Original Article
Original Article

Affiliation:
1University of Lille, Inserm, U1286 – INFINITE – Lille Inflammation Research International Center, ICPAL, 59000 Lille, France
ORCID: https://orcid.org/0000-0001-6336-3923
Affiliation:
2University of Lille, CHU Lille, CNRS, Inserm, UMR9020 – UMR1277 – CANTHER – Cancer Heterogeneity, Plasticity and Resistance to Therapies, 59000 Lille, France
3Oncowitan, Scientific Consulting Office, 59290 Lille, France
Email: christian.bailly@univ-lille.fr
ORCID: https://orcid.org/0000-0002-2973-9357
Explor Drug Sci. 2024;2:277–291 DOI: https://doi.org/10.37349/eds.2024.00047
Received: November 28, 2023 Accepted: January 24, 2024 Published: May 21, 2024
Academic Editor: Fernando Albericio, Universities of KwaZulu-Natal, South Africa; Universidad de Barcelona, Spain
Aim: New microtubule-targeting agents are needed to improve cancer treatment. The recent characterization of the anticancer alkaloid securinine as a tubulin-binding agent prompted us to explore the interaction of related monomeric and dimeric analogues with tubulin. The interaction between the α/β-tubulin dimer and alkaloids fluevirines A–F and flueggenines A–I, isolated from the bush Flueggea virosa (Roxb. ex Willd.) Royle, was investigated using molecular docking.
Methods: Two molecular models were initially compared for the binding of securinine to α/β-tubulin. The pironetin-binding site model (5FNV) was selected for the subsequent docking analysis with all compounds. Empirical energies of interaction (ΔE) were measured and compared.
Results: Fluevirine A has been identified as a potent tubulin binder. This dimeric alkaloid formed more stable complexes with tubulin than the monomeric counterparts, such as fluevirines B–D. The bis-indole derivative fluevirine E also provided more stable complexes than (nor)securinine. The study was extended to the dimeric alkaloids flueggenines A–I and three compounds were identified as potential tubulin binders: the polycyclic product flueggenine B, the norsecurinine-indole hybrid flueggenine E, and the norsecurinine dimer flueggenine I. This later compound proved to be well adapted to fit into the pironetin site of tubulin, extending its two norsecurinine units between the colchicine-binding area and the pironetin site, in close proximity to the pironetin-reactive cysteine-316 residue. Structure-binding relationships were delineated.
Conclusions: The study identifies the dimeric alkaloids fluevirine A and flueggenine I as potential α-tubulin binding agents. For the first time, dimeric alkaloids including two C-C connected norsecurinine units are characterized as tubulin ligands. The study contributes to a better understanding of the mechanism of action of Flueggea alkaloids and should help the design of anticancer analogues targeting the pironetin site of α-tubulin.
Drugs interfering with microtubule dynamics have been used to treat cancers for decades. The disruption of microtubule architecture and functions through the stabilization or destabilization of the microtubule network in cells results in mitotic arrest and then apoptotic cell death [1]. For a long time, microtubule-targeting agents (MTAs) have played a major role in the treatment of advanced cancers, notably the vinca-alkaloids such as vincristine, vinblastine, and vinflunine, and the taxanes such as paclitaxel, docetaxel, and cabazitaxel. MTAs are used to treat both solid tumors and liquid malignancies. Three main microtubule-binding sites have been defined, the vinblastine site, taxol site, or colchicine site, but there are other sites, seven in total, around the α/β-tubulin dimer either promoting microtubule stabilization or depolymerization [2]. Many of the known MTAs are natural products or derivatives such as taxol and the precursor 10-deacetyl baccatin III which are major taxanes extracted from the bark and needles (and endophytes) of the European yew tree Taxus baccata L. [3]. Beyond taxanes and vinca-alkaloids, other natural products capable of interacting with α/β-tubulin heterodimers have been identified and studied, such as the taccalonolides, combretastatins, and curcumin derivatives [4]. There is a need for new drugs that could overcome chemoresistance to taxanes, as well as safer drugs to avoid the toxicities, notably drug-induced peripheric neuropathic pain, which severely restricts clinical applications of MTAs [5–7].
Tubulin and microtubules also represent potential protein targets to treat parasitic infections, such as African trypanosomiasis [8, 9]. The microtubule cytoskeleton remains a valid target to combat certain tropical parasitic diseases but also fungal infections [10]. Moreover, microtubules are associated with diverse central nervous system (CNS) pathologies, notably neurodegenerative diseases such as Alzheimer’s disease (AD). An aberrant phosphorylation of the tau protein causes its dissociation from the microtubules and the formation of neurofibrillary tangles directly implicated in the progression of the neurodegenerative disorder [11]. For these different reasons, the search for novel MTAs remains very active using different strategies: (i) via the rational design and synthesis of new compounds, small molecules, and peptides (e.g., indazole derivatives [12], quinoxaline derivatives [13], and cyclic peptides [14]), (ii) via the repositioning of known drugs characterized as MTAs [15, 16], and (iii) the identification of novel natural products targeting tubulin and/or microtubules [17]. The search for new tubulin binders can be performed using medium/high-throughput drug screening methodologies with defined chemical libraries of natural products, synthetic compounds, and small fragments [18], and also using computational methods and platforms to guide the selection of new compounds [19, 20]. Molecular docking campaigns can be very useful to identify new tubulin-binding scaffolds [21, 22]. Recently, virtual screening procedures have contributed to the identification of new tubulin binders and active anticancer agents [23]. In this frame, our approach using molecular docking has led to the identification of new natural products, such as cryptoconcatones F and L, able to bind to the pironetin site on α-tubulin [24].
The recent discovery of the tubulin binding capacity of the alkaloid securinine (Kd = 9.7 μM) and its known anticancer and anti-metastatic properties [25, 26] prompted us to investigate further its interaction with tubulin and to explore the interaction of monomeric and dimeric derivatives with tubulin using molecular docking. We selected two series of compounds among Securinega alkaloids (Figure 1) [27]. We started with the fluevirines A–F, isolated from the white berry bush Flueggea virosa (Roxb. ex Willd.) Royle, also known as Securinega virosa (Roxb. ex Willd.) Baill. [28, 29]. The identification of the dimeric compound fluevirine A as a good tubulin binder prompted us to extend the investigation with a second series of alkaloids, the flueggenines A–I which all include an indolizidine scaffold derived from the monomeric precursor (–)-norsecurinine. The first two dimers, flueggenines A and B, were isolated in 2006 from the roots of F. virosa [30]. Flueggenines C and D were identified later from the twigs and leaves of the same plant together with other alkaloids [31, 32]. The other flueggenines were also identified in 2015 from the twigs and leaves of F. virosa [33]. The tubulin binding capacity of these 15 compounds was compared using molecular docking.
Two tridimensional structures of a drug-bound α/β-tubulin dimer were retrieved from the Protein Data Bank (PDB; www.rcsb.org) under the PDB codes 1TVK [34] and 5FNV [35]. The GOLD 5.3 software (Cambridge Crystallographic Data Centre, Cambridge, UK) was used to perform molecular docking analysis. Prior to the docking operations, the structure of each ligand was optimized using a classical Monte Carlo (MC) conformational searching procedure via the Biochemical and Organic Simulation System (BOSS) software [36]. For the ligands preparation, the 2D and 3D structures of the natural products (SDF files then converted into PDB or MOL2 files) were obtained from the PubChem compound database (https://pubchem.ncbi.nlm.nih.gov/) and from the original publications describing the compounds (Figure 1). Molecular graphics and analysis were performed using Discovery Studio Visualizer, Biovia 2020 (Dassault Systèmes BIOVIA Discovery Studio Visualizer 2020, San Diego, Dassault Systèmes).
The web server Computed Atlas of Surface Topography of proteins (CASTp) 3.0 was used to identify potential ligand-binding sites on the tubulin dimer (1TVK). CASTp is a convenient geometry-based approach to predict the position of ligand binding sites, with an estimated success rate of 74%, equivalent to that of other programs [37]. The method locates and measures pockets and voids on 3D protein structures based on the alpha shape and the pocket algorithm. It is a convenient method of reproducing the secondary structure distribution of key residues in proteins and identifying residues implicated in cavities or clefts [38, 39]. The molecular modeling software Chimera 1.15 was used for visualization [40].
The colchicine/pironetin binding area within the tubulin dimer structure (5FNV) was considered the potential binding site for the studied alkaloids. During the process, the side chains of the following amino acids within the binding site were rendered fully flexible: Phe135, Ser165, Phe169, Cys200, Phe202, Ser241, Leu242, Phe255, Cys316, Lys352. A docking grid centered in the volume defined by the central amino acid has been defined based on shape complementarity and geometry considerations. In general, up to 100 poses considered energetically reasonable are selected during the search for the correct binding mode of the ligand. The decision to select a trial pose is based on ranked poses, using the fitness scoring function [piecewise linear potential (PLP) score incorporated in GOLD 5.3] [41]. The same procedure was used to establish molecular models for all studied alkaloids.
In general, 6 poses are selected per analysis. The ranking leads to the evaluation of the empirical potential energy of the interaction (ΔE), defined using the expression ΔE(interaction) = E(complex) – [E(protein) + E(ligand)]. The SPASIBA (Spectroscopic Potential Algorithm for Simulating Biomolecular conformational Adaptability) spectroscopic force field is used to calculate the final energy. The required parameters are derived from vibrational wavenumbers obtained in the infrared and Raman spectra of a large series of compounds of diverse chemical nature (organic molecules, amino acids, saccharides, nucleic acids, and lipids). The last step corresponds to a validation using the SPASIBA force field, an essential step to determine the best protein-ligand structure. This force field has been specifically developed to provide refined empirical molecular mechanical (MM) force field parameters [42]. SPASIBA (integrated into CHARMM) empirical energies of interaction are calculated. It is an excellent system for reproducing crystal phase infrared data. SPASIBA has been specifically developed to provide refined empirical MM force field parameters, as described in other studies [43–45]. Using this specific force field for MC simulations achieves the same level of convergence as molecular dynamics (MD), with less computer time [46]. The BOSS program and the MM/generalized Born surface area (MM/GBSA) procedure were used to evaluate free energies of hydration (ΔG), in relation to aqueous solubility [47].
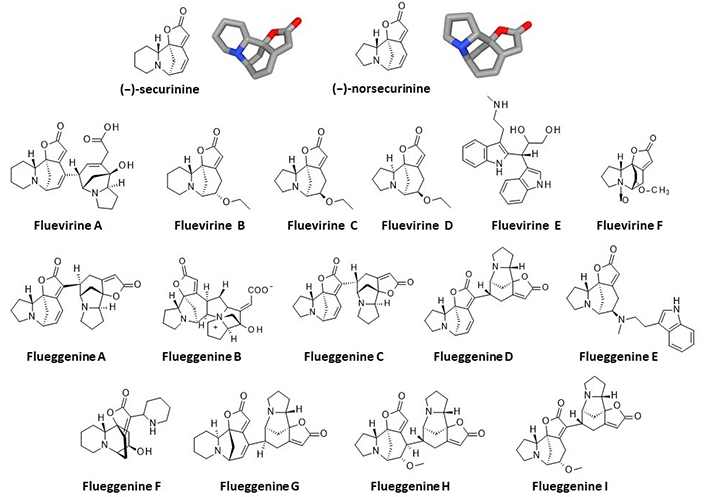
Several crystallographic structures of α/β-tubulin heterodimers are available from the PDB. In their study of securinine binding to tubulin, the authors reported a computational docking analysis of the compound bound to tubulin based on the crystallographic structure of the α/β-tubulin dimer in interaction with the drug epothilone A (PDB: 1TVK). They proposed binding of securinine at the interface of the α/β-tubulin dimer, with the alkaloid engaging a key H-bond with residue Asn228 of the α-tubulin unit [26]. We performed a similar docking analysis with the same structure (1TVK) positioning the alkaloid at the described site to allow the interaction between the compound and Asn228. In this case, the calculated empirical energy of interaction (ΔE) and free energy of hydration (ΔG) were –40.40 kcal/mol and –13.80 kcal/mol, respectively. This model is satisfactory but we consider that it presents a limitation, that is the necessity to remove the GDP molecule present in the cavity and hindering access to Asn228. The crystallographic structure includes a GDP molecule deeply inserted into the cavity and stacked over the tyrosine residue Tyr172 of α-tubulin, as represented in Figure 2. This GDP molecule precludes access to the site. Securinine can be positioned at this site, only if the GDP is removed. For this reason, we considered this binding option as non-optimal and this prompted us to search for an alternative model.
Molecular model of epothilone-bound tubulin dimer, with a close-up view of the GDP binding site (from PDB: 1TVK). The GDP molecule stacks over Tyr172. Residues Gln15 (red), Tyr172 (yellow), Asn228 (blue), and Asn247 (purple) are indicated
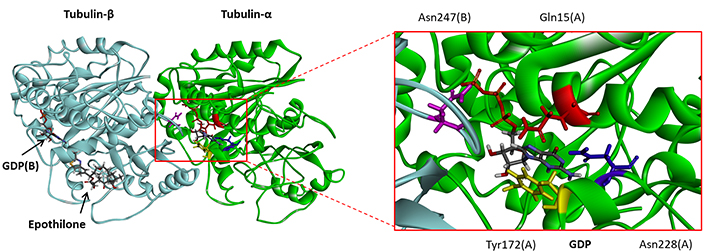
The PDB structure 5FNV provides another model for tubulin, corresponding also to a noncovalent heterodimer composed of one α- and one β-tubulin monomer. The original crystallographic structure refers to the interaction of α/β-tubulin with the α,β-unsaturated lactone derivative pironetin [35]. It is a convenient model to analyze drug binding to tubulin, notably for compounds which bind to the interface between the two facing monomers. There is a large cavity between the two monomers, accessible to small molecules. An analysis of the model with the web server CASTp 3.0 provides a clear definition of the potential binding areas (Figure 3). The main binding area is located between the two monomers, delimiting a large zone of 1,595 Å3 in which bulky molecules can bind. There are also smaller cavities on each subunit, seven in total but they are too small to accommodate dimeric compounds. Securinine can easily enter into the interfacial cavity to engage in contact with the two-facing tubulin units. In this case, the calculated empirical energy of interaction (ΔE) and free energy of hydration (ΔG) were –44.60 kcal/mol and –17.10 kcal/mol, respectively. Therefore, this model offers a better representation of the capacity of securinine to bind to the α/β-tubulin compared to the initial (1TVK) model. For this reason, we selected this second model (5FNV) for the subsequent docking analysis with all studied compounds. There are more than 20 high-resolution tubulin crystal structures available from the PDB, corresponding to the free protein or the protein bound with different types of small molecules, mostly natural products [48]. We considered the pironetin bonding site as a suitable cavity for this type of molecules, as shown recently with dihydropyrone derivatives [24]. Other drug-binding sites on tubulin may be considered as well, but we favored the pironetin site for its capacity to accommodate large, extended molecules.
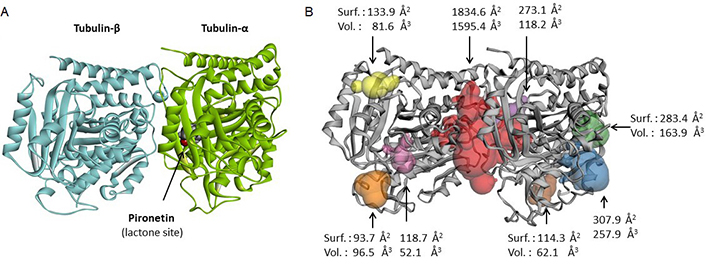
Binding site analysis of tubulin α/β using web server CASTp 3.0. (A) Molecular model of pironetin-bound tubulin dimer (from PDB: 5FNV); (B) the analysis of the tubulin dimer reveals primarily to the large interfacial area at the junction of the α/β units (in red). Small sites can be identified in the periphery of the structure, with the indicated surface (Surf.) and volume (Vol.)
Next, we performed a comparative analysis of the interaction of a dimeric alkaloid with the two tubulin models, 1TVK and 5FNV. The dimeric ligand flueggine B was selected as a test ligand. This dimeric indolizidine alkaloid has revealed potent antiproliferative activities against cultured cancer cells [48]. Molecular models of flueggine B bound to tubulin dimers were elaborated using both structures. The calculated ΔE values were –54.40 kcal/mol and –63.40 kcal/mol with structures 1TVK and 5FNV, respectively. Much more stable complexes were obtained with 5FNV compared to 1TVK. This comparative analysis reinforced our choice of structure 5FNV for the analysis of the flueggenines.
The six fluevirines were docked into the interfacial cavity, as done with securinine. Fluevirine B is a direct analogue of securinine with an additional ethoxyl substituent at position 14. Fluevirines C and D both bear the same 14-ethoxyl group in the norsecurinine series. They differ by the orientation of the hydrogen atom at position 2; fluevirine C is the β anomer whereas fluevirine D corresponds to the α anomer. The tubulin binding capacity of these three compounds is relatively modest, as observed also with the related N-oxide product fluevirine F. This latter product is not well adapted to bind to the target, with a very low hydration free energy (ΔG). The situation is significantly better with the unrelated product fluevirine E which is a totally distinct bis-indolyl compound. Its interaction with tubulin is stabilized by 22 molecular interactions with the protein dimer. The two essential contacts are an H-bond between Asn258 and an indolic NH group, and an aromatic stacking interaction between the other indole group and residue Phe255 (Figure 4). It provided a first observation that dimeric molecules were better adapted for tubulin binding than monomeric compounds. The other dimer in the series is fluevirine A with a norsecurinine-type tricyclic unit connected to a bicyclic unit equipped with an acid side chain (Figure 1). This atypical compound turned out to be a potent tubulin binder, with the implication of its two units in the binding process. There are 25 interactions between the alkaloid and the protein, with in particular two H-bonds with Lys352 (Figure 4). The free energy of interaction (ΔE) calculated with this compound is about 40% superior (more negative) than that measured with the corresponding monomer fluevirine C (Table 1). This observation prompted us to investigate other dimeric alkaloids from F. virosa.
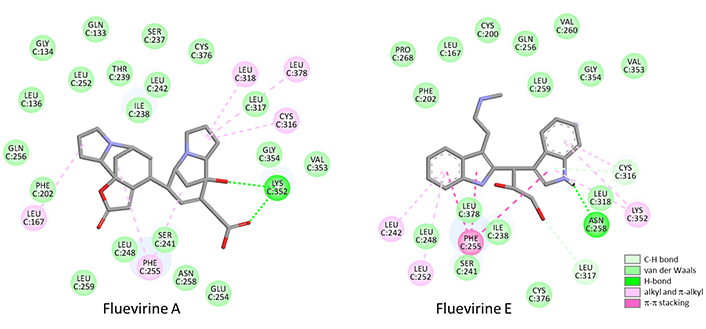
Binding map contacts for fluevirine A and fluevirine E bound to tubulin dimer (5FNV)
Calculated potential energy of interaction (ΔE) and free energy of hydration (ΔG) for the interaction of fluevirines with the tubulin dimer
| Compounds | ΔE (kcal/mol) | ΔG (kcal/mol) |
|---|---|---|
| Securinine | –44.60 | –17.10 |
| Norsecurinine | –44.20 | –17.10 |
| Fluevirine A | –73.90 | –20.90 |
| Fluevirine B | –49.30 | –20.90 |
| Fluevirine C | –52.20 | –14.90 |
| Fluevirine D | –47.70 | –16.85 |
| Fluevirine E | –56.85 | –24.50 |
| Fluevirine F | –50.20 | –12.90 |
The series of flueggenines includes 9 compounds (A–I) and each of them was docked onto the same drug binding site at the interface of the tubulin dimer. The calculated energies are collated in Table 2. Flueggenine A is a dimeric archetype consisting of two norsecurinine units. Its interaction with the tubulin dimer is reinforced compared to the norsecurinine precursor, but it is not as strong as fluevirine A. In this series, the best compound is the atypical polycyclic compound flueggenine B which seems to be well adapted to fit into the pironetin site of tubulin. The tubulin-flueggenine B complex is stabilized by a network of about 20 molecular contacts, including two essential H-bonds with residue Gln256 and Lys352 (Figure 5A, B). The hybrid compound flueggenine E which combines a norsecurinine-type unit with an aminoethyl-indole side chain provided also a good tubulin binder. In this case, the number of ligand-protein contacts was even more important (27 contacts identified). A π-π stacking interaction between the ligand indole ring and residue Phe255 can be evidenced (Figure 5C, D), as observed previously with the bis-indole compound fluevirine E (Figure 4).
Calculated potential energy of interaction (ΔE) and free energy of hydration (ΔG) for the interaction of flueggenines with the tubulin dimer
| Compounds | ΔE (kcal/mol) | ΔG (kcal/mol) |
|---|---|---|
| Flueggenine A | –61.10 | –23.95 |
| Flueggenine B | –77.10 | –18.30 |
| Flueggenine C | –64.70 | –24.90 |
| Flueggenine D | –68.05 | –28.40 |
| Flueggenine E | –72.40 | –29.75 |
| Flueggenine F | –51.35 | –20.30 |
| Flueggenine G | –65.80 | –27.20 |
| Flueggenine H | –65.30 | –19.40 |
| Flueggenine I | –76.40 | –28.00 |
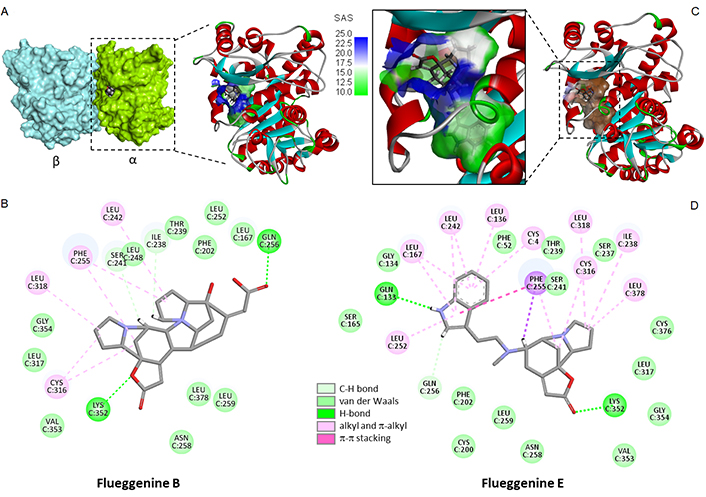
Molecular model of flueggenine bound to tubulin. (A) A surface of flueggenine B bound to the α/β-tubulin dimer and a close-up view of the ligand binding site at the protein interface; (B) the binding map contact for flueggenine B bound to α-tubulin; (C) model of flueggenine E bound to α-tubulin dimer with a close-up view of the compound inserted into the binding cavity, with the solvent-accessible surface (SAS) surrounding the drug binding zone (color code indicated); (D) binding map contact for flueggenine E bound to α-tubulin (color code indicated)
Another interesting compound in the series is flueggenine I which is also a C-C linked norsecurinine-type dimer. Its binding to the tubulin site chiefly implicates the two lactone units of the ligand and H-bonding interaction with residues Lys352 and Leu 242. The natural product showed the best comprise in terms of binding free energy (ΔE) and free energy of hydration (ΔG). Its shape is well adapted to fit into the hydrophobic pocket and here again, there are over 20 molecular contacts between the ligand and the protein (Figure 6). The molecule is relatively compact for a dimer, with a linear shape suitable for inserting between two long α-helices of the protein. Upon binding to the tubulin α-subunit, it is positioned just in front of the GDP binding site on the opposite β-tubulin subunit. Signal transmission between the drug-binding site and the nucleotide site of tubulin can occur [49]. Its molecular interaction with the protein dimer is profound and maximal, as represented in Figure 7. The other flueggenines provided fewer stable complexes than flueggenine I, our favorite ligand in the series (Table 2). The binding configuration observed with all compounds, including the reference compounds securinine and norsecurinine, is shown in Figure S1.
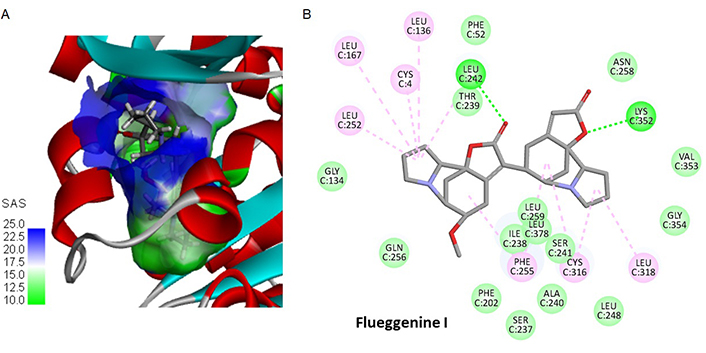
Binding of flueggenine I to tubulin. (A) A detailed view of flueggenine I bound to α-tubulin, with the solvent-accessible surface (SAS) surrounding the drug binding zone; (B) binding map contact for flueggenine I bound to α-tubulin
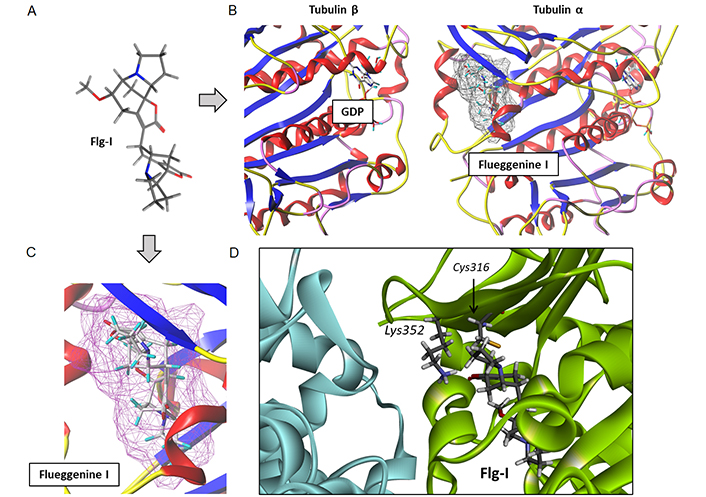
A detailed analysis of the complex between flueggenine I and the α/β-tubulin dimer. (A) Conformation of the ligand alone; (B) binding of flueggenine I to the α-tubulin subunit position the ligand in front of the GDP binding area on the opposite β-tubulin unit; (C) the deep insertion of flueggenine I in the binding cavity and (D) the close proximity between the top norsecurinine unit of flueggenine B and the key residues Lys352 and Cys316 of the pironetin site. Flg-I: flueggenine I
The search for novel MTAs remains an active strategy to try to improve the treatment of cancer and the plant kingdom is still the primary source for MTAs [50]. The great successes of the taxanes and vinca alkaloids make microtubules a durable attractive target for cancer therapeutics [51, 52]. New MTAs are regularly proposed, including synthetic molecules such as stilbene and quinolinamine derivatives [53, 54], synthetic products derived from a marine source, such as plinabulin and zampanolide derivatives [55, 56], and many others. Among the newest MTAs, a promising drug candidate is lisavanbulin as the prodrug of avanbulin, currently evaluated in patients with advanced cancers, notably those with ovarian tumour or glioblastoma [57]. In experimental models, this compound (also known as BAL101553) has shown good brain penetration and robust antitumor activity alone or in combination with radiotherapy or temozolomide-based chemotherapy [58, 59]. It binds to the colchicine site of tubulin [60], which is a conventional and very frequent site encountered with MTAs. Many natural products have been shown to bind to the colchicine site of tubulin, such as the diterpenoid laxiflorin B, combretastatin A-4, and derivatives [61, 62]. They are usually referred to as colchicine binding site inhibitors (CBSIs), considered as promising molecules to overcome multidrug resistance [13, 63, 64].
The use of molecular docking and other in silico methodologies is a convenient approach to identifying new colchicine-site binders. Computational methods are used to guide drug design or to support experimental data, notably inhibition of tubulin polymerization [65–68]. The colchicine site is convenient because it corresponds to a large hydrophobic pocket surrounded by two α-helices and by strands of the two tubulin β-sheets [69]. Its large size enables the binding of extended molecules, notably dimeric or hybrid compounds, such as indole-pyrazole hybrids recently identified as potent MTA targeting the colchicine site of tubulin [70]. Similarly, coumarin-indole, phenyl-indole, furanone-indole, and other indole derivatives with an extended conformation have been shown to bind to the colchicine site [71–75]. Indole derivatives are versatile tubulin inhibitors [76–78]. Therefore, it is not surprising to identify other molecules of this subclass, such as the bis-indole fluevirine E (Figure 4) and the indole-norsecurinine hybrid compound flueggenine E (Figure 5).
Our favorite molecule in the series is flueggenine I identified as a remarkable tubulin binder, at least based on this molecular docking analysis. We are aware that a conventional docking tool, as used here, may suffer from limited performance in terms of pose quality and binding affinity accuracy. There exist better or complementary tools to identify small molecule protein binders [79, 80], but the straightforward molecular modeling approach used here is convenient for screening a homogeneous series of molecules. The approach is useful to help prioritize the molecules to be investigated further, such as flueggenine I. This norsecurinine dimer occupies a narrow groove within the α-tubulin subunit, to establish contact with protein residues belonging to both the colchicine binding site (e.g., Leu242 and Asn258) and the pironetin binding site. In particular, the top part of the molecule is approaching the two residues Lys352 and Cys316 (Figure 7D) which are key elements for pironetin binding. Pironetin binds covalently to Cys316 of α-tubulin [81] and we have previously evidenced a similar behavior with dihydropyrone derivatives in the cryptoconcatones series [24]. Flueggenine I is not a chemically-reactive molecule, but it disposes of one of its two norsecurinine units close to the cystein residue. Its mode of binding to the pironetin site is reminiscent of that presented recently for two xanthanolides isolated from a Chinese Xanthium species. Xanthatin and 8-epi-xanthatin isomers were shown to bind indistinctly into the pironetin and colchicine binding sites [82]. At the pironetin site, the configuration proposed (from an in silico analysis) is directly reminiscent of that observed here with flueggenine I. A structural analogy can be seen between flueggenine I and these xanthanolides which are constituted of a seven-member ring fused with a five-member unit forming a bicyclic core with α-methylene-γ-lactone ring as the key active group. They can also form dimeric structures [83].
Flueggenine I is far from an ideal compound in terms of drug properties [molecular weight: 438.5 g/mol; hydrogen bonds donor: 0; hydrogen bonds acceptor: 5; calculated logP(octanol/water): 0.099; logK(HSA binding): –0.923; calculated logBB (brain/blood): 0.468]. However, the molecule provides a starting point to select other compounds for bioactivity testing, notably among Flueggea alkaloids. The identification of α-tubulin as a potential molecular target for different fluevirine and flueggenine compounds is important to understand the mechanism of action of these little-studied natural products. There are many other Flueggea alkaloids with related structures, such as the fluevirosine, fluevirosinine, flueindolines, and other indolizidine alkaloids [27]. The present study can also serve to guide the design of analogues with a reinforced affinity for tubulin. The total synthesis of flueggenines C, D, and I has been established [84–87]. Now, analogues of flueggenine I can be rationally designed based on the tubulin interaction model proposed here.
CASTp: Computed Atlas of Surface Topography of proteins
MM: molecular mechanical
MTAs: microtubule-targeting agents
PDB: Protein Data Bank
SPASIBA: Spectroscopic Potential Algorithm for Simulating Biomolecular conformational Adaptability
The supplementary material for this article is available at: https://www.explorationpub.com/uploads/Article/file/100847_sup_1.pdf.
GV: Visualization, Software, Data curation. CB: Conceptualization, Investigation, Visualization, Writing—original draft, Writing—review & editing.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
All relevant data is contained within the manuscript and the supplementary files.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 2559
Download: 26
Times Cited: 0