
Abstract
Liver inflammation, injury, and hepatic cell death are caused by external agents (viruses, bacteria, drugs, alcohol, etc.) along with the genetic susceptibility of an individual. Persistent activation of the fibrogenic response in cells leads to liver fibrosis which in turn progresses to cirrhosis and cancer. The dysregulation of the immune system generates reactive oxygen species which in turn induce necrosis of hepatocytes. This process activates hepatic stellate cells and myofibroblasts to produce a huge quantity of collagens, alpha-smooth muscle actin, and extracellular matrix deposition in liver parenchyma. Due to the multifactorial nature of this disease, conventional therapies increasingly attempted combinatorial therapy or polytherapy to target multiple mechanistic sites in order to prevent entry into further complicated irreversible stages. Despite advancements in conventional therapy, several cases aggravate fibrosis (grade 3 to 4) and cirrhosis. The inconsistency in treatment outcomes and limited organ donors for liver transplantation have led to an ever-increasing and challenging demand for alternative therapies. In this review, we analyze the mechanism and causative factors of liver diseases, conventional mode, and alternative therapeutic options. The central to liver diseases are immune dysregulation, hence bioactive agents with immunomodulatory properties should be searched and exploited to meet therapeutic needs. Mesenchymal stem cells (MSCs) with their specialized anti-inflammatory and immunomodulatory properties could be utilized as an effective alternative therapeutic candidate in treating inflammatory liver diseases. MSC-derived exosome further provides an additional immunomodulatory option that could work in tandem with MSC in a synergistic form. In this series, we have reviewed preconditioned and genetically edited MSCs to augment homing, proliferation, and differentiation. Importantly, all the clinical challenges should be noted and addressed before stem cell cytotherapy should be considered safe and effective for patients with liver diseases. Published literature indicated that MSC therapy has the potential to substitute conventional options in the treatment of high-grade fibrosis and cirrhosis.
Keywords
Mesenchymal stem cell, exosome, hepatic stellate cell, collagen, extracellular matrix, cell therapy, liver fibrosis, inflammationIntroduction
Multiple factors cause liver inflammation, injury, and cell death [1–3]. These factors comprise hepatocyte-specific viruses, alcohol, metabolic syndromes, defective bile acid products, and genetic abnormalities [2]. The early stage of liver fibrosis or scarring is an elementary wound-healing response to chronic liver disease (CLD) irrespective of etiologies [1–3]. In this process of tissue healing, quiescent hepatic stellate cells (qHSCs) which are involved in tissue repair under normal circumstances are activated to be transformed into myofibroblasts [1, 3]. The body’s pro-fibrotic and anti-fibrotic mechanisms are in balance in short-term early stage liver injury/scare and damage can be reversed to prevent entry into advanced forms of liver diseases such as cirrhosis [1, 3]. However, long-term chronic liver injury leads to necrosis and apoptosis of hepatocytes. These injured or scared hepatocytes release damage-associated molecular patterns (DAMPs) to activate inflammatory cascades and immune cell infiltration (Figure 1). This process activates the fibrotic phenotype of HSCs to transdifferentiate into myofibroblasts [1–4]. Notably, HSCs are the primary effector cells that lead to the deposition of collagen in the extracellular matrix (ECM) [5, 6]. In addition to HSCs, other cell types such as portal fibroblasts, bone marrow derived myofibroblasts, and epithelial to mesenchymal transition (EMT) also transdifferentiate to form myofibroblasts [4, 5]. Transdifferentiated myofibroblasts in the injured liver produce a large amount of ECM associated with collagen (type I and III) and fibronectin, accumulation of these proteins results in the formation of liver fibrosis [6, 7] (Figure 1). The clear understanding and advancement in the pathophysiology of liver fibrosis indicated it is a multifactorial disease, therefore multiple targets should be “HIT” simultaneously through conventional therapies. Conventional therapy inconsistently reverses only the early stage of liver fibrosis, while late stage (grades 3 and 4) and cirrhosis are difficult to control and treat through conventional therapies [3, 4]. Therefore, alternative treatment options must be explored in order to reduce the disease burden on the society. Based on published literature, this review discusses the immense potential of stem cell therapy for the treatment of liver fibrosis and cirrhosis and how to replace inconsistent conventional treatment options with a mesenchymal stem cell (MSC) regimen.
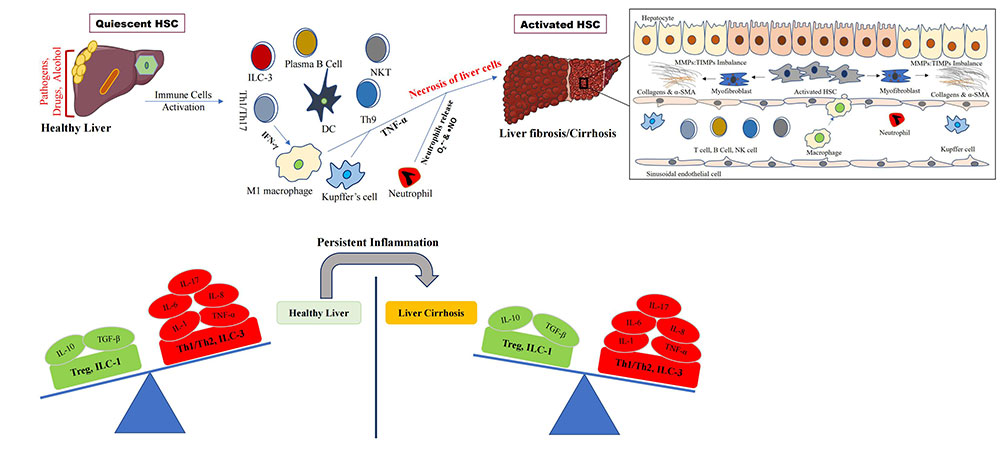
Diverse factors/etiologies (pathogens, drugs, alcohol, etc.) initiate the activation of inflammatory immune cells and liver diseases. Persistent inflammation leads to dysregulation of the immune system that exacerbates liver fibrosis and cirrhosis. Inset picture shows the activation of hepatic stellate cells (HSCs) and myofibroblast to secrete collagen fibers and accumulation in the liver that give rise to fibrosis and cirrhosis. ILC-3: innate lymphoid cell type 3; IFN-γ: interferon-γ; Th1: T helper 1; DC: dendritic cell; NKT: natural killer T cell; IL-6: interleukin-6; TGF-β: transforming growth factor-β; TNF-α: tumor necrosis factor-α; Treg: T regulatory cell; MMPs: matrix metalloproteinases; TIMPs: tissue inhibitors of MMPs; α-SMA: α-smooth muscle actin
Note. Parts of the figure were used from or adapted from pictures provided by Servier Medical Art, licensed under CC BY 4.0
Causative factors of liver fibrosis
Dysregulation of the immune system in liver fibrosis-cirrhosis
HSCs activation and myofibroblast conversion to the development of liver fibrosis are initiated by the inflammatory immune cells such as hepatic macrophages, T and B lymphocytes, natural killer (NK) cells, neutrophils, and platelets [7–9]. The key immune cells associated effectors involved in this process are cytokines, chemokines, and DAMPs [7–9]. There is bidirectional communication between HSCs and immune cells [10, 11]. HSCs regulate immune cell chemotaxis response via secretion of soluble mediators, while immune cells produce cytokines to activate HSCs [10–12]. Leptin, angiotensin II, interleukin-1β (IL-1β), transforming growth factor-β (TGF-β), platelet derived growth factor (PDGF), and tumor necrosis factor-α (TNF-α) are important immune cells associated effectors (soluble factors) involved in fibrogenesis [12, 13]. Several studies suggested the activation of the TGF-β pathway as a central event in the induction of hepatic fibrosis [14, 15] (Figure 1). TGF-β inhibits HSC apoptosis and promotes ECM remodeling into a profibrogenic phenotype [16, 17]. In addition to TGF-β, studies indicated the involvement of other pathways in the activation of HSC, one such pathway is Hippo, its components such as yes-associated protein-1 (YAP1) and protein kinases such as macrophage stimulating 1, 2 (MST1, MST2) have been reported to be involved in the initial HSC activation [12, 18, 19]. There are several reports of upregulation of Wnt/β-catenin [20–22], nuclear factor-kappa B (NF-κB) [23], mitogen activated protein kinases (MAPK) [24], phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) [25], hedgehog (Hh)/Gli [26] signaling pathways and their involvement in activation of HSCs and progression of hepatic fibrosis. Pro-inflammatory cytokine TGF-α inhibits apoptosis and increases proliferation of HSC, furthermore, it also triggers chemokines and intercellular adhesion molecule-1 (ICAM-1) in HSCs [27, 28]. Activated stellate cells secrete several chemokines such as C-C motif ligand 2 (CCL2), CCL5, CXC chemokine ligand 8 (CXCL8), and CXCL9, which are instrumental in recruiting immune cells such as neutrophils, monocytes, etc. at the site of activation [28–31]. Chronic hepatic injury leads to systemic inflammation that induces intestinal epithelial barrier disruption and subsequent permeation of luminal antigens and solutes [32–35]. Several studies suggested that intestinal inflammation due to epithelial barrier destruction contributes to liver fibrosis via toll like receptor 2 (TLR-2) signaling on monocytes [32–35]. The study also indicated the role of enteric TNFR-1 as a mediator in cholestatic liver fibrosis [35]. Pathogen associated molecular patterns (PAMPs) related to microorganisms translocated from the intestine, and DAMPs released from injured organs preferentially activate TLR-4 associated with proinflammatory macrophages 1 (M1), leading to the release of IL-1β, TNF-α, IL-6, IL-12 and interferon-γ (IFN-γ) [34]. The imbalance of liver immune cells such as T regulatory cell (Treg) and T helper (Th) cells such as Th1 and Th17 play a critical role in the occurrence of liver fibrosis and cirrhosis. The combined effect of Th1 and Th17 and their cytokines have been reported in promoting HSC activation via TGF-β signaling, thus indirectly and directly promoting liver fibrogenesis [35–37].
Oxidative stress and liver fibrosis-cirrhosis
Leukocyte infiltration during the inflammatory phase leads to increased production of reactive oxygen species (ROS). An increase in ROS is a key process that drives liver damage and initiates fibrosis. This process intensifies the disruption of cellular lipids, proteins, and DNA that triggers hepatocyte necrosis and apoptosis (Figure 1) [38]. The major source of ROS in the liver is NADPH oxidases (NOXs) located in the membrane of phagocytic cells such as neutrophils and macrophages [38]. ROS facilitates fibrogenic response linked to angiotensin II, PDGF, and TGF-β [39–42]. ROS mediated oxidative stress also influences the expression transcription factor NF-κB. NF-κB is an important modulator of liver fibrosis, due to its effect on wound healing, inflammation, and cell death [23, 43]. In addition to ROS, other reactive mediators such as 4-hydroxynonenal (HNE) released by activated inflammatory cells or hepatocytes also upregulate the expression of critical genes such as procollagen type I, monocyte chemoattractant protein-1 (MCP-1), and tissue inhibitors of matrix metalloproteinases-1 (TIMP-1) involved in liver fibrogenesis [44, 45].
ECM turnover
As we have discussed above a large number of inflammatory factors stimulate the activation and proliferation of HSCs. Long-term accumulation of collagen into the extracellular space increases the ECM and results in liver fibrosis, confirming the chronicity of the disease. The balance between ECM formation and hydrolysis is regulated by the physiological amount of matrix metalloproteinases (MMPs) and TIMPs produced in the liver [46–49]. These enzymes play a pivotal role in both fibrogenesis and fibrolysis [46–49]. ECM turnover is a prominent feature of liver fibrosis, indicating an imbalance between ECM synthesis and degradation [48–49]. The protracted exposure and accumulation of the fibrotic factors lead to liver cirrhosis [48, 49]. There have been contradictory findings of leptin on HSCs activation and fibrogenesis. Leclercq et al. [50] showed that leptin promotes HSC induced fibrogenesis and enhances TIMP-1 expression. On the contrary, leptin also partially suppresses peroxisome proliferator-activated receptor-γ (PPAR-γ), an antifibrogenic nuclear receptor that reverses HSC activation and maintains HSC quiescence [51, 52].
Genetic factors and circulating microRNA
Along with inflammatory factors, genetic factors also play an important role in the initiation of fibrosis. Multiple susceptible genes such as FAH, ASL, ABCB4, ALDOB, GBE1, SLC25A13, and SERPINA1, express abnormally high in individuals predisposed to liver fibrosis [53]. Mutations in these genes trigger liver fibrosis and cirrhosis [53]. Patatin-like phospholipase domain-containing protein 3 (PNPLA3) gene express abundantly in hepatocytes, adipocytes, and HSCs, however, mutations in this gene could be a major predisposing factor in non-alcoholic fatty liver disease (NAFLD) [54]. Additionally, the PNPLA3 I148M allelic variant is positively linked with disease diseases such as NAFLD, non-alcoholic steatohepatitis (NASH), and hepatocellular carcinoma [55]. The role of circulating microRNA in liver fibrosis has been studied, findings suggested that the circulating miR-29 largely induces cell (hepatocyte) apoptosis and regulates ECM accumulation by modulating the PI3K/AKT signaling pathway [56]. Immune activation of TLR-4 inhibits miR-29 to enhance collagen production and fibrosis [56, 57]. Other microRNA such as miR-34, miR-199, and miRNA-200 also promote the progression of hepatic fibrosis by inducing activation of HSCs and ECM deposition [58, 59].
Diagnosis of liver fibrosis
Till date, biopsy is a gold standard method for the diagnosis of liver fibrosis [60]. The biopsy tissues are stained using methods such as hematoxylin-eosin or Sirius red, to positively detect collagen deposition in ECM space or cells to confirm liver fibrosis. However, due to the pain and risk of potential complications of liver biopsy, non-invasive techniques/fibroscan (e.g., elastography scanning), and biomarkers (e.g., aminotransferase to platelet ratio (APRI) are recommended for the diagnosis of liver fibrosis [61, 62].
Conventional treatment options
Early stages (grade 1 and 2) of liver fibrosis are reversible, but when it crosses a threshold (grade 3 and 4), the majority of these cases attend an irreversible state and hence difficult to treat through conventional therapy [63, 64]. Notably, the conventional method of treatment results in inconsistent clinical outcomes due to the multifactorial nature of liver fibrosis. Therefore, multiple targets should be “HIT” simultaneously through conventional therapies [65–67] (Figure 2). Direct “HIT” is an effective approach to reverse liver fibrosis in cases when the underlying disease process is known. A few examples are the eradication of HBV or HCV with potent antivirals, anti-cell death treatment, regulators of lipid metabolism, etc. This process reverses fibrosis, repair liver injury, and improves its function and metabolic equilibrium [66–68]. The situation in which the underlying disease process is not known, indirect “HIT” would be highly effective antifibrotic treatment option [65] (Figure 2).
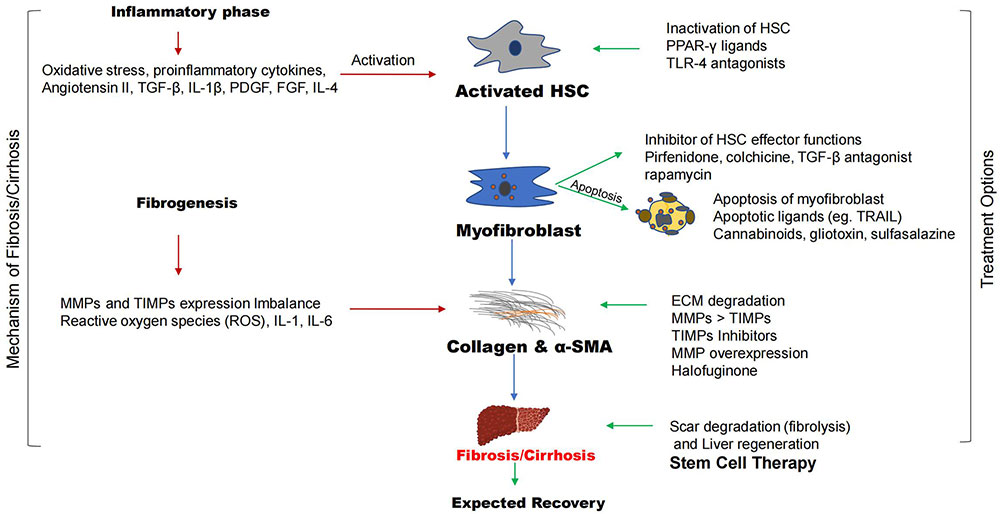
The steps involved in the process of liver fibrosis are called fibrogenesis. The inflammatory phase triggers this process leading to the accumulation of collagen fibers and α-smooth muscle actin (α-SMA) into fibrotic phases. The conventional treatment options involved the degradation of scars or fibers, this process is known as fibrolysis. The conventional drugs can target multiple sites such as inhibition of activated HSCs, apoptosis of myofibroblasts, and inhibition of HSC effector functions, breakdown of collagen, α-SMA to degrade liver scar, and regenerate hepatocytes. TGF-β: transforming growth factor-β; IL-4: interleukin-4; PDGF: platelet derived growth factor; FGF: fibroblast growth factor; HSC: hepatic stellate cell; PPAR-γ: peroxisome proliferator-activated receptor-γ; TLR-4: toll like receptor; TRAIL: tumor necrosis factor-related apoptosis-inducing ligand; MMPs: matrix metalloproteinases; TIMPs: tissue inhibitors of MMPs
Note. Parts of the figure were used from pictures provided by Servier Medical Art, licensed under CC BY 4.0
Antifibrotic combinatorial therapies
Antifibrotic therapies would target multiple target sites in the fibrogenic cascade when the underlying disease process is not known: a) activated immune cells and inflammatory cytokines & chemokines; b) inhibition of collagen synthesis and matrix deposition/degradation; c) modulation of stellate cell activation; d) death or apoptosis of activated HSCs and myofibroblasts; e) modulation of cell signaling pathways. Increasingly, the combinatorial therapy or polytherapy that targets or “HIT” different mechanistic levels are most effective method to treat liver fibrosis or cirrhosis (Figure 2).
Inhibition of activated immune cells
Inhibition of activated immune cells or their secreted inflammatory chemokines and cytokines are important treatment options in liver fibrosis. Antioxidants (astaxanthin) suppress the infiltration of monocyte derived macrophages, activated HSC, liver oxidative stress response, and hepatocyte death by minimizing the expression of proinflammatory cytokines such as TNF-α, TGF-β1, and IL-1β [69, 70]. In addition, targeting chemokines and chemokine receptors should be investigated and included as a potential therapeutic prospect [64]. Tregs have a central role in regulating the immune response, and its dysregulation is involved in the pathogenesis of NAFLD and NASH linked fibrosis [71, 72]. Therefore, immunoregulating Tregs could be one of the important treatment options. The polarization of M2 is a key immunotherapeutic process that induces immunosuppressive Th2 cytokines (IL-4, IL-13, and IL-10) secretion [73, 74]. In addition, chemokine receptors are commonly expressed by immune cells in fibrotic conditions. C-C chemokine receptor 2 and 5 (CCR2 and CCR5) are expressed significantly by monocytes and subtypes of liver macrophages in fibrotic conditions, therefore these receptors can be targeted to ameliorate liver fibrosis [75, 76].
Inhibition of collagen synthesis and matrix deposition/degradation
The imbalance in MMPs and TIMPs results in increased synthesis and decreased degradation of ECM in the liver. This unregulated accumulation of ECM leads to hepatic fibrosis. Studies demonstrated the role of four TIMP proteins in ECM accumulation, among them, TIMP-1 and TIMP-2 are primarily expressed in hepatic tissues [77]. Since hepatic tissue expresses higher levels of TIMP-1 and TIMP-2 in fibrosis, it could be used as a potential biomarker in identifying liver diseases [77, 78]. Therefore, fibroblasts and myofibroblasts are the main cells that express TIMPs during the process of liver fibrosis. Mechanistic studies indicated that TIMP-1 promotes liver fibrosis by inhibiting the activities of MMP-1, MMP-8, and MMP-13 in the liver and reducing the degradation of accumulated collagens [77, 78]. Restoring the balance between MMPs and TIMPs i.e. changing the ratio of MMPs/TIMPs expression can alleviate liver fibrosis. Ideally during the regression of fibrosis; TIMP levels decrease dramatically and MMP activity increases to restore MMPs/TIMPs balance, which reverses the process of liver fibrosis. Therefore, MMPs/TIMPs could be potential target sites to repress the progression of liver fibrosis (Figure 2).
Inhibition of activated HSCs via polytherapy
Apoptosis driven hepatocyte cell death is a primary stimulus for HSC activation in the evolution of liver fibrogenesis [79, 80]. Hence, inhibition of hepatocyte apoptosis would be a target to repress liver fibrosis [81, 82]. Studies indicated that the inhibition of activated HSCs could only be achieved by combinatorial therapy that would work at different mechanistic levels [83]. Several investigations point to the overproduction of angiotensin II in the injured liver, and its role in stimulation of stellate cell activation and fibrogenesis [84] (Figure 2). Therefore, the angiotensin II system represents an attractive anti-fibrotic target. Angiotensin receptor blockers (losartan) have been tested to analyze their effects in advanced liver disease settings [85, 86]. Experimental data revealed the specific role of IFN-γ in the inhibition of HSC fibroblasts ECM synthesis [87]. Preclinical studies also confirmed the role of IFN-γ in the inhibition of multiple aspects of stellate cell activation [88]. The reduced expression of PPAR-γ is associated with HSC activation during liver injury. The activation of this receptor in HSCs by exogenous PPAR-γ ligands is reported to improve liver fibrosis substantially [89, 90] (Figure 2). In the case of liver fibrosis, the concentration of conjugated 12α-hydroxylated bile acids, such as taurodeoxy-cholate and glycodeoxycholate are significantly higher in patients with NASH [91]. One such bile acid receptor, farnesoid-X-receptor (FXR) is widely studied due to its role in lipid and glucose metabolism as well as in inflammation and fibrosis [91]. Lipotoxicity or fatty liver triggers caspase-mediated apoptosis of hepatic cells that induce liver inflammation and injury in NAFLD. Caspase inhibitors such as emricasan could decrease lipotoxicity and liver fibrosis [92].
Modulation of signaling pathways
Modulation of signaling pathways should be considered as an important step for the regression of hepatic fibrosis [93]. Smad proteins are essential intracellular effectors of the TGF-β signaling pathway that have diverse roles in liver fibrosis. Rapamycin, a TGF-β antagonist and inhibitor of HSC effector functions could facilitate regression of fibrosis. Pirfenidone is a small orally bioavailable molecule that inhibited collagen synthesis and exhibited antifibrotic action in animal studies [94, 95]. Colchicine, a compound derived from plants inhibits an important step involved in collagen secretion and deposition i.e. polymerization of microtubules [96]. Similarly, apoptotic ligand TRAIL-mediated HSCs apoptosis is also associated with repression of liver fibrosis [97, 98]. Gliotoxin, an apoptotic drug, inhibits inducible NF-κB signaling activity by preventing IκB degradation [99]. Sulfasalazine is an anti-inflammatory drug that inhibits NF-κB signaling genes and nuclear translocation to repress liver fibrosis. In addition, it also exhibits antioxidant properties and regulates the expression of TGF-β and COX-2 activity [100].
Modulation of gut microbiota-TLR ligands
Modulation of gut microbiota is another method in the treatment of liver cirrhosis. Decontamination of gram-negative bacteria through oral administration of antibiotics decreases TLR ligands involved in the progression of liver fibrosis [101, 102]. Likewise, oral administration of antibiotics reduces alcohol-induced steatohepatitis in rats [103]. However, long-term antibiotic usage may cause unfavorable alteration of the gut microbiome, so this approach should be applied with utmost care to treat liver fibrosis. An alternative approach to the reduction of TLR ligands has also been applied in the form of probiotics to implant beneficial microbiomes in the intestine [104, 105]. It has been reported that the treatment with probiotic Lactobacillus rhamnosus GG significantly reduces the production of hepatic bile acids to decrease liver inflammation and fibrosis in mice [106]. Therefore, selective usage of antibiotics and probiotics could restore effective gut microbiota that improves liver fibrosis.
Reduction of oxidative stress
Minimizing oxidative stress signals could reduce liver injury associated hepatocyte cell death. Apoptosis signal-regulating kinase (ASK1) belongs to the MAPK pathways that relate to hepatic apoptosis, inflammation, and fibrosis, therefore it qualifies as an important target site to control liver fibrosis [107, 108]. The selective ASK1 inhibitor selonsertib improves fibrosis in a murine NASH model [107]. Several studies hypothesized the role of antioxidants (vitamin E precursor, d-alpha-tocopherol, and vitamin D) as putative antifibrotics [109–111]. Thioacetamide-induced hepatic fibrosis rat model alleviates liver injury and the expression of ECM proteins such as TGF-β and α-smooth muscle actin (α-SMA) when treated with vitamin D3 [111]. It is important to indicate that these compounds are not primary antifibrotic rather their effects are secondary in nature [111].
Modulation of NK and mast cell
Following liver inflammation and injury, NK cells are involved in directly eliminating activated HSCs, diminishing myofibroblast differentiation and ECM deposition [112, 113]. So, NK cell mediated elimination of HSCs exhibits an antifibrotic effect. During the process of liver fibrogenesis, mast cells are activated to degranulate and release mediators, such as histamine, tryptase, chymase, TGF-β1, TNF-α, cytokines, etc. [114]. These mediators (especially tryptase) are known to increase fibrogenic factors, such as collagen and laminin to exacerbate liver fibrosis [114, 115]. Mast cell stabilizer could be a better option for the treatment of liver fibrosis.
Genetic intervention
Gene editing tools are used for the treatment of diseases linked to genetic disorders. Noncoding RNAs, such as miRNAs, long noncoding RNAs, small interference RNAs (siRNA), and circular RNAs (circRNA) are important genetic intervening agents. Noticeably, siRNA mediated silencing of CCR2 regulates liver immunity to inhibit the infiltration of profibrotic macrophages and neutrophils in murine fibrotic livers [116]. While circRNA-ASPH suppresses liver fibrosis by binding miR-139-5p by regulating neurogenic locus notch homolog protein 1 (Notch 1) expression [117].
Stem cell-based alternative treatment options
Liver fibrosis is reversible only in the early stages (grade 1 and 2). However, there are inconsistencies in the conventional treatment methods due to the multifactorial nature of the disease that requires multiple drugs concurrently. Once fibrosis attends a threshold (grade 3, 4) stage, the fibrogenic type I collagen forms crosslinks that are typically associated with cell damage and inflammation [64, 118]. Over a period of time, the accumulation of a large number of crosslinked collagen and elastin fibers crosslinked within the tissue beds, making them inaccessible to proteolytic digestion. The majority of these cases are usually in an irreversible state and difficult to treat through conventional therapy. In these patients liver transplantation is the only treatment option [118]. However, due to the shortage of donors and strict regulations, there is ever growing and unmet need for alternative therapeutic options. MSCs with their broad range immunomodulatory, anti-inflammatory, and anti-apoptotic properties have emerged as a promising therapeutic candidate in treating inflammatory liver diseases such as liver fibrosis and cirrhosis [119–121].
MSC characterization and optimization for therapeutic usage
MSCs are multipotential progenitor cells with an intrinsic ability to differentiate into mesodermal or ectodermal cell lineages [121]. Historically, MSCs have been isolated from bone marrow [122, 123], but recently it has been isolated from many other sources, most prominently from adipose tissue, umbilical cord/blood, dental pulp, etc. [124–127]. Interestingly, the first set of studies reported similar biological characteristics among different sources of MSCs [128, 129]. Whereas, the second set of studies reported source-dependent biological differences among MSCs in terms of surface antigen and protein expression, cytokine secretion, differentiation capacity, and immunomodulatory activity [130–132]. The variability and heterogeneity of the cultured MSCs are due to passage number, media usage, donor variation, isolation techniques, and tissue origin of the cells [133, 134]. To address these issues, the International Society for Cellular Therapy (ISCT) recommends a minimum of three conditions to define human MSCs [135]. These include (1) adherence to the plastic surface when maintained under standard culture conditions; (2) positive expression of cluster differentiation 105 (CD105), CD73, CD90 surface markers, and lack of expression of CD45, CD34, CD14, or CD11b, CD79α, or CD19 and human leukocyte antigen-DR (HLA-DR) surface molecules, and (3) Potential to differentiate into osteoblasts, adipocytes, and chondroblasts cell types [135]. The most fascinating and clinically useful feature of MSCs is hypoimmunogenicity. Studies demonstrated that the MSCs express major histocompatibility complex class I (MHC-I) molecules, but no expression of MHC-class II and costimulatory molecules, such as CD40, CD40L, CD80, and CD86 [136–138]. The low-level expression of MHC-I exhibits protective effects towards MSCs by inhibiting NK cell-mediated killing, while the absence of MHC-II expression enhances MSC’s ability to escape immune recognition by CD4 cells. In addition, critical costimulatory molecules needed for MHC’s activation is firmly regulated by inhibitory molecules [139, 140]. The safety and efficacy of MSCs should be investigated extensively for the potential therapeutic use in liver diseases as discussed below.
Immunomodulatory nature of MSCs and its application in liver diseases
MSCs secrete predominant anti-inflammatory cytokines which mediate its immunomodulatory effect [141–144]. Since most liver diseases including liver fibrosis and cirrhosis are inflammatory in nature, so immunoregulatory properties of the MSCs could be exploited to treat these disorders [145, 146]. Apart from the immunosuppressive nature of the MSCs, it also regulates additional multiple disease-causing factors such as signaling pathways, apoptosis, MMPs/TIMPs ratio, etc. The disease alleviating cascading effects of MSCs make it a perfect therapeutic candidate for multifactorial liver diseases. MSCs modulate innate and adaptive immune systems by suppressing B and T-cell activation and proliferation while promoting the secretion of Tregs [145, 146]. MSCs not only suppress dendritic cell maturation but also inhibit the proliferation and cytotoxicity of NK cells [145, 146]. MSCs downregulate T cells by releasing numerous soluble factors (secretomes) such as nitric oxide (NO), prostaglandin E2 (PGE2), indoleamine 2, 3-dioxygenase (IDO), neurotrophin 3 (NT-3) factor, TNF-α, IL-6, IL-10, and HLA-G [139, 140, 147, 148] (Figure 3). MSC-derived PGE2 inhibits TGF-β activated kinase 1 and NOD-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasome activation in hepatic macrophages, thereby reducing inflammatory cytokine production [149]. These factors control the proliferation of different immune cells that upregulate Tregs anti-inflammatory function [139]. MSCs inhibits the proliferation of T cells by directly interacting with T-lymphocytes to facilitate immunosuppressive activity [139, 140, 150]. Adhesion molecules [ICAM-1, VCAM-1, and C-X-C motif chemokine receptor 3 (CXCR3)] present on the surface of MSCs help in adherence to T cells [151]. In addition, MSCs also suppress T cell proliferation by facilitating inhibitory programmed death 1 (PD-1) molecules to bind to PDL-1 and 2 ligands [145]. The combined effect of cytokines (IFN-γ, IL-1α, and IL-4, etc.) secreted by MSCs generates powerful immunosuppressive micro-environment [148]. MSCs also inhibit activation of B cell proliferation, differentiation, and chemotaxis [152]. The exposure of lipopolysaccharides (LPS) to mature B cells results in the expression of B lymphocyte-induced maturation protein-1 (Blimp-1) that induces differentiation of B cells into plasma cells [152]. MSCs suppresses plasma cell generation in co-cultured B cells, mediated via the release of humoral factor(s) that reduces Blimp-1 mRNA expression [152]. Additionally, MSCs inhibit the activation of B cells by reducing immunoglobulin levels [153, 154]. MSCs also efficiently induce polarization of inflammatory M1 into anti-inflammatory M2. This alteration releases anti-inflammatory soluble factors [IL-4, IL-10, IL-13, and IL-1 receptor antagonist (IL-1Ra)] that improve liver injury [155]. Moreover, MSCs also modulate the IL-17 signaling pathway, this decreases profibrogenic IL-17 production in hepatic NKT cells to alleviate liver fibrotic load [156]. Importantly, downregulation of IL-17 increases immunosuppressive and hepatoprotective (IL-10 and IDO) soluble factors [157]. Uniquely, HLA-G5 secreted by MSC expands Tregs [158, 159]. HLA-G5 also suppresses T lymphocyte proliferation after binding to its specific receptor immunoglobulin-like transcript 2 (ILT-2) [160, 161]. In general, MSCs suppress T lymphocyte activation and cytotoxicity, decrease inflammation cures injury, and overall facilitate liver regeneration.
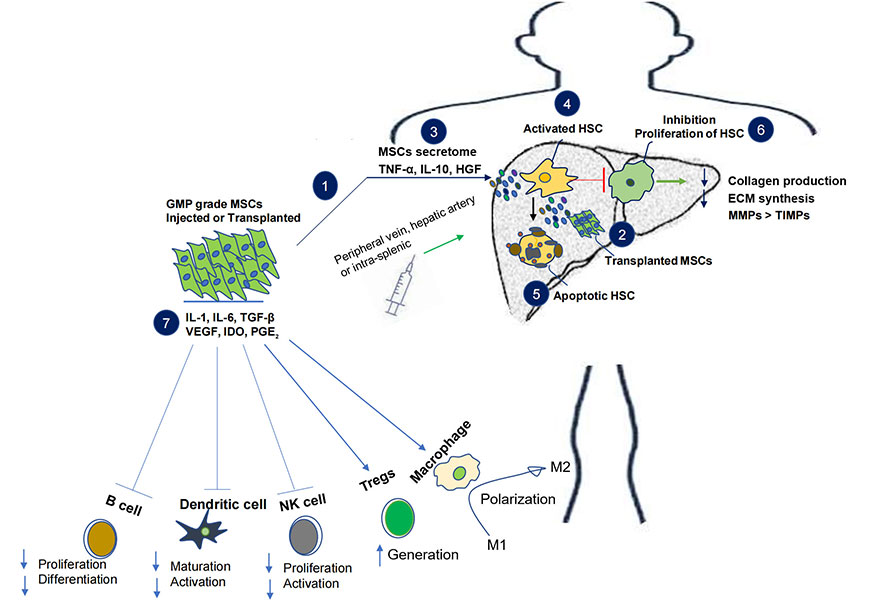
Alternative mesenchymal stem cell (MSC) therapy and its effect on liver fibrosis-cirrhosis. Injection or transplantation of GMP grade MSCs in subjects leads to the release of secretome with anti-inflammatory and immunosuppressive properties. The immunomodulatory role of MSCs includes decrease in B and NK cell proliferation and activation as well as inhibition of dendritic cell maturation. MSCs also enhance the generation of Tregs along with activation and polarization of macrophage 2, together they incline the immune balance towards anti-inflammation and immunosuppression. MSCs secretome in the liver inhibit activation and proliferation of HSCs, it also induces apoptosis of HSCs. Overall MSCs decrease collagen production and ECM synthesis by balancing physiological MMPs and TIMPs. VEGF: vascular endothelial growth factor; TGF-β: transforming growth factor-β; IL-1: interleukin-1; IDO: indoleamine 2, 3-dioxygenase; PGE2: prostaglandin E2; HGF: hepatocyte growth factor; HSC: hepatic stellate cell; NK: natural killer; ECM: extracellular matrix; MMPs: matrix metalloproteinases; TIMPs: tissue inhibitors of MMPs
MSCs transplantation and immune mechanism
MSC transplantation accelerates the regeneration procedure due to its differentiation potential, while its derivative secretome supports regenerative mechanisms. In the graft vs host disease model, apoptosis of infused or transplanted MSCs by host macrophages leads to the release of key soluble factor IDO that initiates immunosuppressive activity [161]. Another study showed, how monocytes initiate rapid clearance of infused MSCs via phagocytosis [162]. The phagocytosis of MSCs by monocytes triggers PD-1 and IL-10 expression, it also induces Foxp3+ regulatory T-cell formation to initiate anti-inflammatory pathways (Figure 3). MSC-derived extracellular vesicles (EVs) (i.e. exosomes) also provide a conducive microenvironment for MSCs transplanted cells to HOME and differentiate [163, 164].
Effect of MSCs on signaling pathways involved in fibrogenesis
There are several signaling pathways that play an important role in the development of liver fibrosis, especially HSCs activation, these include PDGF [165], TGF-β [166], oxidative stress [167], inflammasome-caspase-1 [168], and Wnt/β-Catenin [169] signaling pathways. Numerous studies demonstrated that MSCs produce and secrete diverse bioactive molecules such as cytokines, chemokines, and growth factors that stimulate neighboring cells [145–165]. These secretome and growth factors act on molecular signaling pathways of progenitor cells to stimulate proliferation and differentiation [170]. MSC derived IL-10 lead anti-inflammatory and immunosuppressive effect is regulated through signal transducer and activator of transcription 3 (STAT3)/SOCS3 signaling pathway [171]. PGE2 inhibits inflammasome (NLRP3) activation to reduce inflammatory cytokines production [148]. Transplantation of MSCs and conditioned media significantly inhibit HSCs activation and decrease the expression of fibrotic factors such as α-SMA, collagens, metalloproteinases, TGF-β, and Smad proteins [170]. MSCs induced inhibition of YAP suppress TGF-β-induced fibrotic cascades [172, 173]. Selective depletion of YAP in myofibroblastic or activated HSCs could promote senescence or apoptosis and hence reduce liver injury and repress fibrosis [174, 175]. Distinctively, MSCs-derived TNF-α-stimulated gene-6 (TSG-6) promotes liver regeneration by inhibiting HSC activation [176]. MSC-derived IL-4 promotes polarization and differentiation of M1 to M2 phenotype [177, 178]. In addition, PGE2 significantly increases M2 in the liver via STAT6 and mammalian target of rapamycin (mTOR) pathways to reduce inflammation and liver injury [177, 178]. Heme-oxygenase-1 (HO-1) -MSC derived factor improves liver disease conditions by reducing the infiltration and function of neutrophils and also by activating autophagy (a highly conserved eukaryotic cellular recycling process) through the PI3K/AKT signaling pathway [179–181]. JAK/STAT, Ras/Raf, Wnt/β-catenin along with other signaling pathways discussed above are involved in the expression of the liver fibrotic markers such as α-SMA, collagen I, TLR4, MMPs, and TIMPs [182–187]. MSCs transplantation and conditioned media significantly inhibit HSCs activation and maintain and facilitate the physiological state of fibrotic molecules [185–187]. MSCs also promote apoptosis by increasing the activity of caspase 3/7 and hence inhibit the proliferation of activated HSCs [188].
Antifibrotic role of MSC
There are several reports of MSCs alleviating hepatic fibrosis but still the mechanism is not fully understood [188–191]. However, researchers hypothesized that MSCs reduced the proliferation of activated HSCs and the deposition of collagen by indirect or direct cell to cell contact [191–194]. In the indirect mechanism, MSCs secretomes such as epidermal growth factor (EGF), TGF-β-isoform 3 (TGF-β3), TNF-α, IL-10, IL-13, IL-18, NT-3, and TSG-6 were linked to therapeutic benefits by inhibiting HSCs proliferation and collagen type I synthesis [186]. In addition, hepatocyte growth factor (HGF) and nerve growth factor (NGF) showed anti-fibrotic properties, induced apoptosis of activated HSCs, and promoted proliferation of hepatocytes [192–195]. MSC induced apoptotic cycle, increased expression of pro-apoptotic genes Bax and cleaved caspase-3 protein Bax, along with inhibition of NOX pathway [192]. In the direct mechanism, co-culture inhibited the proliferation of HSCs and expression of α-SMA [193]. It is a well-known fact that MMPs and TIMPs contribute to both the progression and regression of liver fibrosis. MSCs increase the expression of MMPs (MMP-2, -9, -13, and -14) and decrease the expression of TIMPs, especially 1, and 2 [194, 195]. Notably, Wang et al. [195] indicated that MMP-9 is an essential target for treating fibrotic diseases [195]. In addition, MSCs stimulate antioxidant response elements (AREs) in carbon tetrachloride (CCl4) and thioacetamide models of fibrosis [196, 197]. Distinctively, MSCs also showed antifibrotic properties by inhibiting EMT in the fibrosis model [198, 199]. Human UC-MSCs derived exosomes showed antifibrotic effect through inhibition of hepatocytes EMT in animal models [200]. MSCs derived exosomes inhibit EMT via inactivation of the TGF-β1/Smad signaling pathway [200]. Similarly, inhibition of thrombospondin-1 secretion (MSC-derived soluble factor) also decreases active TGF-β and subsequent attenuation of liver damage [201].
MSCs transplantation, homing, and transdifferentiation
There are several MSCs transplantation routes into the liver that correspond to intravenous, intrahepatic, intra-peritoneal, intrasplenic, and portal vein injection. The peripheral vein and hepatic artery are the most commonly used transplantation routes [119]. After transplantation, homing is the critical procedure for successful therapeutical outcome. Homing of MSC is classified into 2 different types: i) localized transplantation at the injury site, non-systemic type; and ii) release of homing-promoting molecules from the injured tissue, systemic type [202]. Hepatic differentiation of MSCs is influenced by specialized growth factors such as vascular endothelial growth factor (VEGF), HGF-1, leukemia inhibitory factor (LIF), and keratinocyte growth factor (KGF) [203, 204]. Differentiation of MSCs into hepatocyte-like cells (HLCs) are considered substitute sources for liver regeneration [203]. ECM surrounded with injured liver tissues have been used as the location for MSC engraftment and differentiation [119, 205]. Several studies in vivo have suggested that human MSCs can differentiate into HLCs when transplanted to the injured sites [205–212]. Studies indicated that MSC-based hepatocyte like derived cells could replace damaged tissues in liver diseases [205–212]. Studies indicated that only a small percentage (~1%) of hepatocytes differentiate from MSC after transplantation [213]. Inefficiency of homing and smaller differentiation percentage lead researchers to MSC-sourced secretomes called exosomes that exhibit high reparability and low immunogenicity [163, 164, 214].
MSC-derived exosomes and its therapeutic potential
Human MSC conditioned media (MSC-CM) fractioned to obtain a cell-free supernatant, called EVs or exosomes [215, 216]. Notably, exosomes are easier to obtain through conventional isolation methods like ultracentrifugation or via commercially available kits and can be stored at –20°C to –80°C [215, 216].
However, like MSC, there is no consensus on the exosome preparation methodology, therefore International Society for EVs (ISEV) set minimal criteria to characterize purified exosomes with specific surface marker proteins such as CD9, CD63, CD81, Alix, and TSG-101 expression [217, 218]. Exosomes encapsulate mRNA, miRNA, and assorted proteins that can modulate the biological function of target cells [219, 220] (Figure 4). Recent research has shown that MSCs produce a large amount of EVs. EVs exert their therapeutic effects through mechanisms involving the regulation of cell signaling, intercellular communication, and cellular metabolism [221–223]. A wide array of therapeutic effects such as suppression of renal tubular injury [216], acute myocardial infarction (AMI) [224], sepsis [225], and Alzheimer’s disease [226] has been attributed to MSC exosomes (MSC-exo). In light of this, MSC-derived exosomes are a promising alternative strategy for the treatment of liver diseases.
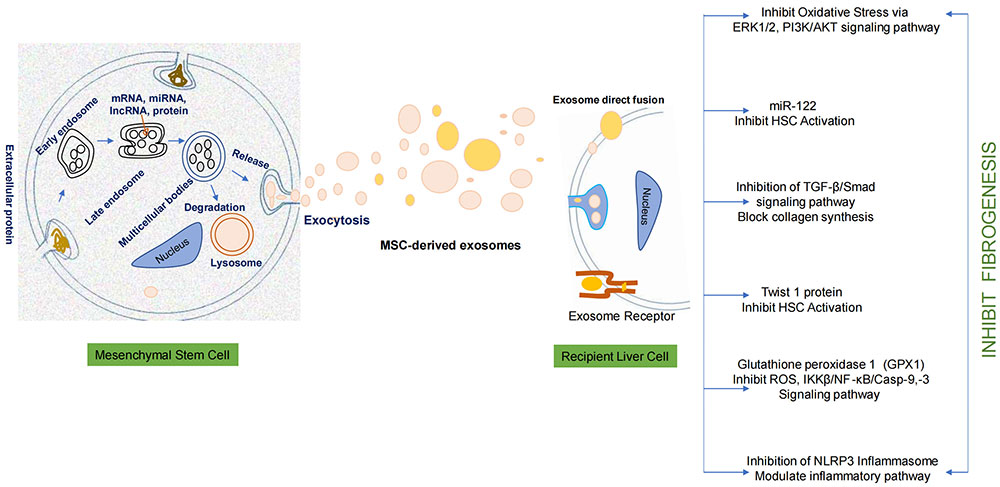
Mesenchymal stem cell releases a cellular product called exosome in cell culture media. These exosomes demonstrate antifibrotic potential once it interact with the recipient liver cell. Exosomes exhibit hepatoprotective effect-antifibrotic effect by inhibiting oxidative stress pathway, TGF-β/Smad pathway, IKKβ/NF-κB/Casp-9, -3 pathway, and NLRP3 inflammasome pathway. The encapsulated bioactive products in exosomes inhibit HSC activation. The overall effect leads to the inhibition of fibrogenesis. ERK1/2: extracellular signal-regulated kinases 1/2; HSC: hepatic stellate cell; TGF-β: transforming growth factor-β; ROS: reactive oxygen species; IKKβ: IκB kinase β; NF-κB: nuclear factor-kappa B; NLRP3: NOD-like receptor thermal protein domain associated protein 3
Role of MSC-derived exosomes in liver diseases
Studies showed that exosomes mediate intercellular communication between MSCs and injured organ sites and show therapeutic potential in liver diseases such as acute liver damage, hepatic fibrosis, and cirrhosis [227–231]. Additionally, exosomes have the ability to penetrate deep tissues avoid immune attacks, and facilitate the delivery of their therapeutic cargo into target cells [232, 233]. Hence, MSC-exo treatment could be a promising alternative to MSCs-based treatment in liver diseases. Therapeutic role of MSC-exo in liver diseases includes hepatoprotection through modulation of inflammatory signaling pathways such as extracellular signal-regulated kinases 1/2 (ERK1/2) and insulin-like growth factor-1 receptor (IGF-1R)/PI3K/AKT [234, 235]. Several studies indicated that exosomes rich in miR-122 inhibit HSCs activation [235]. Exosomes mediated inhibition of activated HSCs leads to downregulation of genes like IGF-1R, cyclin G, etc. involved in proliferation and collagen maturation [234, 235]. Exosomes encapsulated enzyme glutathione peroxidase (gpx1) inhibit ROS and IKKβ/NF-κB/casp-9/-3 signaling pathways in liver cells to alleviate liver fibrosis [234, 235]. In the CCl4 induced liver failure study, gpx1 exosomes elicit antioxidant and anti-apoptotic effects [234]. These exosomes regulate antifibrotic action through the inactivation of the TGF-β1/Smad signaling pathway to prevent expression of collagen I and III, and EMT. Exosomes pre-treated with TNF-α also inhibit activation of the NLRP3 inflammasome pathway that reduces ALT, AST, and pro-inflammatory cytokine levels and promotes tissue repair [236]. In addition, qHSCs release exosomes containing Twist1 protein, which in turn increase miR-214 concentration to inhibit neighboring HSC activation via a decrease in the expression of cellular communication network factor 2 (CCN2) [235]. In contrast, exosomes containing miR-19a derived from HCV infected hepatocytes promote activation of HSCs via SOCS3/STAT3/TGF-β pathway [227]. Therefore, it is noteworthy that exosomes from stem cells inhibit HSC activation and liver fibrosis progression, however, exosomes obtained from disease conditions may promote fibrogenesis [237–254] (Table 1). Still, there are limitations in the treatment with exosomes due to batch differences, purity, drug delivery, and off target effects. Therefore additional studies are required to characterize exosome cargo content, safety profile, effective therapeutic dosages, and delivery methods in order to target liver diseases effectively [255, 256].
Exosomes derived from various cells have different functions
| Source of exosomes | Action on liver fibrosis | Reference (s) |
|---|---|---|
| Human umbilical cord MSC | Inhibition | [238, 239] |
| Adipose tissue-derived MSC | Inhibition | [241] |
| Bone marrow derived MSC | Inhibition | [243, 246, 254] |
| Amnion-derived MSC | Inhibition | [244] |
| Embryonic stem cell-derived MSC | Inhibition | [245] |
| Human liver stem cell | Inhibition | [247] |
| Human induced pluripotent stem cell | Inhibition | [248] |
| HCV infected hepatocyte | Promotion | [242] |
| Cholangiocytes | Promotion | [249, 250] |
| Neighboring quiescent HSC | Inhibition | [240, 251] |
| NK Cell | Inhibition | [252] |
| M1 macrophage | Promotion | [253] |
Adapted with permission from [230]. © 2023 The Authors
Preconditioning of MSCs
MSCs culture in hypoxic condition
Preconditioning of MSCs is a very important adaptative procedure for the cells to undergo proper homing, proliferation, and differentiation [257]. Hypoxia is one of the most important methods of exposing stem cells to low oxygen concentration for proper growth, maintenance, pluripotentiality, differentiation, and function [257–260]. The primary mediator of hypoxic adaptation is hypoxia inducible factor (HIF) [260]. Hypoxia also stimulates specific gene expressions; such as Glut-1, EPO, and VEGF that correspond to glycolysis, erythropoiesis, and angiogenesis. VEGF produced by stem cells in hypoxic conditions directly affects the surrounding environment [257, 258]. Precondition MSCs cultured in hypoxic conditions (oxygen range: 4–10%) will adapt well during transplantation (Figure 5).
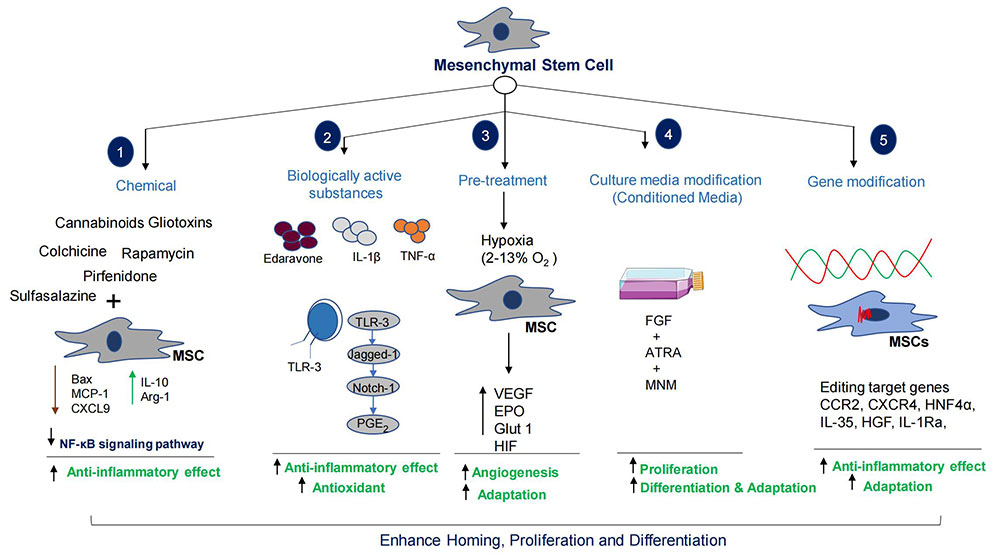
Preconditioning of MSCs enhances homing and differentiation potential. The combinatorial therapy increases MSCs therapeutic capacity many folds. HGF: hepatocyte growth factor; MSC: mesenchymal stem cells; MCP-1: monocyte chemoattractant protein-1; CXCL9: CXC chemokine ligand 9; IL-10: interleukin-10; NF-κB: nuclear factor-kappa B; TLR-3: toll like receptor 3; PGE2: prostaglandin E2; TNF-α: tumor necrosis factor-α; VEGF: Vascular epithelial growth factor; EPO: erythropoietin; Glut 1: glucose transporter 1; HIF: hypoxia inducible factor; FGF: fibroblast growth factor; ATRA: all trans retinoic acid; MNM: Modified neuronal media; CCR2: C-C chemokine receptor 2; CXCR4: C-X-C motif chemokine receptor 4; HNF4α: hepatocyte nuclear factor 4α
Note. Parts of the figure were used from pictures provided by Servier Medical Art, licensed under CC BY 4.0
Gene editing of MSCs
There is a promising effect of stem cells in the treatment of liver diseases, however homing and differentiation are key issues in clinical applications [261]. Importantly, HOMING and migration of MSCs are affected by a variety of factors such as cell number, microenvironment, and route of administration [261]. Studies indicated that only a small percent (~1%) of transplanted MSCs differentiate into HLCs [211–213]. Studies reported that targeted editing of MSCs genes could enhance differentiation into HLCs, secretion of chemokine factors, growth factors release, immunomodulatory effects, and anti-fibrotic, anti-apoptotic, and anti-oxidant activities [261, 262]. The verified genes that could enhance the therapeutic potentials of MSCs are c-Met, CCR2, CXCR4, hepatocyte nuclear factor 4 α (HNF4α), IL-35, HGF, IL-1Ra, forkhead box A2 (Foxa2), and VEGF165, etc. (Figure 5) [261–267]. Several studies have shown essential roles for the CXCL12-CXCR4 axis in the survival, homing, and improved colonization of stem cells [259–265]. Gene edited MSCs overexpressing chemokines (CCR2, and CXCR4) are more likely to reach the injured liver and facilitate recovery [263–265]. Importantly, HGF secreted by MSCs is a ligand of c-Met, a tyrosine kinase receptor. HGF is the ligand of c-Met, a tyrosine kinase receptor family member. The HGF/c-Met axis is crucial in the proliferation, regeneration, development, protection, scattering process, and differentiation of bone marrow MSCs (BMSCs) into hepatocytes [267, 268]. However, the insufficient capacity of stem cells to reside in the damaged liver has been a concern for their therapeutic properties. The overexpression of c-Met protein in BMSCs using genetically engineered lenti-c-Met-GFP vectors (c-Met-BMSCs) demonstrated increased migration activity of c-Met-BMSCs against the control BMSC group associated with HGF [267]. Overexpression of HNF4α enhances the therapeutic potential of MSCs by increasing the expression of IL-10 and polarization towards M2 [269]. The overexpression of IL-1Ra in MSCs, promotes liver regeneration and inhibits hepatocyte apoptosis [270]. Chae et al. [271] reported that overexpression of Foxa2 in MSCs enhances hepatocyte-like differentiation that alleviates acute liver failure (ALF) [271]. The above studies showed how genetic manipulation increases the regenerative capacities of MSCs in liver diseases. Injection of engineered MSCs can attenuate activation of HSCs, collagen deposition, inflammation, apoptosis, and fibrotic processes.
Pretreatment of MSCs
It has been reported that before transplantation, pretreatment of MSCs with different stimuli improves therapeutic efficacy in liver diseases (Figure 5). Edaravone, IL-1β TNF-α, etc. are the pretreatment agents or factors used in the experimental studies. Edaravone enrich antioxidant levels in MSCs that significantly improve liver tissue repair and regeneration by increasing MSCs’ homing, proliferation, apoptosis, and secretion of HGF [272]. Nie et al. [272] reported that IL-1β (20 ng/mL) pretreatment could enhance homing ability of MSCs by increasing the expression of CXCR4 [272]. In addition, Zhang et al. [237] found that TNF-α (1 ng/mL) pretreatment of MSCs could secrete therapeutic exosomes to suppress NLRP3 activation in macrophages [273] (Figure 5).
Combinatorial therapy of MSCs and stimulant
The study reported that combination therapy of MSCs and IL-1Ra (2 mg/kg) synergistically regulate inflammation and apoptosis in ALF conditions [274]. Likewise, transplantation of human umbilical cord blood MSCs with granulocyte colony stimulating factor (G-CSF) improves liver injury by regulating inflammation, oxidative stress, and hepatocyte apoptosis [275]. Combination therapy of adipose derived stem cells (ADSCs) with eugenol (a natural compound), inhibits NF-κB activation, promotes cell cycle arrest, and reduces inflammatory cytokines [276]. Effective homing of ADSCs also resulted in decreased expression of genes involved in inflammation including inducible NO (iNOS), MCP-1, CD163, TNF-α, macrophage inflammatory protein-1α (MIP-1α) and MIP-1β, TGF-β, and M2 polarization [276]. Additionally, combination therapy improves micro-environment around MSCs due to enhance secretion of paracrine factors, to influence overall clinical outcomes.
MSC-based clinical studies and its efficacy and safety
Late stage liver fibrosis and cirrhosis are progressive liver diseases, and there are no conventional treatment options [277]. Numerous studies indicated the potential of MSCs in clinical application [278, 279]. The preclinical animal studies have demonstrated the efficacy, safety, and feasibility of MSC in the treatment of liver cirrhosis [280–283]. In one such study, a 7-day consecutive tail vein injection of UC-MSCs significantly improved liver function in CCl4-treated mice [284]. Apart from preclinical studies, a large number of clinical trials have demonstrated the beneficial effect of MSCs administration in CLD patients [285–306]. Clinical trials have shown that infusion of MSCs can improve liver function profiles without obvious adverse effects [285–306]. In addition, transplanted MSCs further improve other complications such as hepatic encephalopathy, ascites, spontaneous bacterial peritonitis, and liver failure [307]. Furthermore, chronic hepatitis C or B-induced cirrhotic patients treated with MSCs showed improved MELD scores, ascites, and peripheral edema [288, 289, 296, 299, 301, 305]. In addition, after 2 and 4 weeks post-MSC transplantation, patients showed reductions in IL-6, IL-17, and TNF-α levels and a rise in IL-10 levels [285–289]. Moreover, in MSC transplanted groups, CD4 and Tregs concentration were higher, whereas CD8 T and B cells were markedly decreased [307]. Importantly, MSC therapy reduces the proportionate collagen area [299]. Wang et al. [296] reported a reduction in alkaline phosphatase and gamma-glutamyl transferase in ursodeoxycholic acid-resistant primary biliary cholangitis (PBC) settings [296]. The preclinical and clinical studies demonstrated that MSCs are safe and effective for treating liver diseases. Unfortunately, no standard cellular dosage regimen is currently available for clinical MSC treatment [306, 307]. However, larger and double-blinded controlled trials are required across different liver cirrhosis etiologies with a wider geographical distribution to reliably evaluate the effectiveness and dose regimen of MSCs.
Autologous or allogeneic MSC: which one is better?
Both autologous and allogeneic MSCs have been studied in preclinical and clinical studies [255]. Though, autologous MSCs are relatively easy to culture, there is a time critical aspect of expanding enough cells for transfusion. Moreover, it is also difficult to obtain sufficient adipose tissues for autologous MSCs culture and expansion from thinner patients [255]. Elderly or aged patients possess less regenerative properties i.e. minimum proliferation and differentiation of MSCs. Apart from logistical challenges, there is less quality assurance and a high cost of preparation for a single recipient. In contrast, allogeneic MSCs are usually obtained from young healthy donors, readily available, easy to culture, expand, and cryopreserved. Importantly, it can be quickly thawed prior to administration [255].
Hazard and safety profiles of MSC therapy
Though MSC regenerative therapy for liver diseases has been found to be safe and effective, still strict vigilance and caution are required to exclude any side effects i.e. the probability of carcinogenesis and viral transmission. Since MSCs secrete various growth factors, there is a possibility of tumor cell growth and angiogenesis with the increase in the number of cell culture passages [308]. Animal studies also suggested telomeric deletions with the increase in the number of passages [309]. Notably, there are no reported cases of malignant transformation of human MSCs observed in clinical trials [310, 311]. Extra care and precaution should be taken in the exclusion criteria during MSCs use for therapeutic purposes. The presence of viruses and endotoxins as well as chromosomal integrity must also be analyzed before MSCs transplantation to ensure the safety of the procedure [310, 311].
Conclusions
The inconsistencies in the conventional treatment outcomes and limited availability of organs for liver transplantation have led to a growing and unmet demand for alternative therapies. In this prospect, MSC-based therapy has been most studied and investigated. A primary mechanism of action has been proposed as paracrine effects via their immunomodulation. Studies have shown that MSC-derived cytokines such as IL-10, IL-4, HGF, IDO, PGE2, TSG-6, and HO-1 have anti-inflammatory and immunosuppressive effects. Immunomodulatory pathways include ECM degradation in liver parenchyma, repairing fibrotic tissues to improving liver function. However, HOMING and differentiation of transplanted cells remain a challenge. Priming MSCs has emerged as a novel strategy to enhance their therapeutic efficacy by preconditioning. Preconditioning such as hypoxia, combination therapy, and genetic engineering prepare these cells for optimal homing and differentiation in challenging in vivo environments.
Additionally, MSC-derived exosomes exhibit therapeutic benefits, that have the potential to circumvent any risk posed by MSCs and will be safer to use in clinical practices to treat liver diseases. Future prospective: furthermore, MSCs priming or preconditioning warrant further research to harness their full potential. Smart editing of MSCs using CRISPR-Cas9 to target primary causative factor. Smart editing of exosomes via incorporation of nanomedicine, miRNA, drugs, and anticancer agents as a deliverable that could target specific sites. Furthermore, standardized protocols for the production of MSCs and exosomes should be optimized to overcome the remaining challenges and pave the way for stem cell therapy to be applied in wider clinical practices at affordable cost.
Abbreviations
| ADSCs: | adipose derived stem cells |
| AKT: | protein kinase B |
| ALF: | acute liver failure |
| ASK1: | apoptosis signal-regulating kinase |
| Blimp-1: | B lymphocyte-induced maturation protein-1 |
| BMSCs: | bone marrow mesenchymal stem cells |
| CCL2: | C-C motif ligand 2 |
| CCl4: | carbon tetrachloride |
| CCR2: | C-C chemokine receptor 2 |
| CD105: | cluster differentiation 105 |
| circRNA: | circular RNAs |
| CLD: | chronic liver disease |
| CXCL8: | CXC chemokine ligand 8 |
| CXCR3: | C-X-C motif chemokine receptor 3 |
| DAMPs: | damage-associated molecular patterns |
| ECM: | extracellular matrix |
| EMT: | epithelial to mesenchymal transition |
| EVs: | extracellular vesicles |
| Foxa2: | forkhead box A2 |
| gpx1: | glutathione peroxidase |
| HGF: | hepatocyte growth factor |
| HLA-DR: | human leukocyte antigen-DR |
| HLCs: | hepatocyte-like cells |
| HNF4α: | hepatocyte nuclear factor 4 α |
| HO-1: | heme-oxygenase-1 |
| HSCs: | hepatic stellate cells |
| ICAM-1: | intercellular adhesion molecule-1 |
| IDO: | indoleamine 2, 3-dioxygenase |
| IFN-γ: | interferon-γ |
| IGF-1R: | insulin-like growth factor-1 receptor |
| IL-1β: | interleukin-1β |
| IL-1Ra: | interleukin-1 receptor antagonist |
| M1: | macrophages 1 |
| MAPK: | mitogen activated protein kinases |
| MCP-1: | monocyte chemoattractant protein-1 |
| MHC-I: | major histocompatibility complex class I |
| MIP-1α: | macrophage inflammatory protein-1α |
| MMPs: | matrix metalloproteinases |
| MSC: | mesenchymal stem cell |
| MSC-exo: | mesenchymal stem cell exosomes |
| MST1: | macrophage stimulating 1 |
| NAFLD: | non-alcoholic fatty liver disease |
| NASH: | non-alcoholic steatohepatitis |
| NF-κB: | nuclear factor-kappa B |
| NK: | natural killer |
| NLRP3: | NOD-like receptor thermal protein domain associated protein 3 |
| NO: | nitric oxide |
| NOXs: | NADPH oxidases |
| NT-3: | neurotrophin 3 |
| PD-1: | programmed death 1 |
| PDGF: | platelet derived growth factor |
| PGE2: | prostaglandin E2 |
| PI3K: | phosphatidylinositol 3-kinase |
| PNPLA3: | patatin-like phospholipase domain-containing protein 3 |
| PPAR-γ: | peroxisome proliferator-activated receptor-γ |
| qHSCs: | quiescent hepatic stellate cells |
| ROS: | reactive oxygen species |
| siRNA: | small interference RNAs |
| STAT3: | signal transducer and activator of transcription 3 |
| TGF-β: | transforming growth factor-β |
| Th: | T helper |
| TIMP-1: | tissue inhibitors of matrix metalloproteinases-1 |
| TLR2: | toll like receptor 2 |
| TNF-α: | tumor necrosis factor-α |
| Treg: | T regulatory cell |
| TSG-6: | tumor necrosis factor-α-stimulated gene-6 |
| VEGF: | vascular endothelial growth factor |
| YAP1: | yes-associated protein-1 |
| α-SMA: | α-smooth muscle actin |
Declarations
Author contributions
ZH: Conceptualization, Writing—original draft, Writing—review & editing.
Conflicts of interest
The author declares no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2025.
Publisher’s note
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.