
Abstract
Normal hepatobiliary function depends on an adequate bile flow from the liver through the biliary tree to the gallbladder, where bile is stored and concentrated, and from the gallbladder to the duodenum when it is required for the digestive process. Interruption of this secretory function results in partial or complete cholestasis, which is accompanied by important repercussions due to the lack of bile acids in the intestine and their regurgitation from hepatocytes to blood together with potentially toxic compounds that are normally eliminated in bile. The presence of active and selective transporter proteins located at both poles of the plasma membrane of hepatocytes, cholangiocytes, and epithelial cells of the ileal mucosa, together with the ability of hepatocytes to synthesize bile acids from cholesterol, enables the so-called bile acid enterohepatic circulation, which is essential in liver and gastrointestinal tract physiology. The presence in the ducts of the biliary tree of agents reducing their luminal diameter by external compression or space-occupying obstacles, either in the duct wall or its lumen, can result in total or partial obstructive cholestasis. The clinical impact and management of cholestasis are different depending on the intrahepatic or extrahepatic location of the obstacle. Thus, surgical interventions can often be helpful in removing extrahepatic obstructions and restoring normal bile flow to the duodenum. In contrast, hepatocyte or cholangiocyte damage, either global, restricted to subcellular compartments, or more specifically affecting the elements of the canalicular secretory machinery, may result in hepatocellular cholestasis or cholangiopathies. In these cases, bile flow interruption is usually partial and, except for extremely severe cases when liver transplantation is required, these patients often treated with pharmacological agents, such as ursodeoxycholic acid (UDCA) and rifampicin. The present review gathers updated information on the etiopathogenesis and pathophysiological aspects of different types of cholestasis.
Keywords
Bile acid, bile flow, biliary tree, cholangiocyte, hepatocyte, secretion, transportIntroduction
Cholestasis is defined as the lack of normal generation of bile by hepatocytes and cholangiocytes or its reduced ability to reach the duodenum, which occurs as the result of convergent flow through the biliary tree and the gallbladder-connecting ducts. This alteration can be primarily or secondarily linked to many acute or chronic liver diseases [1]. During the last few decades, our understanding of the physiology and pathophysiology of the biliary system has dramatically increased, since the time, by the middle of the twentieth century, when the accepted concept of bile formation by hydrostatic forces was replaced by the more accurate one of osmotic filtration-driven bile flow [2, 3] until the modern availability of tools capable of exploring molecular and genetic aspects of cholestasis. Many excellent scientists have contributed to the progress in this field of hepatology [4]. With this special issue on cholestasis, we want to pay homage to those who have made this extraordinary scientific jump possible and are no longer among us.
Altered biliary secretion is commonly diagnosed following well-established clinical practice algorithms (see, for instance, the one shown in Figure 1). Elevated serum bile acids are the most sensitive marker of cholestasis. Interpreting the results from serum bile acid analyses must consider the existence of gender-associated differences in bile acid pool composition [5, 6]. However, this has little impact on total bile acid serum levels and hence in establishing the threshold between normocholanemia and hypercholanemia, which is commonly fixed at 10 μmol/L. Although, for technical reasons, hyperbilirubinemia is more commonly used as a biomarker for cholestasis. However, the elevation of serum bilirubin levels does not always indicate cholestasis. For instance, whereas conjugated hyperbilirubinemia is commonly due to impaired bile secretion, elevated unconjugated bilirubin in neonates could be due to immaturity-associated low expression of the conjugating enzyme uridine 5’-diphospho (UDP)-glucuronosyltransferase (UGT). Criteria for the diagnosis of neonatal cholestasis include serum total bilirubin > 5 mg/dL and direct (conjugated) > 20%, or serum total bilirubin < 5 mg/dL but direct bilirubin being > 1 mg/dL. If the situation persists for more than 14 days, an evaluation by a pediatric hepatologist is required. Cholestasis affects 1/2,500 neonates born at term. Most frequent causes of neonatal cholestasis are biliary atresia (25–35%), hereditary disease (25%), metabolic disorders (20%), α1-antitrypsin (A1AT) deficiency (A1ATD, 2%), Alagille syndrome (2%), porta-cava shunt (2%), mitochondriopathies (2%), and biliary sludge (2%) [7].
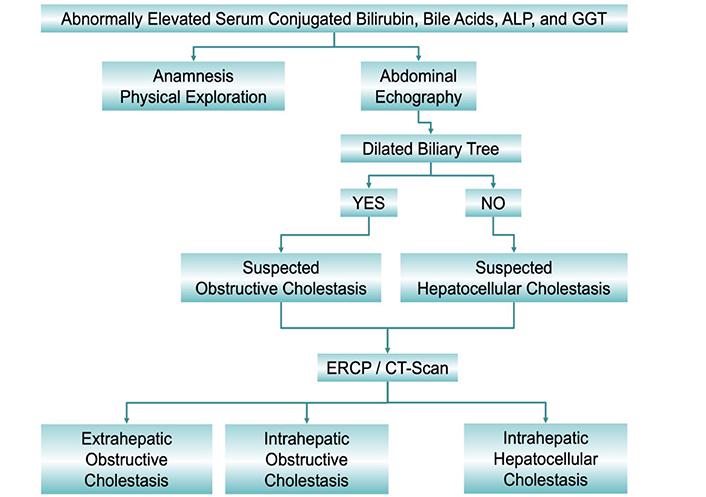
Classification of cholestasis according to biochemical data, image information obtained by abdominal echography, endoscopic retrograde cholangiopancreatography (ERCP) and computed tomography scan (CT-Scan), and the anatomical location of the cause. ALP: alkaline phosphatase; GGT: γ-glutamyltransferase
Cellular and molecular bases of bile formation
The liver performs multiple functions in the organism, such as those related to the metabolism of carbohydrates (i.e., glucose homeostasis, catabolism of hexoses, gluconeogenesis, glycogenolysis, and glycogenogenesis), lipids (i.e., cholesterol synthesis and catabolism, production of fatty acids and triglycerides, synthesis and metabolism of plasma lipoproteins, β-oxidation of fatty acids and production of ketone bodies during fasting), proteins (i.e., synthesis of plasma proteins, the biotransformation of ammonium into urea, catabolism of purine and pyrimidine bases, interconversion of non-essential amino acids), and other compounds, such as heme and porphyrins. Moreover, the liver is involved in hemostasis by controlling the synthesis of fibrinogen and coagulation factors III, V, VII, IX, and XI and in the storage of multiple substances such as glucose in the form of glycogen, vitamin B12, iron, and copper. Another essential role of the liver is the elimination from the body of many endogenous (e.g., bilirubin) and xenobiotic (e.g., phytosterols) compounds. This function is tightly related to detoxification phase I and phase II reactions taking place prior to exporting the biotransformed substances into bile [8].
The hepatobiliary system in charge of bile formation begins at a broad network of tiny canaliculi formed by the apical membrane of hepatocytes, which are connected through biliary ductules or canals of Hering, with intrahepatic and extrahepatic bile ducts constituting the biliary tree, to which the gallbladder is attached [9]. The latter is essential to concentrate bile components by water reabsorption [10], which favors that bile acids secreted into the duodenum could form mixed micelles with dietary fat in order to stimulate their digestion and favor the intestinal uptake of lipids and fat-soluble vitamins. When the normal supply of bile to the duodenum through the common bile duct (CBD) is entirely blocked or partially impaired, the result is complete or partial cholestasis, respectively. In addition to the lack of bile in the intestinal lumen, cholestasis is also often accompanied by regurgitation toward blood across the basolateral membrane of hepatocytes of cholephilic compounds, i.e., compounds that are normally secreted in bile [11, 12]. Here we will review the updated information on the different types of cholestasis classified based on their etiopathogenesis and pathophysiological features.
Accordingly, before starting, we should remind a few basic morphological and physiological concepts regarding bile flow [13]. This process relays on the polarity of epithelial cells in charge of bile secretion, i.e., hepatocytes in the liver parenchyma and cholangiocytes in the biliary tree, and the presence in their basal and apical plasma membranes of specific transporters (Figure 2, Table 1). Bile flow is initially generated at the canalicular space, which is formed by the opposed apical (canalicular) membranes of two or, less frequently, three hepatocytes [14]. The surface of this membrane is less than 1% of the global hepatocyte plasma membrane, but two essential characteristics make it possible for the canalicular membrane to play an essential role in bile formation, namely, a) the presence of an osmotic barrier formed by tight junctions (TJs) located between adjacent hepatocytes, and b) the existence in the canalicular membrane of specific and active transporters (Figure 2, Table 1). Among them, the ATP-binding cassette (ABC) protein bile salt export pump (BSEP, gene symbol ABCB11), is considered the major driving force to generate canalicular bile flow by active transport into the canalicular lumen of monoanionic bile acids [15]. These molecules generate the osmotic gradient accounting for the flux of water toward the biliary space, hence resulting in the so-called bile acid-dependent fraction (BADF) of bile flow [16]. This is added to the osmotic flux of water attracted by other osmolytes also secreted into the canaliculi [e.g., bicarbonate, glutathione (GSH), organic anions, and cations unmodified or conjugated with GSH, sulfate, or glucuronic acid] by canalicular transporters, such as multidrug resistance protein 1 (MDR1), multidrug resistance-associated protein 2 (MRP2), multidrug and toxin extrusion 1 (MATE1), and anion exchange protein 2 (AE2), known as the bile acid-independent fraction (BAIF) of bile flow [16].
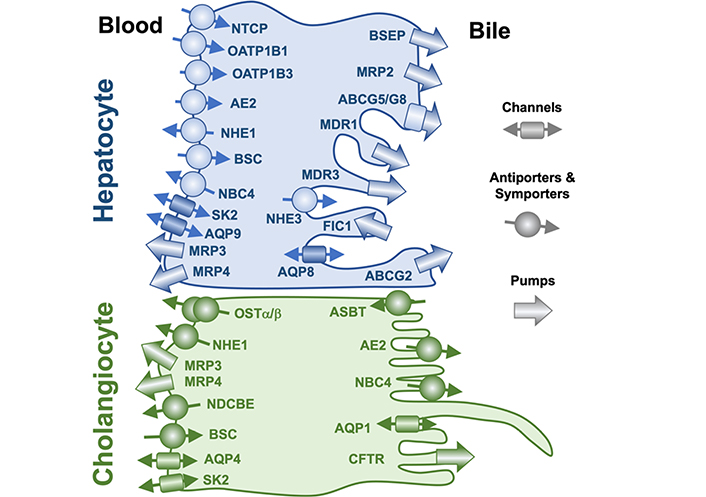
Localization at the apical and basolateral plasma membrane of hepatocytes and cholangiocytes of proteins involved in bile formation. Nomenclature for proteins depicted here and their natural substrates are listed in Table 1. ABCG5: ATP binding cassette subfamily G member 5; AQP9: aquaporin 9; ASBT: apical sodium-dependent bile acid transporter; BSC: bumetanide-sensitive Na+-K+-2Cl– cotransporter; CFTR: cystic fibrosis transmembrane conductance regulator; FIC1: familial intrahepatic cholestasis type 1; NBC4: sodium bicarbonate cotransporter 4; NDCBE: sodium-driven chloride/bicarbonate exchanger; NHE1: sodium–hydrogen antiporter 1; OSTα/β: organic solute transporter α/β; SK2: Ca2+-activated small conductance K+ channel 2
Plasma membrane transporters and channels involved in hepatobiliary function
| Cells | Membrane | Protein | Gene | Substrates |
|---|---|---|---|---|
| Hepatocytes | Basolateral | NTCP | SLC10A1 | Bile acids |
| OATP1B1 | SLCO1B1 | Bile acids, organic anions | ||
| OATP1B3 | SLCO1B3 | Bile acids, organic anions | ||
| MRP3 | ABCC3 | Bile acids, organic anions | ||
| MRP4 | ABCC4 | Bile acids, organic anions | ||
| NBC4 | SLC4A5 | Na+/HCO3–symporter | ||
| NHE1 | SLC9A1 | Na+/H+ antiporter | ||
| SK2 | KCNN2 | K+ channel | ||
| BSC | SLC12A2 | Na+/Cl– symporter | ||
| AQP9 | AQP9 | Water channel | ||
| Hepatocytes | Canalicular | BSEP | ABCB11 | Bile acids |
| MRP2 | ABCC2 | Organic anions | ||
| MDR1 | ABCB1 | Several | ||
| BCRP | ABCG2 | Bile acids, steroids | ||
| MDR3 | ABCB4 | Phosphatidyl choline | ||
| FIC1 | ATP8B1 | Phosphatidyl serine | ||
| ABCG5/G8 | ABCG5/G8 | Cholesterol | ||
| NHE3 | SLC9A3 | Na+/H+ antiporter | ||
| AQP8 | AQP8 | Water channel | ||
| Cholangiocytes | Basolateral | OSTα/β | SLC51A/B | Bile acids, organic anions |
| NHE1 | SLC9A1 | Na+/H+ antiporter | ||
| NDCBE | SLC4A8 | Cl–/HCO3– antiporter | ||
| SK2 | KCNN2 | K+ channel | ||
| BSC | SLC12A2 | Na+/Cl– symporter | ||
| MRP3 | ABCC3 | Bile acids, organic anions | ||
| MRP4 | ABCC4 | Bile acids, organic anions | ||
| AQP4 | AQP4 | Water channel | ||
| Cholangiocytes | Apical | ASBT | SLC10A2 | Bile acids |
| AE2 | SLC4A2 | Cl–/HCO3– antiporter | ||
| CFTR | ABCC7 | Cl– channel | ||
| NBC4 | SLC4A5 | Na+/HCO3– symporter | ||
| AQP1 | AQP1 | Water channel |
BCRP: breast cancer resistance protein; HCO3–: bicarbonate; KCNN2: potassium calcium-activated channel subfamily N member 2
While flowing downstream the biliary tree, canalicular bile is modified by cholangiocytes through reabsorption of some bile components and the secretion of a bicarbonate-rich fluid. The continuous bile acid output across the canalicular membrane is followed by their efficient re-uptake by the intestine, mainly the ileum, and subsequent recovery from portal blood by hepatocytes through the also highly efficient sodium-dependent symporter Na+-taurocholate co-transporting polypeptide (NTCP), located at the basal (sinusoidal) plasma membrane of these cells [17, 18]. In humans, there is also a minor contribution to bile acid uptake by members of the organic anions transporting polypeptide (OATP) family, mainly OATP1B1 and OATP1B3 [19]. The daily fecal loss of bile acids from this enterohepatic circulation (≈ 5% of the total pool) is replaced by de novo synthesis from cholesterol of primary bile acids, mainly cholic acid (CA) and chenodeoxycholic acid (CDCA) [11, 12].
Obstructive cholestasis
The first consideration in classifying cholestasis is the anatomical location of its etiology. Extrahepatic cholestasis is due to a reduction of the lumen of the biliary ducts at any place of the extrahepatic biliary tree. The obstruction can be partial or complete depending on the relative magnitude of the obstacle vs. the duct diameter and its anatomical location, i.e., a) outside the duct (extraparietal), as it occurs, for instance, in patients suffering from tumors located in the head of the pancreas that compress the CBD; b) in the wall of the duct (parietal) due to scars or tumors; or c) in the lumen (intraluminal) due to gallstones. Very often, depending on the etiology and anatomical location, obstructive cholestasis tends to be almost complete, and surgical intervention to remove the obstacle is the recommended procedure. The most frequent etiological causes of these obstructions are gallstones, stenosis, atresia, parasitosis, choledochal cyst, ascending cholangitis, primary sclerosing cholangitis (PSC), secondary sclerosing cholangitis (SSC), and carcinoma of the pancreas or biliary tract. Some of them can also appear inside the liver, causing intrahepatic obstructive cholestasis, which often results in partial interruption of bile flow, except if the obstacle is located near the hepatic hilum.
Extrahepatic obstructive cholestasis
Choledocholithiasis is the most common cause of obstruction affecting extrahepatic bile ducts [20]. Regardless of gallstone composition, i.e., cholesterol (the most frequent ones) [21], biliary pigments, or mixed gallstones [22], the mechanical repercussions of the obstacle are similar in all extrahepatic obstructive cholestasis. Distal biliary stricture is a narrowing in the distal half of the extrahepatic bile duct that makes it difficult for bile to reach the duodenum [23]. Atresia is the cause of very aggressive neonatal cholestasis leading to the destruction and obliteration of the extrahepatic bile ducts in the first weeks of life [24]. In these patients, the onset of biliary fibrosis, which accompanies persistent cholestasis and the retention of biliary components, is very rapid [24].
Some parasitic infections are the cause of extrahepatic cholestasis. For instance, Echinococcus granulosus, a species of tapeworm that induces the formation of hydatid cysts in the liver, often causes compression of the bile ducts and can lead to more severe complications due to biliary tree lesions with increased risk of duct rupture [25]. Fasciola hepatica infects humans only accidentally, as this parasite has livestock as its usual hosts [25]. When reaching the liver, nonspecific biliary tree dilatation together with decreased bile flow appears. Ascaris lumbricoides, usually residing in the small intestine of infected hosts, can migrate through the ampulla of Vater upward to the CBD and cause obstructive jaundice along with episodes of biliary colic [26].
Choledochal cyst is a rare manifestation of hepatobiliary fibropolycystic disease [27]. This is due to malformations during embryonic development of the ductal plate, causing excessive proliferation of the biliary epithelium. As a consequence, fibrogenesis is activated, and fluid-filled cystic spaces in the liver parenchyma and bile ducts are formed, which are also accompanied by gallstones. Altogether, these factors can contribute to hindering the normal bile flow toward the duodenum [27].
Ascending cholangitis is a fatal condition caused by ascending bacterial infection reaching the biliary tree from the small intestine [28]. Choledocholithiasis is the leading predisposing cause because the presence of gallstones impairs the flushing function of bile flow, hence favoring the ascending infection, which leads to inflammation and partial or complete CBD obstruction [28].
Pancreatic carcinoma is one of the most aggressive tumors. Most patients develop obstructive jaundice due to the obliteration of extrahepatic bile ducts caused by the local compression of the tumor mass [29]. The highest probability of CBD obstruction is associated with the localization of the tumor in the head of the pancreas. Benign (cholangiomas) or malignant [cholangiocarcinomas (CCAs)] tumor masses generated in the biliary tree can also obstruct the CBD if they are located at the perihilar region of the biliary tree, i.e., the area of confluence of intrahepatic ducts [30], or closer to the duodenum [31].
Intrahepatic obstructive cholestasis
Gallstones can also be found in the intrahepatic bile ducts and hence obstruct them. Moreover, liver tumors, such as intrahepatic CCA [32], can also obliterate intrahepatic ducts hindering normal bile flow. Intrahepatic pyogenic abscesses, i.e., accumulation of inflammatory cells, mainly neutrophils and tissue debris, commonly caused by bacterial or parasitic liver infections, can obstruct intrahepatic bile ducts [33].
Clonorchis sinensis and Opisthorchis viverrini are two species of food-contaminating liver flukes that infect bile ducts [34]. Besides intrahepatic biliary obstruction, these parasites induce a chronic inflammatory state that contributes to duct obliteration and produces oxidative DNA damage in the infected biliary epithelium favoring malignant transformation [34].
Cholestasis due to pathological events affecting hepatocytes and cholangiocytes
Intrahepatic non-obstructive cholestasis is caused by impaired bile formation due to loss of cellular function at specific subcellular levels or by global dysfunction of hepatocytes. In the case of intrahepatic cholestasis, either due to obstruction or hepatocellular impairment, the reduction in bile flow is commonly not complete because other non-obstructed routes could drain the bile from the biliary tree or not all hepatocytes are affected, respectively. Hence, usually, the hepatobiliary function is not entirely abolished. Consequently, surgical intervention is not the recommended treatment, except in cases where liver transplantation is the only option for these patients.
Cholestasis associated to cholangiopathies
Chronic diseases generated by cholangiocyte alterations are known as cholangiopathies. These can be caused by restricted damage to local cholangiocytes, genetic and epigenetic events, and posttranscriptional mechanisms, such as changes in protein expression due to microRNAs. Owing to the pivotal role of these cells in biliary physiology, cholangiopathies are accompanied by cholestasis. In some cases, such as in biliary atresia, the pathogenic mechanism accounting for cholestasis is obvious. However, in other cholangiopathies, the interference with bile flow is more complex [35].
Primary biliary cirrhosis (PBC) is characterized by nonsuppurative inflammation and destruction of the interlobular bile ducts. These patients, mainly women (90%) have reduced expression of the bicarbonate transporter SLC4A2 located at the apical membrane of cholangiocytes, which reduces their ability to carry out biliary bicarbonate secretion. This negatively impacts bile formation and reduces the protection of the bicarbonate umbrella against bile salt-induced toxicity in cholangiocytes [36].
In PSC, there is chronic inflammation affecting both intrahepatic and extrahepatic bile ducts. This results in strictures that account for the description of the disease as obliterative cholangitis. These changes cause hindered transit of bile through the biliary tree, hence intrahepatic and extrahepatic obstructive cholestasis and progressive liver dysfunction [37, 38].
SSC is clinically comparable to PSC, although it is caused by processes indirectly damaging the biliary tree, such as recurrent pancreatitis, bile duct neoplasms, and congenital anomalies of the biliary tree [39].
Cystic fibrosis (CF) is a systemic disease caused by mutations in the transmembrane conductance regulator gene (ABCC7) that encodes an ABC protein (CFTR) playing an essential role in chloride transfer across the apical membrane of different epithelial cells, including cholangiocytes. Thus, inactivating mutations account for CF, the most frequent autosomal recessive genetic disease in white people. The absence of functional CFTR causes water efflux abnormalities responsible for increasing the density of secretions in different tissues. In the liver, the only cells expressing CFTR are cholangiocytes. The lack of this protein at the apical membrane of cholangiocytes impairs water and bicarbonate secretion leading to cholestasis together with less alkaline bile [40].
Global hepatocyte dysfunction
In clinical practice, one common type of cholestasis is due to global impairment of liver parenchyma function, which can even be associated with the death of hepatocytes due to many different causes. Commonly, following liver injury, cell proliferation and tissue regeneration are induced and carefully controlled. However, this process requires the differentiation of proliferating cells into polarized hepatocytes to become active bile forming cells, which means that during this time, the liver ability to generate bile is reduced.
Alcoholic hepatitis
Alcoholic liver disease (ALD) includes a spectrum of hepatic alterations, ranging from steatosis to alcoholic hepatitis, which increases the risk of cirrhosis [41]. Patients with chronic ALD frequently manifest clinical or histological evidence of inflammation, fibrosis, cholestasis, and eventually liver failure [42]. In cases of severe disease, the patients usually show jaundice, fever, and elevated serum levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT, AST/ALT ratio > 2), GGT, ALP and bilirubin [43]. Although evidence of cholestasis is typically found in all stages of alcoholic hepatitis, its severity has been correlated with histological changes, such as parenchymal necrosis, portal inflammation, and fibrosis stage. Hence, the degree of cholestasis is a prognostic factor for mortality in these patients [44].
Viral hepatitis
The range of clinical manifestations in patients with viral hepatitis is vast, from subclinical “inactive state” to severe acute hepatitis. In chronic cases, there is an enhanced risk of cirrhosis and, subsequently, hepatocellular carcinoma. Intrahepatic cholestasis is an uncommon feature in viral hepatitis, which can be interpreted as an indicator of disease progression or viral reactivation. Thus, in some patients with chronic hepatitis B, sudden development of cholestasis has been associated with hepatitis B virus (HBV) reactivation, which can be typically triggered by anticancer chemotherapy, immunosuppression, or organ transplantation. Viral reactivation is characterized by an increased HBV replication and a substantial increment in serum aminotransferase activity [45]. Besides, about 20–40% of chronic hepatitis B patients who undergo liver transplantation develop fibrosing cholestatic hepatitis, characterized by the rapid development of severe jaundice, associated with marked hyperbilirubinemia and progressive hepatocellular failure [46, 47]. In contrast, fibrosing cholestasis in hepatitis C patients after liver transplantation is rare (2–5%), but when it occurs, the condition is severe due to the high risk of progression to liver failure. The clinical situation is characterized by elevated levels of hepatitis C virus (HCV) RNA both in serum and liver tissue, with hyperbilirubinemia and high levels of serum ALP and GGT, whereas those of AST and ALT are mild or moderate. Histologic features are similar to those found in patients with fibrosing cholestatic hepatitis B [48–50]. Although hepatitis A virus (HAV) causes a benign and self-limited infection, this may occasionally (less than 5% of cases) cause a cholestatic syndrome characterized by marked serum elevation of total bilirubin (up to 40 mg/dL), pruritus, fever, diarrhea, and weight loss, which usually resolves spontaneously [51–55].
Autoimmune hepatitis
Autoimmune hepatitis (AIH) is a chronic inflammatory liver disease characterized by hypergammaglobulinemia, elevated serum AST and ALT levels, the presence in the liver of autoantibodies, and inflammation typically including lymphocytic infiltrates, and interface hepatitis [56]. Patients with AIH may exhibit features of the so-called “overlap syndromes”, which include PBC (7–13%), PSC (6–11%), and cholestatic syndrome in the absence of features of PBC or PSC (5–11%) [57]. The presence of antimitochondrial antibodies (AMAs) and high levels of ALT/AST and bile duct injury are essential for the diagnosis. Thus, AIH patients with AMA, interface hepatitis, and injured or even destructed bile ducts are diagnosed as suffering from AIH-PBC. AIH cases characterized by the absence of AMA, together with interface hepatitis and high levels of AST/ALT, γ-globulin, immunoglobulin G (IgG), and GGT, are diagnosed as AIH-PSC patients. Whereas, in AIH-cholestatic syndrome, a mixture of features is found, including AMA-negativity with high levels of AST/ALT, γ-globulin, IgG, and GGT, as in AIH-PSC together with destructive cholangitis as in AIH-PBC [57–60]. Nevertheless, symptoms of AIH overlap syndromes are often imprecise and heterogeneous, which means that reaching an accurate diagnosis to choose the best therapeutic strategy is not always evident.
Total parenteral nutrition-associated cholestasis
Total parenteral nutrition (TPN) is a life-saving treatment for acute and chronic intestinal insufficiency patients. However, several pathological disorders, such as liver steatosis, steatohepatitis, and cholestasis, have been associated with prolonged TPN. The incidence of cholestasis in patients receiving TPN is higher in infants than in adults (40–60% vs. 15–40%) [61]. The pathogenesis of this condition, characterized by increased levels of serum total bilirubin (> 2 mg/dL), GGT, ALP, AST, and ALT [62–64], is multifactorial. Thus, during prolonged TPN, several changes occur in the liver directly related to the differences in nutrient composition with a normal diet. These changes are also dependent on whether the TPN is administered through the hepatic artery or the portal vein, which determines different hemodynamic conditions in the liver acini. Rarely, cholestasis does not resolve and may progress to severe hepatobiliary damage and death [65].
Subcellular hepatocyte damage
In some cases, the impaired hepatocyte secretory function is associated with specific changes in subcellular compartments (e.g., peroxisomes) or mechanisms (metabolic pathways), resulting in cellular dysfunction and hence directly or indirectly alteration of canalicular bile formation.
Metabolic disorders
Aberrant activity of metabolic pathways in hepatocytes due to inherited mutations has been associated with neonatal cholestasis in several congenital syndromes. One of them is A1ATD, a rare, autosomal dominant disorder that affects approximately 1 in 2,000–5,000 births. In this condition, reduced levels of serum A1AT are accompanied by lung emphysema and liver damage [66], which explains why A1ATD is the most common indication for liver transplantation in children [67]. The disease is caused by mutations in the SERPINA1 gene, which encodes A1AT. This glycoprotein produced mainly in the liver is the major circulating protease inhibitor. Mutated A1AT cannot be efficiently secreted by the hepatocyte, resulting in low levels of circulating A1AT and its intracellular accumulation, leading to hepatocellular injury. In particular, the homozygous Z allele (Glu342Lys) of A1AT has been associated with hepatocyte damage because it encodes a misfolded protein that accumulates in their endoplasmic reticulum (ER), triggering stress of this organelle, mitochondrial dysfunction, and, eventually, cell death. Usually, the main clinical feature is cholestatic jaundice prolonged for the first two months of life. In most patients, cholestasis is reversible and does not progress into more severe liver disease, but in other patients, liver transplantation is required [66].
Neonatal cholestasis can also be due to type 1 tyrosinemia and galactosemia. These are also rare inherited (autosomal recessive) disorders accounted for by the deficient ability to metabolize tyrosine or galactose, respectively. The former appears acutely in neonates and is characterized by acute liver failure, hyperbilirubinemia, ascites, and hypoglycemia. Galactosemia first appears during the first few weeks of life after ingestion of breast milk or milk-based formulas that contain lactose, whose hydrolysis results in glucose and galactose. The inability to normally metabolize galactose results in intrahepatic accumulation of toxic metabolites, which leads to hypoglycemia, diarrhea, vomiting, and jaundice [68].
Alterations in peroxisomal biogenesis
Inborn errors affecting the biogenesis of peroxisomes are the cause of Zellweger syndrome, a rare disorder characterized by a marked reduction or absence of functional peroxisomes [69]. Mutations affecting the peroxisomal biogenesis factor or peroxin (PEX) family of genes, especially in PEX1 or PEX6 genes, encoding proteins essential for peroxisomal assembly, are the cause of Zellweger syndrome [70]. Peroxisomal dysfunction leads to the abnormal accumulation of very long-chain fatty acids in different tissues and C27-bile acid species in hepatocytes. Consequently, patients suffer from neurological disorders, polycystic kidney or chronic liver disease with hepatomegaly, and conjugated hyperbilirubinemia. Cholestasis associated with bile acid metabolism disorders will be addressed more in detail in a separate review included in this special issue.
Impaired intracellular trafficking and membrane sorting of proteins
Several cases of idiopathic familial intrahepatic cholestasis have been associated with mutations in genes involved in the trafficking and sorting of proteins to the hepatocyte apical or basolateral membranes. This is a complex intracellular process requiring the coordinated function of recycling endosomes and the Golgi apparatus [71].
In patients with arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome, a rare multisystemic disease in which neonatal patients suffer from cholestasis without elevation of serum GGT activity [72], mutation in VPS33B [73] and VIPAS39 [74] genes have been identified. They encode two proteins that work together in an endosomal recycling system accounting for the apical delivery and retrieval from the canalicular membrane of proteins, such as BSEP. Inactivating mutations in these genes impair the control of BSEP insertion/function, reducing the intracellular levels of the encoded proteins by misfolding and degradation of the mutant variants or hindering protein-protein interaction [71].
The AP1S1 gene encodes the sigma 1A subunit of the clathrin adaptor protein complex AP-1, which is involved in sorting cytoplasmic proteins into clathrin-coated transport vesicles. AP1S1 mutants are related to MEDNIK syndrome, characterized by mental retardation, enteropathy, deafness, peripheral neuropathy, ichthyosis, and keratodermia [75]. More precisely, patients with the homozygous p.Asp322GlyfsX17 frameshift AP-1 variant suffer from intrahepatic cholestasis and copper overload. Although the molecular mechanism that links AP-1 with impaired bile secretion has not been unambiguously elucidated, this is likely related to altered BSEP expression/trafficking due to copper-induced farnesoid X receptor (FXR)-mediated deregulation [71].
The loss of the cytoskeletal protein SCYL1, involved in the retrograde traffic of proteins in the interface between the ER and the Golgi apparatus, affects the morphology of the latter and has been associated with deficiencies in the N-glycosylation of proteins, such as BSEP, that may reduce their function. Mutations in the SCYL1 gene have been found in patients with neonatal cholestasis and idiopathic acute liver failure [76].
Ubiquitin 53-specific peptidase (USP53) contributes to the maintenance of canalicular membrane polarity. The absence of active USP53 due to inactivating mutations in the USP53 gene alters the targeting of canalicular transporters. Consistently, these mutations have been found in children with a severe progressive cholestatic disease [77].
The MYO5B gene encodes the “actin-based unconventional molecular motor protein myosin Vb”, which is closely linked with the ER-associated Rab family of proteins. These play a crucial role in regulating protein trafficking in polarized epithelial cells. Missense mutations in MYO5B have been identified in patients with intrahepatic cholestasis without elevation of GGT serum levels [78, 79]. MYO5B variants can also cause microvillus inclusion disease (MVID) [78], a severe congenital diarrheal and malabsorption disorder requiring life-long TPN [80]. These patients also frequently suffer from cholestasis in the absence of abnormal GGT levels [81]. Liver biopsies of MVID patients with intrahepatic cholestasis revealed a reduced expression and impaired targeting of BSEP and, in some cases, also MRP2 and the MDR3 [78, 79, 82]. In particular, the missense MYO5B mutant myosin-Vb-P660L has been suggested to account for the deficient insertion of bile canalicular transporters, which results in impaired bile secretion [82]. Recently, inactivating variants of the KIF12 gene, which encodes kinesin family member 12, have been identified as the cause of altered intracellular MRP2 trafficking toward the canalicular membrane [83].
Alterations in the cytoskeleton of parenchymal cells
The integrity of the hepatocyte cytoskeleton is essential for intracellular transport of lipids and proteins, membrane recycling, regulation of paracellular permeability, and bile flow through the bile canaliculi. Thus, disruption of the microtubular system, disturbance of the actin microfilament network, alterations in TJ, and changes in cytokeratin intermediate filaments, can result in impaired bile formation [84]. For example, some cholestatic drugs (e.g., chlorpromazine, cytochalasin B) and toxins (e.g., colchicine) known to alter actin microfilaments may cause cholestasis by preventing the translocation of transport proteins to the canalicular membrane [84].
TJs between adjacent parenchymal liver cells contribute to the barrier that isolates bile from blood. They are also involved in maintaining the polarized phenotype of hepatocytes and cholangiocytes. Although the importance of inter-cholangiocyte and inter-hepatocyte TJ integrity is still poorly understood [85], there is evidence suggesting a critical role of altered TJ in cholestatic diseases. Thus, experimental data obtained in laboratory animals revealed abnormalities in the localization of TJ proteins (TJPs) occludin and zonula occludens-1 (ZO-1) after bile duct ligation (BDL) [86, 87]. Further investigations revealed that inactivating mutations in the TJP2 gene encoding the TJP2 or ZO-2 have been identified in patients with cholestatic liver disease [88, 89]. TJP2 is expressed in many cells, including hepatocytes, where it tethers transmembrane TJPs to the cytoskeleton to preserve the structure of bile canaliculi. The loss of function in this protein disrupts TJ leading to leaky bile ducts and bile canaliculi, allowing bile reflux towards hepatic parenchyma [77, 90]. Newborns with TJP2 deficiency present progressive intrahepatic cholestasis with jaundice and pruritus. The situation can eventually worsen to liver failure or hepatocellular carcinoma development during childhood [91]. Hereditary cholestasis in patients suffering from neonatal ichthyosis and sclerosing cholangitis (NISCH) syndrome has been associated with defective TJ due to mutations in the CLDN1 gene encoding the TJ component claudin-1 [92].
Aggressions to the canalicular membrane and bile ducts
Since the canalicular membrane plays a pivotal role in bile generation several factors, such as drugs (e.g., steroids), porphyria, Hodgkin’s disease, genetic alterations associated with some types of familial cholestasis, impairing the composition, physical characteristics and even the integrity of this membrane can result in reduced bile flow.
Erythropoietic protoporphyria (EPP), the most common porphyria in children, with an incidence of 2 cases to 5 cases in 1,000,000 individuals, is a disorder caused by autosomal recessive mutations in the gene encoding ferrochelatase (FECH), the heme pathway terminal enzyme [93]. Most affected patients (98%) are heterozygous for a loss-of-function variant that markedly decreases FECH activity and a second low-expression pathogenic variant that reduces the FECH activity by about 50% [93]. FECH deficiency leads to protoporphyrin IX (PPIX) being released from the bone marrow into the circulation. PPIX is taken up from the blood by hepatocytes and the vascular endothelium. In skin endothelial cells, PPIX is activated by sunlight, triggering the formation of reactive oxygen species causing dermal injury. Therefore, patients usually suffer photosensitivity characterized by tingling, burning, pain, and itching after exposure to the sunlight. On the other hand, when PPIX accumulates in the liver, owing to its poor water solubility, it adheres to membranes and precipitates in hepatocytes and bile canaliculi, leading to architectural changes. Consequently, PPIX accumulation results in decreased bile flow, cholelithiasis, fibrosis, and, ultimately, liver failure [93, 94]. Indeed, in early EPP, ultrastructural damage in bile canaliculi associated with protoporphyrin crystals has been described [95]. Bile canaliculi dilatation has also been described in these patients [96, 97]. Approximately 20–30% of EPP patients have mild liver dysfunction, typically with slight elevations of the liver enzymes. A small proportion of them (5%) may develop more advanced liver disease and motor neuropathy [98]. In these cases, severe liver damage could be difficult to treat and sometimes requires liver transplantation. In experimental models, PPIX causes marked cholestasis [99] with impaired biliary lipid secretion [100]. Bile acids with detergent ability, such as taurocholic acid, have been found to enhance PPIX metabolism and ameliorate biliary function [100, 101].
Hodgkin’s lymphoma (HL) is a type of lymphoma that originates from lymphocytes. Common symptoms of this cancer include painless swelling of lymph nodes in the neck, armpits, or groin, fatigue, fever, and severe itching. Hepatic dysfunction is uncommon, and in sporadic cases, intrahepatic cholestasis and vanishing bile duct syndrome (VBDS) due to the loss of intrahepatic bile ducts have been reported in HL patients [102].
Xenobiotics and endogenous substances-induced hepatocellular cholestasis
Cholestatic disorders are the most severe clinical manifestations of drug-induced liver injury (DILI). More than 1,000 drugs have the potential to cause cholestatic DILI [103]. Several compounds can induce cholestasis by impairing hepatocellular functions, such as inhibiting transporter expression and function, inducing cytoskeleton alterations, or disrupting the biliary epithelium [104, 105]. The existence of some genetic variations in transporter genes can predispose to cholestasis caused by xenobiotics and endogenous substances [105].
Although inhibition of MRP2 by drugs, such as the antibiotic fusidate, may contribute to cholestasis and jaundice [106], the most frequent target for drug-induced cholestasis is BSEP. This canalicular transporter can be cis-inhibited in a competitive manner by many drugs, such as cyclosporine A, rifampicin, bosentan, troglitazone, and glibenclamide [107]. Besides, other compounds, such as steroid hormones, for instance, estrogen and progesterone metabolites, can trans-inhibit BSEP function, which results in hepatocellular cholestasis. Moreover, the estrogen metabolite estradiol-17β-D-glucuronide (E17G) induces acute cholestasis by a mechanism that also involves the enhanced internalization of canalicular BSEP and MRP2 via clathrin-mediated endocytosis [108].
Sulfated progesterone metabolites can directly trans-inhibit BSEP [109]. Besides, they can also act indirectly by downregulating FXR [110]. Estradiol and other non-steroid compounds can also inhibit BSEP expression [111]. Altogether, this evidence supports the concept that elevated levels of estrogens and progesterone in pregnancy are involved in developing intrahepatic cholestasis of pregnancy (ICP), a not uncommon disorder affecting some women during the second–third trimester of gestation. The maternal symptoms are characterized by pruritus and elevated serum bile acid levels [112].
Other hepatocellular effects driven by cholestatic drugs that can affect the canalicular membrane function are changes in membrane fluidity (e.g., cyclosporin A), disruption of TJs (e.g., rifampicin and E17G), or constriction/dilatation of bile canaliculi (e.g., chlorpromazine, flucloxacillin, and bosentan) [104].
Inborn errors affecting the expression/function of canalicular secretory machinery
This group of cholestasis initially included those characterized by the specific failure in one of the ATP-dependent transporters located at the canalicular membrane and directly involved in bile secretion. These alterations are due to inborn errors, which justifies that they are classified as familiar cholestasis. Until the ‘70s, these rare diseases were diagnosed by the pediatricians as idiopathic neonatal hepatitis in children with mild to very severe heterogeneous symptomatology, which markedly depends on the affected gene and the degree of subsequent dysfunction. These aspects are the bases to classify them into two groups, benign recurrent intrahepatic cholestasis (BRIC) and progressive familial intrahepatic cholestasis (PFIC). BRIC is characterized by recurrent crises of severe pruritus, with weight loss, but without progression to severe liver damage or cirrhosis. In contrast, patients with PFIC suffer from recurrent episodes of cholestasis at the beginning that later becomes permanent and of progressive severity. This condition is accompanied by steatorrhea, hepatomegaly, and growth retardation. The clinical situation frequently progresses to liver cirrhosis, eventually requiring liver transplantation.
According to etiological criteria, PFIC/BRIC has traditionally been classified into three subtypes. The ATP8B1 gene encodes the translocase FIC1, which is involved in maintaining the asymmetry between both leaflets of the canalicular membrane by continuous inwardly directed aminophospholipid transfer. This determines the appropriate physical environment required for the efficient function of transmembrane transporters, mainly BSEP. FIC1 deficiency (PFIC1/BRIC1) was the first described case of fatal familial intrahepatic cholestasis. This was initially named Byler disease, based on the name of a common ancestor of all first patients in the Amish kindred where the disease was identified, Jacob Byler [113]. If FIC1 does not work correctly, lipid distribution in the canalicular membrane is abnormal, leading to altered membrane fluidity and impaired bile formation [114]. Inborn errors in the ABCB11 gene resulting in the loss of BSEP function lead to cholestasis (PFIC2/BRIC2), characterized by reduced biliary bile acid secretion, regurgitation toward blood, and accumulation in the hepatocyte with the consequent cellular damage. The ABC protein MDR3 (ABCB4 gene) is responsible for phospholipid transfer, mainly phosphatidylcholine, from the inner to the outer leaflet of the canalicular membrane, where it is available for buffering the detergent activity of secreted bile acids and hence forms mixed micelles. The lack of efficient MDR3 fails to protect hepatocytes and cholangiocytes from bile acids. Consequently, canalicular and apical ductular membranes are attacked and damaged, resulting in secondary impairment of bile formation (PFIC3/BRIC3), characterized by elevated GGT release into plasma [115–117].
The development of more sensitive analytical methods, like LC-MS/MS, has allowed identifying newer types of PFIC. Thus, the existence of inborn errors in bile acid metabolism leading to the generation of molecular species with unsaturated steroid ring and non-shortened sidechain (C27-bile acids) able to impair hepatocyte function has been described. Inhibition of de novo synthesis with endogenous C24-bile acids together with the hepatoprotective activity of ursodeoxycholic acid (UDCA) could be beneficial in these patients (Figure 2) [118].
Considering clinical practice, due to the low incidence of PFIC, even the most frequent forms, when biochemical analyses make the pediatrician suspect cholestasis, it is always mandatory to rule out more frequent diseases, such as hepatitis, PBC, Alagille syndrome, sclerosing cholangitis, and others (Figure 3). Afterward, the presence of elevated serum levels of GGT is a clear suggestion for PFIC3. UDCA hepatoprotective activity and hypercholeretic effects are beneficial for these patients by neutralizing and diluting more toxic bile acid species. Treatment with UDCA is also recommended in the case of other PFIC types with partial preservation of biliary bile acid output. Although these patients show moderately elevated serum levels of bile acids due to impaired BSEP function and subsequent regurgitation toward blood, these are mature C24 saturated steroids. Partly preserved bile acid output justifies the beneficial effect of bile drainage and cholestyramine, which favors the elimination of bile acids from the body and reduces hypercholanemia. Extrahepatic manifestations constitute a valuable clue for PFIC1 diagnosis. Since FIC1 is also expressed in a few extrahepatic tissues, its absence has an impact on the structure and function of other organs, such as the intestinal epithelium, the exocrine pancreas, and the inner ear, resulting in diarrhea, pancreatitis, and deafness, respectively. As BSEP is selectively expressed in hepatocytes, patients with PFIC2 do not show extrahepatic manifestations. In these patients, hypercholanemia is accounted for by normal C24 bile acids. Cholestasis-associated symptoms could be treated with rifampicin but not phenobarbital, which is not indicated in pediatric patients. In most severe cases of PFIC patients, pharmacological treatments are merely palliative because they eventually require liver transplantation. In the case of impaired bile secretion due to the accumulation of cholestatic bile acid species, since the secretory machinery is not constitutively impaired but only inhibited, the pharmacological correction of the altered metabolic pathway restores the function. Moreover, intestinal sequestering of endogenous bile acids using resin cholestyramine [119] or dilution of the secreted by treatment with exogenous UDCA [118] have been reported as strategies to reduce the signs of liver damage.
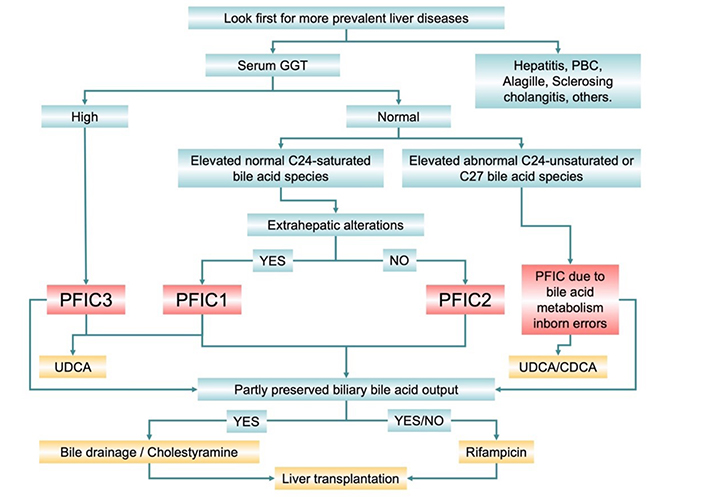
Diagnostic algorithm and suggested treatment of most frequent forms of intrahepatic familial cholestasis (PFIC)
Next-generation sequencing has allowed identifying several very infrequent forms of PFIC. Thus, PFIC4 has been found in patients with loss-of-function mutations in TJP2, which affects the integrity of TJs in the canalicular membrane, as described above [77, 90]. Familial cholestasis due to inactivating mutations in the NR1H4 gene, encoding FXR, a nuclear receptor acting as a bile acid-sensor that regulates the expression of many genes involved in bile acid homeostasis, such as ABCB11 and ABCB4, has been classified as PFIC5 [120, 121]. PFIC5 is extremely rare and is characterized by neonatal cholestasis with normal GGT levels, elevated serum bilirubin, elevated serum α-fetoprotein (AFP) levels, undetectable expression of BSEP in the bile canaliculi, and vitamin K independent coagulopathy [121]. Histological liver examination revealed intralobular cholestasis with ductular reaction, hepatocellular ballooning, giant cell transformation, and fibrosis with progression to micronodular cirrhosis [122]. Additional newly identified forms of PFIC include those commented above due to mutations in USP53 (PFIC7) [77] and MYO5B (PFIC10) [78, 79].
The canalicular ABC pump MRP2 (gene ABCC2) transports a broad range of endogenous compounds into bile, including anionic conjugates, such as bilirubin glucuronides and xenobiotics. MRP2 deficiency causes Dubin-Johnson syndrome (DJS), an autosomal recessive disorder characterized by conjugated hyperbilirubinemia and chronic jaundice. Although patients with DJS do typically not present other features of hepatobiliary diseases, recent studies have reported neonatal intrahepatic cholestasis in DJS patients [123–125]. The exact mechanism by which a genetic defect in ABCC2 might lead to cholestasis remains unknown. Although MRP2 is involved in the biliary secretion of di-anionic bile acid species, under physiological conditions, these molecules constitute only a small fraction of the bile acid pool. Nevertheless, there is evidence of a link between MRP2 and cholestasis-associated events [91, 126, 127].
Experimental models to study cholestasis
In the search for a better understanding of the physiology and pathophysiology of the biliary function, several experimental models have been used over the last decades [128]. Without any doubt, the most popular and ancient one is BDL, firstly carried out by Malpighi in cats in 1687 and more recently incorporated into modern experimental physiology by Cameron and Oakley [129]. Commonly performed in rats and less frequently in mice, BDL mimics complete extrahepatic obstructive cholestasis. The Mdr2–/– mouse has often been used as an experimental model mimicking PSC. Its popularity is based on its high reproducibility and ease of handling, advantages to which must be added that it is a very well-characterized model characterized by numerous studies carried out by many different groups [130]. Other models intend to cause progressive liver damage with accompanying cholestasis by administration of either 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC), 2,4,6-trinitrobenzene sulfonic acid (TNBS) or lithocholic acid (LCA) [128]. Besides, several genetic manipulations have been carried out to generate animal models suitable to study PBC. Among these attempts, several mice models, such as NOD.c3c4, IL-2Rα–/–, dnTGFβRII, Scurfy, Ae2a,b–/–, ARE-Del–/–, and MRL/lpr have been developed. Their characteristics and advantages have been recently gathered in a helpful review on this issue [128].
Conclusions
Impaired bile flow from the canalicular lumen to the duodenum is a common feature in a large variety of hepatopathies. Cholestasis can be either the consequence of altered function and structure affecting the liver parenchyma and the biliary tree or the primary cause of several liver diseases. The development of more modern and sophisticated technologies regarding metabolomic, proteomic, and genomic analyses has permitted the identification of the etiology of cholestatic disease previously diagnosed as idiopathic. Moreover, the possibility to advance in our understanding of the pathogenic mechanism has fostered the investigation in search of more efficient and selective therapeutic strategies aimed at either correcting the underlying dysfunction, ameliorating the collateral alterations, which can prevent further life-threatening liver damage, or at least, improving the quality of life of these patients. The current understanding and the expected advances in this field permit us to be optimistic about the possibility of carrying out better personalized pharmacological or gene therapy-based treatments of cholestasis in the near future.
Abbreviations
| A1AT: | α1-antitrypsin |
| A1ATD: | α1-antitrypsin deficiency |
| ABC: | ATP-binding cassette |
| AE2: | anion exchange protein 2 |
| AIH: | autoimmune hepatitis |
| ALP: | alkaline phosphatase |
| ALT: | alanine aminotransferase |
| AMAs: | antimitochondrial antibodies |
| AST: | aspartate aminotransferase |
| BDL: | bile duct ligation |
| BRIC: | benign recurrent intrahepatic cholestasis |
| BSEP: | bile salt export pump |
| CBD: | common bile duct |
| CFTR: | cystic fibrosis transmembrane conductance regulator |
| DJS: | Dubin-Johnson syndrome |
| EPP: | erythropoietic protoporphyria |
| ER: | endoplasmic reticulum |
| FECH: | ferrochelatase |
| FIC1: | familial intrahepatic cholestasis type 1 |
| FXR: | farnesoid X receptor |
| GGT: | γ-glutamyltransferase |
| MDR1: | multidrug resistance protein 1 |
| MRP2: | multidrug resistance-associated protein 2 |
| NTCP: | Na+-taurocholate co-transporting polypeptide |
| OATP: | organic anions transporting polypeptide |
| PBC: | primary biliary cirrhosis |
| PEX: | peroxisomal biogenesis factor or peroxin |
| PFIC: | progressive familial intrahepatic cholestasis |
| PPIX: | protoporphyrin IX |
| PSC: | primary sclerosing cholangitis |
| TJPs: | tight junction proteins |
| TJs: | tight junctions |
| TPN: | total parenteral nutrition |
| UDCA: | ursodeoxycholic acid |
| USP53: | ubiquitin 53-specific peptidase |
Declarations
Author contributions
MA: writing – review & editing; SOR and AMC: writing – original draft; JJGM: supervision.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This study was funded by the CIBERehd (EHD15PI05/2016) and “Fondo de Investi-gaciones Sanitarias, Instituto de Salud Carlos III”, Spain (PI16/00598 and PI19/00819, co-funded by European Regional Development Fund/European Social Fund, “Investing in your future”); Spanish Ministry of Economy, Industry and Competitiveness (SAF2016-75197-R); “Junta de Cas-tilla y Leon” (SA063P17); AECC Scientific Foundation (2017/2020), Spain; “Proyectos de Investigación Modalidad C2”, University of Salamanca (18.K137/463AC01 and 18.K140/463AC01); “Centro Internacional sobre el Envejecimiento” (OLD-HEPAMARKER, 0348_CIE_6_E), Spain and Fundación University of Salamanca, Spain (PC-TCUE18-20_051); Fundació Marato TV3 (Ref. 201916-31). SOR was supported by a postdoctoral contract (FPU) funded by the “Junta de Castilla y Leon” (SA074P20). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2022.