphological and functional modification (activated-HSCs) that leads to tissue repair or fibrogenesis depending upon the nature, severity, and duration of the injury. The early “pre-inflammatory stage” of liver damage, initiates significant changes in HSC, which include changes in gene expression, epigenetic modification, activation of hedgehog signaling, and autophagy. These changes may occur because of HSC lipotoxicity or external stimuli [54, 55]. Primary changes in gene expression and the phenotype of HSCs make them more susceptible to the effects of cytokines, and other intra-, and extrahepatic stimuli. Some may result from a complex interaction between resident liver cells, infiltrating inflammatory cells, several locally acting signals, and the interplay between the ECM and cells [44].
Activation of quiescent-HSC (Q-HSC) into an activated form, which is called myofibroblast-HSC (MF-HSCs) after an initial hepatic injury is required for either outcome, resolution, or fibrosis. Several factors have been implicated in the activation of the quiescent HSC. Among them are but not limited to hepatocellular necrosis resulting from oxidant stress, apoptosis, necroptosis, and soluble growth factors. The secretion of cytokines and growth factors [(transforming growth factor-β (TGF-β), platelet-derived growth factor (PDGF)] by neutrophils, Kupffer cells (the resident macrophages of the liver) bone-marrow-derived monocytes, and Th17 cells can promote HSC activation [56]. Liver sinusoidal endothelial cells (LSECs), when injured, may exhibit a rapid loss in fenestrations and expression of pro-inflammatory molecules including intercellular adhesion molecule-1 (ICAM-1), vascular endothelial growth factor (VEGF) and adhesion molecules [57]. Above all, hedgehog ligands and hedgehog signaling play a pivotal role in the activation of Q-HSC, and intracellular metabolic, genetic, and epigenetic changes.
MF-HSCs perform two key functions; transformation into progenitor cells with the ability to replicate and differentiate into hepatocytes, cholangiocytes, or HSC along with angiogenesis as a process of regeneration and secondly initiate fibrogenesis and modulate its progression or regression [41, 42]. Moreover, MF-HSCs have been implicated in IR development which is predominantly mediated by their inflammatory properties. This undeniably links their activation to diabetes and MASLD development [52, 53]. After initial HSC activation, non-parenchymal cell-directed pro- or anti-fibrogenic responses control activated HSCs’ transit into spontaneous resolution via reversion and apoptosis or passed into a perpetuated state that results in the maintenance of a pro-inflammatory and pro-fibrogenic microenvironment as well as liver degrading ECM accumulation. After a causative liver injury has halted, fibrosis starts regression, and activated HSCs induce apoptosis or revert into a quiescent state. PPAR-γ expression in HSCs is associated with HSC reversal. Some activated HSCs become senescent, resulting in loss of profibrogenic property in which p53 plays a role. Moreover, angiogenesis contributes to both fibrosis development and regression [58].
MF-HSCs are characterized by the metabolic switch of a source of energy as a need of rapidly proliferating cells (Warburg effect) [43]. Because of the transition from quiescent to the activated form of HSC, the cell loses the oxidative phosphorylation and converts pyruvate into lactate. Secondly, activated HSCs also depend upon glutaminolysis with resultant α-ketoglutarate (α-KG), the end-product of glutaminolysis, which helps to replenish the TCA cycle to satisfy the high bioenergetic and biosynthetic demands of MF-HSCs. Both of these metabolic changes have been linked to enhanced hedgehog signaling. As a consequence of these metabolic changes, a lipid droplet is reconfigured into a new lipid droplet. Both old and new lipid droplets can be identified in MF-HSC. The new lipid droplets, considerably smaller than old lipid droplets, have less reticulum endoplasm but are enriched in triacylglycerol, and are peripherally located. Diacylglycerol O-acyltransferase 1 (DGAT1) and adipose triglyceride lipase (ATGL) engage in the synthesis and degradation of newly created triacylglycerol, respectively. Another characteristic feature of MF-HSC is an increased concentration of cellular free cholesterol. Increased MF-HSC accumulation of free cholesterol enhances PDGF-BB-induced HSC proliferation by extracellular signal-regulated kinases (ERKs), p38, c-Jun N-terminal kinases (JNK), and protein kinase B (AKT) phosphorylation. In addition, it is involved in the impairment of mitochondrial respiration. Moreover, MF-HSC free cholesterol accumulation upregulates toll-like receptor 4 protein (TLR4) expression, thereby sensitizing cells to TGF-β-induced activation through down-regulation of TGF-β-pseudoreceptor BAMBI. Along with HSC activation, subsequent upregulation of both sterol regulatory element-binding protein 2 (SREBP2) and miR-33a signaling results in further free cholesterol accumulation and exaggerates liver fibrosis in a positive feedforward loop. Lastly, MF-HSCs are characterized by the upregulation of markers of fibrogenesis; α-SMA, TGF-β, and collagen type I. Succinate secreted by hepatocytes binds with the G-protein coupled receptor 91 (GPR91) and is the mechanism behind this upregulation of fibrogenic markers. Repression of succinate-GPR91 signaling by LY2405319, an analog of fibroblast growth factor 21 (FGF21), as well as metformin inhibits HSC activation [43].
After the initial phase of activation of Q-HSC, MF-HSCs turn into the perpetuation phase leading to fibrogenesis. This phase is characterized by proliferation, migration, enhanced markers of fibrogenesis, contractability, chemokine secretion, and production and deposition of ECM. MF-HSCs proliferate and localize at the liver injury site, secreting ECM [59]. Myofibroblasts also release VEGF, directly promoting the proliferation of HSCs [60]. TGF-β secreted by Kupffer cells also plays a role in fibrogenesis inducing their activation into myofibroblasts and collagen type I and III synthesis [61–63]. Macrophages release PDGF, a powerful mitogen for HSCs. The PDGF signaling pathway is vital to HSC activation and liver fibrosis development [64].
In summary, the concepts of hepatic tissue injury, and its ability to repair and heal have advanced in the past couple of decades. Hepatic fibrosis is the consequence of severe and persistent exposure to the injury. Elimination of exposure to the pathogen(s) is associated with regression of fibrosis. Intervention with agents that interfere with hedgehog signaling, lactate accumulation, succinate, and growth factors are potential novel therapeutic targets.
The role of liver injury in the causation of metabolic dysfunction
Chronic liver injury independent of underlying etiology with advanced fibrosis has little functional reserve for the adequate maintenance of metabolic functions as described above. Lipoprotein metabolism is impaired in the setting of cirrhosis. Hepatic impairment severity assessed by total bilirubin, INR, and albumin directly correlates with lipoprotein metabolism. As a consequence, abnormal plasma concentrations of total cholesterol, high-density lipoprotein (HDL) cholesterol, low-density lipoprotein (LDL) cholesterol, and VLDL cholesterol are observed. These changes are secondary to characteristic abnormalities in plasma enzymes responsible for lipoprotein remodeling, including lecithin-cholesterol acyl transferase (LCAT), hepatic lipase, and phospholipid transfer protein (PLTP). HDL-C and enzymes participating in HDL maturation and metabolism are decreased in cirrhosis patients. There is a shift in the composition of HDL in patients with cirrhosis toward the larger HDL2 subclass, with a reduction in small HDL3 particles. The latter is associated with diminished cholesterol efflux capacity, which is an independent predictor of mortality [65].
Natural history of MASLD to cirrhosis and hepatic decompensation: factors affecting the progression
Hepatic steatosis is an adaptive physiological mechanism to an excessive FFA influx and is not necessarily harmful according to experimental data. However, cellular lipotoxicity is responsible for the manifestation and advancement of hepatocellular injury, inflammation, HSC activation, and ECM accumulation, characteristic of the phenotype of MASH [66].
Paired liver biopsy studies have revealed disease severity in the absence of intervention methods may remain stable, progress, or even regress over time. Such studies have shown that around 25% of MASLD patients displayed signs of MASH progression and bridging fibrosis. In a meta-analysis of biopsy-proven cohorts, the one-fibrosis-stage progression rate of hepatic steatosis patients was 14 years and seven years for MASH patients. However, individual differences have been described with accelerated disease progression being dependent on the presence and the diversity of the risk factors [67, 68]. Furthermore, Sanyal et al. [69] found that the development of cirrhosis or decompensation over two years occurs in one in five MASH patients with bridging fibrosis or cirrhosis. Several studies have utilized serial biopsies to identify the best methods for fibrosis progression prediction. Currently, baseline fibrosis is the main predictor of fibrosis progression. The usage of the NAS score at baseline for fibrosis progression assessment is debated. Nonetheless, a novel therapeutic agent must exhibit efficacy in both MASH resolution and fibrosis regression [69, 70].
Conclusions
MASLD is a very complex and heterogeneous disease with significant interindividual variation. It correlates strongly with cardiovascular disease risk, development of steatohepatitis, cirrhosis, and hepatic malignancy. Several factors play a role in causing MASLD, and this creates significant heterogeneity, making it challenging to evaluate, treat, and monitor the disease’s progress. Most importantly, this heterogeneity has thus far prevented the development of a breakthrough medication. There is an unmet need to subtype MASLD based on risk factors and pathogenetic processes. Evaluation of a patient with MASLD requires assessment of several factors including clinical, biochemical, hormonal, genetic mutations, and histological factors. Such a comprehensive evaluation includes measuring hepatic fat, intra-abdominal visceral fat, baseline hepatic fibrosis, severity of IR, characterizing dyslipidemia and DM, assessing glycemic control of diabetes, conducting genetic testing for PNPLA3 and TM6SF2 gene mutations, and obtaining PRS. Moreover, the liver tissue staining for CD36 and α-SMA may aid the assessment of the rate of adipose tissue-derived fatty acid hepatic uptake and fibrosis risk.
Abbreviations
| BMI: | body mass index |
| CVD: | cardiovascular disease |
| DM: | diabetes mellitus |
| DNL: | de novo lipogenesis |
| ECM: | extracellular matrix |
| FFA: | free fatty acids |
| FXR: | farnesoid X receptor |
| GPR91: | G-protein coupled receptor 91 |
| HCC: | hepatocellular carcinoma |
| HDL: | high-density lipoprotein |
| HSC: | hepatic stellate cell |
| IR: | insulin resistance |
| MASH: | metabolic dysfunction associated steatohepatitis |
| MASLD: | metabolic dysfunctions-associated steatotic liver disease |
| MF-HSCs: | myofibroblast-hepatic stellate cells |
| MODY: | maturity-onset diabetes of the young |
| PDGF: | platelet-derived growth factor |
| PNPLA3: | patatin-like phospholipase domain-containing 3 |
| PPAR: | peroxisome proliferator activator receptor |
| PRS: | polygenic risk score |
| Q-HSC: | quiescent-hepatic stellate cell |
| SLD: | steatotic liver disease |
| SNPs: | single nucleotide polymorphisms |
| TGF-β: | transforming growth factor-β |
| TM6SF2: | transmembrane 6 superfamily member 2 |
| VEGF: | vascular endothelial growth factor |
| α-SMA: | alpha-smooth muscle actin |
Declarations
Author contributions
SH: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. AJ: Writing—original draft, Writing—review & editing.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
It was supported by Liver Institute PLLC. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2024.