
Abstract
Leishmanial diseases, caused by various species of the protozoan parasite Leishmania, continue to pose a significant global health challenge. Medicinal drugs have been at the forefront of combating these diseases, offering hope for afflicted populations. This review article provides: (1) a comprehensive analysis of current knowledge and the evolving landscape of heterocyclic drug therapies for leishmanial diseases; (2) focusing on the mechanism of drug action; (3) therapeutic effects; (4) side effects; (5) potential future directions. The review begins by outlining the critical importance of heterocyclic drugs in treating leishmanial diseases. It highlights the diverse array of drugs used to combat Leishmania and elucidates the unique mechanisms underlying their efficacy. These mechanisms include disruption of cellular processes within the parasite, interference with DNA replication, and modulation of host immune responses. In addition, the article delves into the effects and side effects of drug therapy, providing an in-depth analysis of their impact on patients. It emphasizes the need for a fine balance between effective parasite clearance and minimizing adverse effects, stressing the importance of continuous research to refine drug regimens and reduce drug resistance. The review also explores various therapies for leishmanial diseases, from chemotherapy to immunotherapy, and discusses their advantages and limitations. Furthermore, it discusses ongoing research efforts aimed at developing novel drug formulations, such as liposomal and nano-carrier-based delivery systems, to enhance drug efficacy and reduce toxicity. This article crucially focuses on future perspectives in heterocyclic drug therapies for leishmanial diseases. It emphasizes the importance of interdisciplinary research and integrating emerging technologies, such as genomics and proteomics, to identify new drug targets and strategies for disease control. The potential for combination therapies and immunomodulators to improve treatment outcomes and combat drug resistance will also discussed.
Keywords
Leishmania , leishmaniasis, parasites, heterocyclic drugs, therapiesIntroduction
Leishmaniasis represents a cluster of human and animal maladies initiated by Leishmania, a protozoan parasite belonging to the Trypanosomatidae family. Over 20 Leishmania species distributed globally have been identified as the causative agents for human infection, transmitted via the bite of phlebotomine sandflies during their hematophagic feeding. Distinct Leishmania species give rise to varying clinical presentations of the disease, resulting in at least three distinct syndromes: visceral leishmaniasis (VL), commonly known as kala-azar; cutaneous leishmaniasis (CL); and mucocutaneous leishmaniasis (MCL) [1].
Leishmaniasis manifest in three primary clinical forms: VL, representing the gravest form due to its high fatality rate in the absence of treatment; CL, the most prevalent form characterized by skin ulcerations; and MCL, which affects the mucous membranes of the mouth, nose, and throat [1, 2].
Globally, leishmaniasis ranks among the top 10 neglected tropical diseases, afflicting over 12 million individuals. Leishmaniasis is prevalent in 99 countries, with CL occurring in 89 countries and VL in 80 countries. In 71 countries, both clinical forms, CL and VL, are endemic. Of the nations reporting 85% of CL cases, three are in the Americas: Brazil, Colombia, and Peru. Four countries, India, Sudan, Brazil, and Kenya, are responsible for 68% of VL cases worldwide. Leishmania-human immunodeficiency virus (HIV) co-infection is documented in 42 countries, intensifying the disease’s burden due to its complicating effects on clinical management and treatment.
Within the Americas, cases of CL have been reported spanning from the southern United States to northern Argentina, excluding the Caribbean islands, Chile, and Uruguay. CL and VL are prevalent in 21 countries, with CL being endemic in 19 and VL in 13. Between 2001 and 2021, a cumulative total of 1,105,545 cases of CL and mucosal leishmaniasis (ML) were reported to the Pan American Health Organization (PAHO), averaging 52,645 cases per year. During the same period, 69,665 new cases of VL were documented, with an annual average of 2,488 cases and a case fatality rate approaching 8%, which is considered the highest rate compared to other continents [2].
Leishmaniasis is an affliction induced by protozoan parasites, transmitted via the bites of female phlebotomine sandflies infected with the parasitic agents. The disease predominantly afflicts economically disadvantaged populations, with associations observed in instances of malnutrition, population displacement, substandard housing conditions, compromised immune systems, and limited financial resources. The global burden of leishmaniasis is substantial, with an estimated annual incidence ranging from 700,000 to 1 million new cases. It is important to note that only a minority of individuals harboring the parasites responsible for leishmaniasis will eventually manifest clinical symptoms of the disease. This is current data from WHO January 2023 report [3].
A pronounced prevalence of leishmaniasis is predominantly observed within economically disadvantaged populations residing in the most impoverished nations, where elimination initiatives are characterized by insufficient financial support, limited attention from pharmaceutical companies, and deficient healthcare infrastructure [4–6]. These variables collectively facilitate the dissemination and expansion of the disease. In a global context, leishmaniasis maintains endemicity in regions spanning Asia, the Middle East, North Africa, East Africa, the Mediterranean, South and Central America [7].
Types of diseases
Cutaneous leishmaniasis (CL).
Mucocutaneous leishmaniasis (MCL).
Visceral leishmaniasis (VL).
Cutaneous leishmaniasis (CL)
CL, the most prevalent form of the disease, is characterized by distinct parasite species, categorized into the Old World (encompassing Southern Europe, the Middle East, Asia, and Africa) represented by Leishmania tropica, Leishmania major, and Leishmania aethiopica, and the New World leishmaniasis (occurring in Latin America) with Leishmania mexicana and Leishmania braziliensis.
Typically, cutaneous manifestations of the disease manifest as skin ulcers on exposed body areas, such as the face, arms, and legs. In some cases, the disease can generate a significant +number of lesions, sometimes reaching up to 200, leading to severe disability and invariably resulting in permanent scarring. This condition may lead to pronounced social prejudice. The incubation period for this form of the disease spans approximately 2 weeks to 6 weeks.
Diffuse CL is exceptionally rare, even in regions where leishmaniasis is endemic. It is primarily induced by L. mexicana and L. aethiopica, as David and Craft [8] outline. The disease commences with the appearance of a small, painless pimple at the site of inoculation. Subsequently, it develops diffuse, non-ulcerative, erythematous violet-colored macules, nodules, and plaques densely infiltrated with amastigotes. The regions most affected by this form of leishmaniasis typically include the face, upper and lower extremities, and buttocks [8].
Mucocutaneous leishmaniasis (MCL)
MCL stands in stark contrast to CL, being a life-threatening condition that necessitates prompt intervention. This affliction is instigated by specific Leishmania species within the Viannia subgenus, namely Leishmania Viannia braziliensis, Leishmania Viannia amazonesis, Leishmania Viannia panamensis, and Leishmania Viannia guyanensis. The clinical progression of this malady is contingent upon the interplay between the host’s cell-mediated immunity and the virulence of the infecting parasite.
Patients afflicted with MCL frequently bear scars from previous episodes of CL. The initial stages of MCL commence with erythema and ulceration of the nostril. Subsequently, cartilaginous structures are relentlessly deteriorating in the facial region, upper airway, and nasopharyngeal mucosa. Secondary infections, disfigurement, and airway obstruction follow this. Diagnosis of this condition necessitates the identification of intracellular amastigotes and biopsy [9].
Visceral leishmaniasis (VL)
VL is attributed to the etiological agent Leishmania donovani. Post-kala-azar dermal leishmaniasis (PKDL) represents the dermatological sequel to VL. PKDL exhibits a spectrum of manifestations, ranging from hypopigmented macules to infiltrated papules. The development of PKDL is contingent upon various factors, including the specific Leishmania species involved and the geographical region. In some cases, PKDL lesions can serve as reservoirs for the parasites. The diagnosis primarily relies on epidemiological patterns and clinical assessments. The gold standard diagnostic methods encompass the culture of tissue samples and the examination of slit smears [9].
Morphology of Leishmania
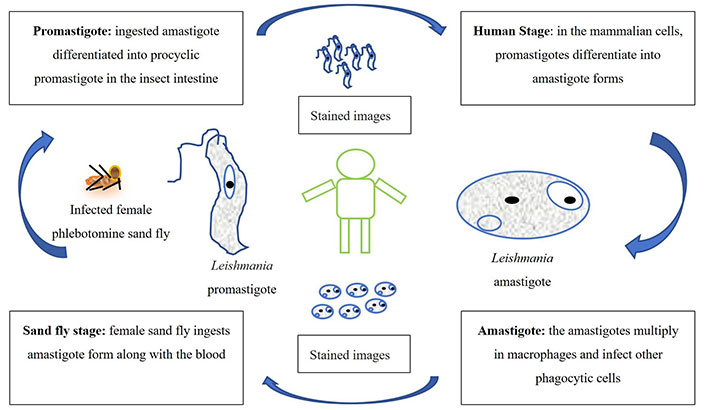
Leishmania manifests in two distinct forms: the amastigote and the promastigote. Amastigote forms predominantly reside within the vertebrate host, whereas the insect vector primarily harbors promastigote forms. During the blood-feeding process, sandflies introduce infectious promastigotes into a susceptible mammal. Once inside the host’s tissues, promastigotes are phagocytosed and transform into amastigotes, which multiply through binary fission within the host’s phagocytic cells. The parasite continues infecting additional phagocytic cells at the initial site of skin infection or within secondary lymphoid organs [10–12].
In CL cases, sandflies become infected with active skin lesions. In contrast, in VL, infection occurs through parasitemia in the host’s bloodstream due to sand fly feeding. Within the sand fly’s midgut, the parasites undergo a transformation back into promastigote forms. These promastigotes subsequently migrate from the midgut and become heavily laden with the parasite.
The life cycle of Leishmania is schematically represented in Figure 1. The amastigotes depicted in Figure 1 are typically 2–4 μm in length and exhibit an oval or round morphology. These amastigotes are usually found within monocytes, polymorphonuclear leukocytes, and endothelial cells. When stained with Giemsa, the cytoplasm appears blue, while the nucleus takes on a pink or dark red hue. The rod-shaped kinetoplast is stained in dark red, shiny red, or purple. Amastigotes are non-motile and acquire nutrients through osmosis, relying on tissue as their nutrient source. They are aerobic organisms and multiply longitudinally through binary fission within macrophages. The division begins with separating the kinetoplast and blepharoplast, followed by dividing the nucleus and cytoplasm. Notably, a large nucleus is proximal to the cytoplasm, with the kinetoplast positioned adjacent to the nucleus. Additionally, vacuoles, blepharoplast, and axonemes are present in the cytoplasm. The flagellum does not protrude freely from the cell. In Leishmania species, there is only a single mitochondrion, and a Golgi apparatus and lysosome, which facilitate parasitic nutrient acquisition through various enzymatic activities (Figure 1) [10–12].
Current therapies and field-deployable therapies
Chemotherapies
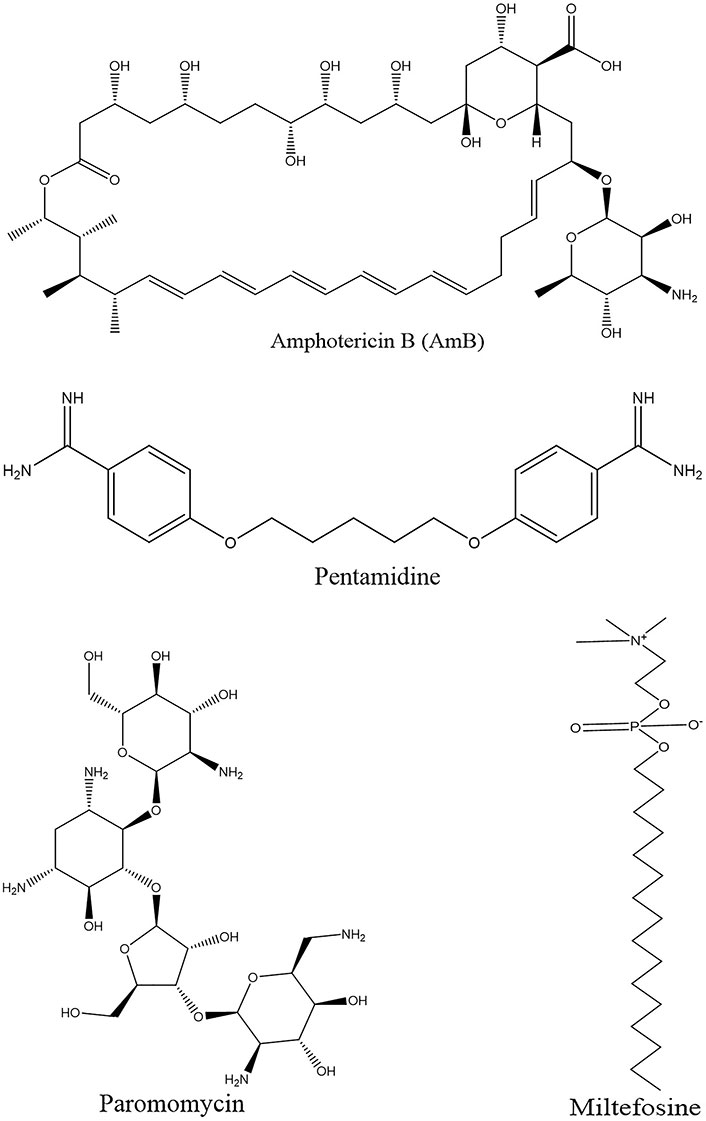
Historically, pentavalent antimony (SbV) has traditionally held the position of the primary drug treatment for leishmaniasis. However, its use is marred by a range of adverse effects, including cardiotoxicity, cirrhosis, pancreatic toxicity, and the potential for the emergence of drug resistance. Subsequently, as second-line therapies, amphotericin B (AmB, and its lipid formulations) have become prominent in the treatment arsenal [13, 14]. Miltefosine has recently been adopted to treat VL and CL. This particular drug offers the advantage of oral administration, demonstrating good efficacy and shorter treatment duration. Nonetheless, its use is constrained by concerns related to teratogenicity and the potential for the development of drug resistance.
In addition, existing drugs repurposed for leishmaniasis treatment include AmB, miltefosine, paromomycin, and pentamidine (Figure 2). Azole antifungal agents have also been investigated for their efficacy against Leishmania, with itraconazole emerging as superior to ketoconazole and fluconazole in inhibiting the growth of most Leishmania strains. A multicenter trial revealed the effectiveness of paromomycin in Indian patients with VL; however, it exhibited reduced efficacy in a Sudanese population. Pentamidine is administered intramuscularly or intravenously but lacks an oral formulation. It offers the advantage of a shorter treatment course and although its effectiveness varies across different Leishmania species, its use may be linked to dysglycemia and other adverse effects [14, 15].
Chemotherapy in leishmaniasis: current drugs, limitations, and challenges
Antimonials
SbV has been employed as a first-line chemotherapeutic approach, administered parenterally as stibogluconate, a practice established in 1945. Trivalent antimony (Sb3+), commonly administered as emetic tartar, initially served as a primary monotherapy for VL treatment, with continued application in canine leishmaniasis management. However, due to its heightened toxicity, Sb3+ was supplanted by SbV (Sb5+) compounds such as urea stibamine. Urea stibamine has since emerged as a preferred therapeutic agent for VL, demonstrating efficacy while mitigating the adverse effects associated with Sb3+, particularly in the context of the Indian subcontinent. It is important to note that SbV is a prodrug that requires conversion into its trivalent form to exert activity against the parasitic organism. This conversion is mediated through interactions between the host and intracellular amastigotes [16].
The precise mechanism of action of SbV has yet to be entirely elucidated. However, it is hypothesized that its mechanism of action encompasses a spectrum of biochemical impacts, including the inhibition of DNA topoisomerase 1, disruption of the distinctive glutathione (GSH) molecule characteristic of trypanosomatids (referred to as trypanothione), and interference with glycolytic enzymes. Selective accumulation of the drug within intracellular parasites is facilitated by modulation of the aquaglyceroporin AQP1 gene transporter, augmented production of thiols, and upregulation of ABC transporters, such as LABCI4 and MRPA [17, 18].
SbV is available in two different formulations, Glucantime® and Pentostam®, with an approximate efficacy rate of 90%. Nevertheless, the limited use of antimonials can be attributed to associated side effects and toxicity concerns, as well as the emergence of resistance and therapeutic failures in certain regions. Patients undergoing antimonial treatment commonly experience local pain at the site of intramuscular injection, along with severe side effects that may encompass cardiotoxicity, pancreatitis, hepatotoxicity, and nephrotoxicity [18–20].
Amphotericin
It is a polyene antifungal agent derived from the fermentation of Streptomyces nodosus parenthesis. It treats leishmaniasis and operates by selectively binding to ergosterol, a component found in Leishmania cell membranes [21]. AmB demonstrates efficacy against various Leishmania species and is recommended for use in pregnant women and patients coinfected with HIV. The success rate of AmB treatment exceeds 90%; however, akin to SbV, it is associated with considerable toxicity and is relatively expensive [22].
The fundamental basis for the leishmanicidal efficacy of AmB in clinical settings lies in the absence of ergosterol in mammalian cells. This condition is pivotal in preventing the development of significant drug resistance. Lipid formulations of AmB, including AmBisome®, Amphocil®, and Abelcet®, are recognized for their diminished toxicity relative to non-liposomal AmB [23].
A phase III clinical trial conducted in Bahia, Brazil, centered on disseminated leishmaniasis, an emerging manifestation of CL in the Americas. The trial demonstrated a therapeutic success rate of 75%, with doses surpassing 30 mg/kg of liposomal AmB. Administration of liposomal AmB is correlated with reduced adverse reactions and is deemed a preferable primary treatment option in Brazil [24].
In a murine model of VL, both the polymeric micelle system and AmBisome® demonstrated notable reductions in parasite burden and induced the development of a pathogen-specific Th1 immune response, all while avoiding hepatic or renal impairment. In contrast, treatment with AmB deoxycholate and Glucantime® led to significant toxicity in the infected animals. From an economic standpoint, affordable alternatives such as low-cost liposomal AmpB (Fungisome®) and other drug delivery systems like albumin microspheres, niosomes, chitosan, and nanodisks may offer sustainable solutions for regions with limited financial resources [25, 26].
Miltefosine
Miltefosine, an alkyl phosphocholine derivative, was originally identified as an antineoplastic agent for the treatment of cutaneous tumors. It induces apoptosis in tumor cells. Its mechanism of action involves perturbing cell membrane architecture by inhibiting phospholipid metabolism and affecting the synthesis of phosphatidylcholine and phosphatidylethanolamine, resulting in a reduction in intracellular choline levels [27].
The in vivo antileishmanial efficacy of miltefosine has been established, and it is acknowledged as a dependable chemotherapeutic agent for leishmaniasis, exhibiting clinical effectiveness comparable to that of AmB. Miltefosine stands out as the only orally administered medication for leishmaniasis treatment, demonstrating a remarkable efficacy rate of 95% in a clinical trial conducted in India. Its utilization is also recommended in Ethiopia and South America [28, 29].
However, a notable limitation of miltefosine administration is its extended half-life (t1/2) exceeding 120 hours, and its teratogenic potential. This prolonged half-life poses a constraint, particularly for women of reproductive age, potentially restricting its usage during fertile periods. Additionally, the efficacy of miltefosine in murine models varies depending on the Leishmania species under treatment. For instance, susceptible BALB/c mice infected with L. braziliensis and L. amazonensis have demonstrated disease recurrence, implying the drug’s variable effectiveness across different forms of leishmaniasis, possibly necessitating higher doses for treatment [30, 31].
Since miltefosine is orally administered, the potential for the development of drug resistance is elevated, particularly in regions where self-medication practices without prescriptions are common, such as in India. Indeed, the emergence of miltefosine resistance has been demonstrated under laboratory conditions with L. donovani, and instances of a loss of clinical efficacy have been reported and validated in laboratory experiments [32].
Pentamidine
Pentamidine is administered via intravenous or intramuscular routes in patients unresponsive to SbV therapy. Its use in the context of VL has been associated with significant toxicity, including cardiotoxicity, hypotension, and the development of irreversible insulin-dependent diabetes mellitus. The drug exerts its action by binding to kinetoplastid DNA upon entry into the cell through arginine and polyamine transporters. Moreover, resistance to pentamidine has been reported, with such resistance being linked to the upregulation of drug efflux mechanisms, resulting in reduced intracellular and mitochondrial levels of aromatic diamines [33].
Paromomycin
Paromomycin, monomycin, and aminosidine are aminoglycoside antibiotics derived from Streptomyces krestomuceticus. Its mechanism of action involves interference with protein synthesis within the ribosome containing 16S ribosomal RNA of the target organism and inhibition of respiration. Paromomycin demonstrates efficacy against various protozoan parasites, including Entamoeba and Giardia, and has also exhibited leishmanicidal activity [34].
A phase III clinical trial with paromomycin, administered at a dose of 15 mg/kg (equivalent to 11 mg base) for 21 days, resulted in a cure rate of 95%, leading to its approval for the treatment of VL in India in 2006. However, subsequent reports indicated the drug’s ineffectiveness in treating PKDL [34, 35].
The utility of paromomycin as a standalone therapy is impeded by the development of resistance, not withstanding its cost-effectiveness and minimal toxicity profile. Present chemotherapy modalities for leishmaniasis face multiple challenges, encompassing elevated expenses, toxicities, the emergence of drug resistance, routes of administration, treatment duration, and instances of clinical ineffectiveness (Figure 2) [36].
The emergence of substantial drug resistance against agents such as AmB and miltefosine has intensified, particularly in endemic regions experiencing outbreaks. While these medications have demonstrated clinical efficacy in achieving a cure, complete parasitological eradication still needs to be consistently attained, indicating ongoing disease persistence within the population. This occurrence has been documented in cutaneous and visceral Leishmania infections. It is likely linked to the compromised immune status of affected individuals, thereby contributing to relapses, notably prevalent in endemic regions such as India and elsewhere. This highlights the compromised immune response consequent to parasite infection and suggests that current therapeutic agents may inadequately induce a durable memory response for disease eradication [37, 38].
Drug resistance and significance of combination therapy
Drug resistance is a phenomenon characterized by reduced or ineffective drug efficacy, even in the absence of prior drug exposure. Reports from the Indian subcontinent indicate that parasite resistance mainly originates in areas with anthroponotic transmission. The emergence of unplanned urbanization in these regions has led to developing drug-resistant parasites. Coinfection with HIV in leishmaniasis results in poor treatment outcomes, increased relapse rates, and the potential emergence of drug resistance [39, 40].
The implications of drug resistance on treatment efficacy are stark. Sodium stibogluconate, meglumine antimoniate, and generic formulations, long-standing standard treatments for VL, were rendered obsolete in India and Nepal by 1995 due to their pronounced therapeutic inefficacy. Clinical isolates of L. donovani from endemic areas exhibited approximately threefold lower susceptibility in vitro compared to isolates from treatment-responsive patients [40, 41].
Drug resistance in Leishmania stems from diminished drug levels within the parasite, facilitated by the aquaglyceroporin AQP1 (the principal channel for antimony uptake), or escalated drug efflux facilitated by the ABC transporter ABCC3 (MRPA). Antimony-resistant parasites demonstrate heightened concentrations of thiols, including cysteine, trypanothione, and GSH, attributable to gene overexpression or amplification involved in their biosynthesis, cysteine, trypanothione, and GSH are components of trypanothione, the main intracellular thiol in Leishmania. Antimony resistance can also occur due to the inhibition of drug reduction or inactivation of the active drug [42].
AmB, another treatment, affects the parasite’s membrane sterol, ergosterol. Resistance to AmB has been reported in 20% of Indian patients who were prescribed the drug for VL patients refractory to antimonials. Gene amplification in Leishmania alters drug binding affinity to the plasma membrane by modifying sterol composition [43].
Miltefosine-treated parasites exhibit a notable decline in mitochondrial membrane potential and cytochrome c oxidase activity. Miltefosine binds to the plasma membrane and is internalized through the endocytic pathway via flippase activity mediated by the miltefosine transporter (MT) and its noncatalytic subunit Ros3 miltefosine can be excluded through exocytosis or flippase activity facilitated by the ABC transporter subfamilies ABCB and ABCG. In vitro, susceptibility to miltefosine varies among species and clinical isolates of the pathogen, likely due to variations in substrate specificity, the activity of the MT-Ros3 machinery, cell proliferation rate, biochemical targets, drug metabolism, and plasma membrane composition [44]. Recent findings suggest that the disparity between in vitro susceptibility to miltefosine and clinical effectiveness indicates a need for more correlation. In vitro, resistance to miltefosine can arise through mechanisms involving escalated drug exposure or chemical mutagenesis, potentially linked with impairments in drug internalization regulated by the MT-Ros3 axis. Upon exposure to miltefosine, mutations occur within the MT and Ros3 genes, with a higher mutation frequency observed in MT genes. Inactivation of MT leads to the development of a resistant phenotype in animal models of both VL and CL, highlighting the critical role of MT activity for the efficacy of miltefosine in vivo [44, 45].
Pentamidine, employed in the treatment of CL, has exhibited resistance mechanisms entailing modifications in intracellular arginine and polyamine levels, diminished pentamidine accumulation within mitochondria, and heightened drug efflux, potentially mediated by the ABC transporter PRP1 [46, 47]. Functional cloning utilizing Cos-Seq uncovered a putative protein that mitigates pentamidine resistance in promastigotes. Resistance to paromomycin correlates with modifications in membrane fluidity, lipid metabolism, and mitochondrial function. An identified paromomycin resistance gene encodes a hypothetical protein featuring leucine-rich repeats, imparting resistance to pentamidine. Paromomycin susceptibility demonstrates notable variability among clinical isolates of distinct parasite species [47].
Treatment failure in leishmaniasis constitutes a multifaceted challenge influenced by factors such as improper drug administration, variations in parasite susceptibility among patients, and the continuous emergence of novel isolates. Pharmacokinetics and the individual immune response to specific medications also significantly contribute to this phenomenon. Additionally, the presence of Leishmania RNA virus 1 (LRV-1) within the Viannia subgenus poses an additional obstacle by modifying the host immune response, thereby affecting the efficacy of drug interventions [48–50].
Given the steady emergence of resistance to all antileishmanial drugs, a consensus on adopting combination therapy has been considered the preferred treatment option against fatal VL and other forms of leishmaniasis. Combination therapy aims to reduce the individual drug dose and treatment duration, improve compliance, and minimize toxicity [50, 51]. Different combination regimens, including SbV plus paromomycin and AmB plus miltefosine, have been tested, showing high cure rates and advantages such as reduced treatment cost, shorter therapy duration, lower toxicity, and limited development of drug-resistant pathogens.
Patients with VL and HIV coinfection, who experience comorbidities and high fatality rates, have been treated successfully with combination therapies, such as liposomal AmB plus miltefosine, leading to lower relapse rates compared to monotherapy with liposomal AmB [52, 53].
Combination chemotherapy
To mitigate the emergence of drug resistance, enhance patient adherence to treatment, abbreviate the treatment duration, and consequently lower the overall cost of therapy, combination chemotherapy regimens have been devised [53, 54]. These regimens encompass a range of combinations, such as liposomal AmB with miltefosine, liposomal AmB with paromomycin, miltefosine combined with paromomycin, and sodium stibogluconate/meglumine antimoniate in conjunction with paromomycin [54].
Thermotherapies and cryotherapies
Various forms of thermotherapies and cryotherapies are employed to treat leishmaniasis.
Thermotherapy
Thermotherapies encompass various techniques employing battery-operated devices to administer heat infrared light, lasers, hot water immersions, or radiofrequency (RF) directly to lesions, thereby effectively inhibiting parasite proliferation and eliminating infections. Carbon dioxide lasers present an alternative method of heat therapy applied directly to the infection site, requiring minimal treatment sessions and demonstrating cost-effectiveness in field settings [55]. These thermotherapeutic approaches are characterized by their favorable safety profiles, minimal off-target effects, and applicability to patients with contraindications to conventional treatments, as well as to regions with limited healthcare infrastructure. The efficacy of thermotherapies varies depending on parasite species and lesion size. While they have shown safety and efficacy, combination therapies with chemotherapeutic agents may still be advisable, particularly for patients unable to receive standard chemotherapy due to factors such as pregnancy or immunosuppression [55, 56].
The Leishmania species responsible for cutaneous diseases are susceptible to heat and cannot thrive or survive at temperatures exceeding 39°C. Consequently, thermotherapy has been explored as a potential treatment option for CL lesions. One form of thermotherapy, RF therapy, has been applied to CL patients. A study conducted in Guatemala reported a cure rate of 73% for CL patients treated with RF therapy, which matched the rates achieved with systemic SbV drugs [57]. In RF therapy, heat is evenly delivered to a depth of 4 mm, effectively heating and eliminating the amastigote forms of Leishmania residing in the upper dermal layers while preserving the surrounding skin. Although two randomized studies revealed that RF thermotherapy had a lower cure rate than systemic SbV drugs, it proved to be cost-effective and associated with fewer adverse events. Administering thermotherapy once every three weeks resulted in a cure rate of 73% while increasing the frequency to once weekly elevated the cure rate to 81% [58, 59].
Cryotherapy
Cryotherapy is also utilized to treat CL lesions. It involves the direct application of liquid nitrogen or, in resource-constrained settings, carbon dioxide solids at temperatures lethal to the parasites. Cryotherapy is safe and effective with few side effects, but cure rates vary due to factors including parasite species and lesion size. Although not always capable of achieving sterile immunity or complete parasite clearance. These methods are particularly beneficial for patients who cannot undergo conventional chemotherapies, such as pregnant individuals or those with compromised immune systems [60].
Cryotherapy induces damage to infected cells and amastigotes through the formation of intracellular and extracellular ice crystals, and alterations in cell membranes, all of which occur at temperatures below freezing. This cryonecrosis process leads to the release of antigenic substances that stimulate an immune response, ultimately promoting the healing of other lesions [61]. Cryotherapy viable option for treating CL, particularly in light of the various drawbacks associated with chemotherapy. The treatment has demonstrated excellent efficacy in patients with skin lesions ranging from 10 mm to 30 mm in size, those with fewer lesions, and individuals who have had the condition for less than three months [62, 63].
When combined with intralesional sodium stibogluconate, cryotherapy has proven to be highly effective, resulting in the complete healing of CL lesions in 100% of cases. In another study, a combination treatment approach involving itraconazole and cryotherapy led to an 80.9% improvement in CL lesions, and reduced the risk of liver toxicity, as the dosage of itraconazole could be lowered [64, 65].
In summary, the current treatments for leishmaniasis do not consistently result in sterile immunity or complete parasite clearance. Achieving these outcomes is crucial because latent infections can lead to later reactivation and additional clinical complications, as observed in cases like PKDL and MCL [66, 67]. Such cases contribute to ongoing parasite transmission, threatening leishmaniasis elimination efforts in many regions. Additionally, the persistence of parasites following treatment increases the risk of transmission through other means, such as blood transfusions and organ transplants [68]. Therefore, it is essential to address the barriers to achieving sterile immunity and understand the mechanisms by which Leishmania can evade current treatments when considering future directions in leishmaniasis therapies [69].
Structure- and ligand-based drug design: antileishmanial drug discovery
Structure-based drug design (SBDD)
Applying combinatorial chemistry and high-throughput screening (HTS) in drug discovery emphasizes the importance of in-silico or computational methods in identifying novel medications. These methods utilize protein structures obtained through techniques like NMR and X-ray crystallography. Computational tools are used to model proteins and perform virtual screening of inhibitor molecules. The passage also mentions the significance of SBDD in improving drug discovery, optimizing treatment efficacy, avoiding drug resistance, and minimizing toxicity. It briefly touches on the use of SBDD in the context of trypanosomatids and Leishmania, specifically focusing on a pyrazolopyrimidine-class medication targeting Leishmania cyclin-dependent kinase 12 (CDK12) [70].
Lead compounds from this series exhibited efficacy in a mouse infection model when administered orally at 25 mg/kg twice daily for ten days. This treatment demonstrated comparable effectiveness to the widely-used drug miltefosine, leading to a remarkable 99% reduction in parasite levels. In a separate study, a compound known as LASSBio-1386, classified as an N-acylhydrazone derivative, was effective against L. amazonensis promastigotes. Importantly, it exhibited low toxicity in macrophages, indicating its safety. In vitro experiments with LASSBio-1386 resulted in reduced infection rates of Leishmania within macrophages and a decrease in the number of intracellular parasites. Furthermore, in vivo, testing on BALB/c mice infected with L. amazonensis revealed reductions in lesion size, parasite load, and improvements in histological features compared to control groups [71, 72].
Molecular dynamics and docking studies were conducted to explore potential molecular interactions. These investigations focused on Leishmania’s phosphodiesterase (PDE) B1 (PDB code: 2R8Q) and LASSBio-1386. Computational research suggested that LASSBio-1386 may exert its action against Leishmania by altering leishmanial PDE activity.
Ongoing computational analyses dedicated to discovering and proposing new chemotherapy drugs are a testament to indicating the immense potential of computational methods in expediting the development of anti-trypanosomatid drugs. This approach holds significant promise and has the potential to revolutionize pharmaceutical industry, enhancing performance across diverse therapeutic areas, particularly in managing complex diseases [73].
The incorporation of computational methodologies stands as a pivotal advancement in the pursuit of novel therapeutics. Chemoinformatics tools are categorized within its sphere into SBDD and ligand-based drug design (LBDD) strategies [74, 75]. SBDD utilizes the three-dimensional coordinates of molecular targets to explore and refine interactions between ligands and receptors. This strategy depends on X-ray crystallography to elucidate the three-dimensional structure of diverse drug targets. Molecular docking, including structure-based virtual screening (SBVS), evaluates prospective ligands according to their binding mode and energetic attributes. Structure-activity relationships (SAR) derived from these analyses contribute to improving the affinity and other properties of receptor-ligand interactions [76].
The exploration of numerous macromolecular targets within Leishmania for drug discovery is a significant and engaging area of research. These targets span pathways and biomolecules that are pivotal for parasite survival and physiological functions during host-parasite interactions. Notable targets include pteridine reductase 1 (PTR1), responsible for the pteridine salvage pathway and folate metabolism [76]. Cysteine proteases (CPs), specifically cathepsin-L-like endopeptidase CPB2.8, constitute another key focus. A novel quinalidine derivative, targeting the mitochondrial enzyme NADH dehydrogenase (NDH2), has demonstrated potential as a candidate.
In-depth analyses have utilized SBVS involving 53 leishmanial proteins and molecular dynamics simulations. Notably, L. major dihydroorotate dehydrogenase (LmDHODH) emerged as a promising target [77, 78]. Several LmDHODH inhibitors displayed in vitro leishmanicidal activity against L. panamensis intracellular amastigotes, paralleling the effects of the reference drug AmB. Importantly, these inhibitors exhibited limited toxicity toward human macrophages, underscoring their potential for further development and exploration in animal model studies.
Topoisomerase 1 derived from L. donovani, denoted as L. donovani topoisomerase 1 (LdTop1) represents an additional molecular target in the context of SBDD [78]. LdTop1 plays a pivotal role in the introduction of single-strand breaks in DNA, facilitating alterations in DNA topology, which are indispensable for fundamental cellular processes like gene replication and transcription [79]. Through a combination of scaffold hopping and bioisosteric modifications, a series of inhibitors specific to LdTop1 were identified. Camptothecin and edotecarin, established inhibitors of topoisomerase 1, served as the reference starting points for molecular design. Using molecular docking studies based on the crystal structures of both LdTop1 and the human ortholog, six compounds were meticulously chosen for their compatibility with LdTop1. Subsequent evaluation demonstrated their leishmanicidal activity against L. donovani promastigotes while exhibiting no toxicity towards mammalian cells. Furthermore, molecular docking analysis predicted the structure of a ternary complex involving LdTop1 and DNA, offering insights into the critical structural features of the novel analogs responsible for their leishmanicidal properties without adversely affecting host cell cytotoxicity.
Concurrently, SBDD additionally recognized tryparedoxin peroxidase from Leishmania as a promising molecular target, owing to its function in reducing hydroperoxides generated by infected macrophages, crucial for parasite viability [80]. Molecular docking analysis, employing the X-ray structure of the enzyme from L. major, denoted as L. major tryparedoxin peroxidase (LmTXNPx), informed the selection and design of a series of N,N-disubstituted 3-aminomethyl quinolones exhibiting leishmanicidal characteristics. These compounds hold promise as potential drug candidates for combating leishmaniasis.
Ligand-based drug design
When access to the X-ray three-dimensional structure of the target receptor is limited, an LBDD approach is employed to forecast potential drug candidates. This methodology relies on the utilization of information about the structure, molecular properties, and biological activity of small molecules. LBDD involves the development of chemometric models that establish correlations between molecular characteristics, also known as molecular descriptors and relevant pharmacokinetic and pharmacodynamic parameters associated with the target of interest. This process entails the derivation of quantitative SAR (QSAR) and quantitative structure-property relationships (QSPR), aiming to identify molecular features closely linked to the desired target properties [81].
The LBDD protocol, when integrated with SBDD techniques, has previously been documented in the realm of drug discovery for leishmaniasis. Within the LBDD framework, QSAR and QSPR models are harnessed to make predictions concerning the activity and ADMET (absorption, distribution, metabolism, excretion, and toxicity) parameters of potential drug candidates. Additionally, ligand-based virtual screening (LBVS) techniques are applied to search for novel compounds that align with the desired pharmacological properties [82].
Therapeutic vaccines and immunotherapies
Therapeutic vaccines represent a promising post-exposure treatment option for Leishmania infections. These vaccines are designed to enhance the host’s immune response, aiming to reduce the burden of parasites and resolve established diseases. Consequently, therapeutic vaccines offer a promising avenue for immunotherapy against leishmaniasis. Historically, clinical trials have shown that therapeutic vaccines can achieve higher cure rates for CL compared to chemotherapies while offering advantages in terms of treatment duration, overall outcomes, and limited adverse effects, with most side effects localized to the lesion site. Immunotherapy has proven effective in trials targeting PKDL, a challenging condition to treat with traditional chemotherapies. However, one limitation of therapeutic vaccines in field-related settings is the requirement for booster doses or multiple administrations [83].
In addition to therapeutic vaccines, immunomodulators such as toll-like receptor (TLR) agonists and cytokines have been experimentally evaluated against leishmaniasis. These agents have been administered both individually and in combination with other drugs. Nonetheless, further investigation is needed to explore their efficacy, and cost considerations remain a concern.
Currently, first- and second-generation therapeutic vaccines are undergoing clinical trials. Although they show promise, there is room for improvement, and they are likely to become an essential tool in the future of field-deployable leishmaniasis treatments [83, 84].
Immunotherapy is a fundamental concept that involves modulating the immune response using biological substances or molecules for prophylactic and therapeutic purposes. This approach enhances the host’s natural defenses, restores impaired immune functions, and mitigates excessive host responses, directly or indirectly.
Immunotherapy in leishmaniasis encompasses various interventions, including vaccines, interferons (IFNs), and protein immunomodulators, used individually or in combination. Vaccines which come in different forms, ranging from whole-killed parasites to recombinant proteins produced via genetic engineering techniques are the key area of ongoing research. The development of third-generation vaccines is a testament to the dynamic nature of this field.
IFNs represent naturally occurring cytokines that can be produced using recombinant DNA technology. They have various biological functions, including immunosuppressive actions. For instance, IFN-c can stimulate macrophages to eliminate intracellular Leishmania parasites. IFN-c-1b protein is administered alongside sodium antimony gluconate and has demonstrated good tolerance and efficacy, particularly in patients with VL resistant to SbV therapy. When used in untreated cases of VL, IFN-c has been shown to accelerate parasitological control and enhance the clinical efficacy of SbV treatment. Combining IFN-c with SbV therapy can expedite the elimination of parasites in patients with VL [84].
Immunomodulators include the protein aggregate magnesium-ammonium phospholinoleate-palmitoleate anhydride, which has been found to improve clinical symptoms in canine VL and significantly reduce parasite levels in the skin.
Furthermore, combination therapy, a treatment approach that involves immunotherapeutic and chemotherapeutic agents holds significant potential. This approach synergistically activates the immune system directly acting on the infectious agent.
Leishmanization (LZ) vaccine
LZ is a historical immunization method that references its use in vaccination. LZ involves the intradermal administration of a low dose of live and virulent L. major parasites, leading to developing a single skin lesion [84]. Remarkably, LZ has demonstrated a high level of efficacy, providing protection exceeding 90% against subsequent reinfection. This approach was previously employed in various Middle Eastern countries and the former Soviet Union. However, except in Uzbekistan, an endemic region where LZ continues to be practiced, its use has waned due to various safety concerns. These concerns include the potential for HIV transmission, the use of immunosuppressive medications, ethical considerations, the unpredictability of persistent skin lesions, and the persistence of parasites (Table 1 and 2) [84, 85]. The overarching objective in the field is to develop a vaccine with enhanced safety profiles capable of inducing protective immunity without the manifestation of skin lesions, even in individuals with compromised immune systems. Notably, vaccination using live parasites triggers a more robust Th1-type immune response compared to vaccination with inactivated parasites, which provides only limited protection. However, the precise mechanisms underlying these differences remain poorly elucidated [86].
| Types of vaccines | Contents |
|---|---|
| Leishmanization | Live and virulent L. major or L. tropica |
| Killed vaccines | Killed Leishmania species |
| Live genetically modified vaccines (avirulent) | BT1 (biopterin transporter 1) |
| Ldp27 (L. donovani amastigote specific protein p27) | |
| HSP70-II nul (heat-schok protein) | |
| KHARON1 | |
| Cen (centrin) | |
| Recombinant and subunit vaccines | Recombinant LEISH-F1, LIESH-F2 and LEISH-F3 |
| LdA2, Ldp27, eIF-2, NH (nucleoside hydrolase), CPA (cysteine protease A), CPB, SMT (sterol 24-c-methyltransferase), H1 (histone-1), HSP, LACK | |
| Sand fly saliva antigens: LJM19, LJL143, and PdSP15 | |
| DNA vaccines | A2, LACK, TSA + LmSTI1, gp63 (glycoprotein 63), KMP-11 (kinetoplastid membrane protein 11), CPB, NH36, LeIF, gp63 + HS |
| Semian adenovirus expressing NH and SMT | |
| Live non-pathogenic vaccines | Avirulent Leishmania tarentolae |
| Name of vaccine | Essential component/antigen | Adjuvant and their mechanism of action |
|---|---|---|
| Leishvaccine | Whole-killed promastigotes of L. amazonensis | BCG (Bacillus Calmette Guerin); CD4+, CD8+, B cell activation |
| ALMϼ (autoclaved-killed L. major) | Autoclave-killed L. major | BCG; T cell activation |
| Leishmune | FML (fucose-mannose ligand) | Saponin; T cell activation |
| CaniLeish | LiESP (L. infantum excreted-secreted protein) | Saponin; induction of Th1 cell |
| GALMα (gentamycin-attenuated L. major) | GALM | T and B cell activation |
| LEISH-F1 | TSA + LmSTI1 + LeIF | MPL-SE (monophosphoryl lipid A-stable emulsion); T cell activation |
| LEISH-F2 | LEISH-F2, designed from LEISH-F1 | MPL-SE; T cell activation |
| LEISH-F3 | NH + SMT (nucleoside hydrolase; sterol 24-c-methyltransferase) | GLA (glucopyranosyl lipid A)-SE; T cell activation |
| Leish-Tec | L. donovani A2 protein | Saponin; T cell activation |
| SMTγ + NHμ | NH + SMT | GLA-SE; T cell activation |
| ChAd63-KH | KMP-11 + HASPB (kinetoplastid membrane protein 11; hydrophilic acylated surface protein B) | Broad CD8+ T cell activation |
Furthermore, a study carried out in Iran underscored the profound impact of LZ, showing a significant decrease in disease incidence. In a hyper-endemic region of the country, the disease incidence was reduced to between 1/6 and 1/8 of its original levels. This compelling outcome led to the recommendation of LZ for individuals at high risk of contracting the disease.
Saliva vaccine
Sand fly vectors, specifically those belonging to the Phlebotomus and Lutzomyia genera, are of significant scientific interest, as they induce immune responses that function as adjuvants through the action of specific immunogenic proteins. Examples of such proteins include LJM19 and LJL143, derived from Lutzomyia longipalpis, and PdSP15 from Phlebotomus duboscqui. Typically, sand fly saliva is recognized for its capacity to enhance Leishmania spp. infections [99]. However, prior exposure to sand fly saliva has been observed to provide protective effects against parasitic infections. Carregaro et al. [100] conducted a study on mice, demonstrating that administering sand fly salivary gland extract resulted in increased production of IFN-γ and IL-12 at the site of inflammation, and pre-exposure to sand fly saliva effectively shielded mice from parasitic infections. The authors concluded that developing novel saliva-based vaccine strategies could play a pivotal role in preventing the establishment of Leishmania within the host. Immunization with a vehicle that expresses salivary proteins from the sand fly vector, such as PpSP15, using carriers like Lactococcus lactis and Leishmania tarentolae, has been shown to confer protection against CL or VL [89, 99].
First-generation (whole-killed or fractioned parasite) vaccines
First-generation vaccines were developed utilizing whole-killed parasites, partially purified fractions, or excreted products derived from the parasite. However, these early vaccines have predominantly been supplanted by LZ. The inactivation of these parasites was accomplished through diverse methods, including prolonged in vitro culture, exposure to temperature, pressure, γ radiation, and chemical treatments. While killed parasite vaccines encompass a heterogeneous array of parasite antigens and can confer substantial protection against infection by simulating natural infection processes, investigations have demonstrated that they often induce a weaker Th1-type immune response in comparison to live parasites, with results frequently exhibiting inconsistency. This complexity underscores the need for further research and development. To address these shortcomings, adjuvants such as Bacillus Calmette Guerin (BCG) have been integrated into numerous studies, and alternative routes of administration, including mucosal routes, have been explored [100, 101].
Whole-killed vaccines, typified by the Leishvaccine, consist of whole-killed promastigotes derived from the L. amazonensis strain (IFLA/BR/1967/PH8) in conjunction with BCG. These vaccines have exhibited notable efficacy in combating canine leishmaniasis, demonstrating augmentation of cytokine expression, innate immunity, and adaptive immune responses [102]. Clinical trials conducted during phases I and II to evaluate the suitability of the Leishvaccine for human use have substantiated its safety and immunogenicity. However, it did not achieve the requisite efficacy threshold during phase III trials. Similarly, the administration of autoclaved-killed L. mexicana, in combination with BCG as an adjuvant, resulted in comparatively modest levels of leishmanin skin test (LST) conversion, indicative of cellular immune response activation. Nevertheless, individuals who did exhibit LST conversion experienced a noteworthy reduction in leishmaniasis incidence [103].
Similarly, autoclaved-killed L. major (ALM), representing the old-world species, when administered alongside BCG, elicited LST conversion in approximately one-third of healthy participants. Again, a notable decrease in leishmaniasis incidence was observed among those individuals who exhibited LST conversion [103].
Second-generation (subunit or genetically modified parasite) vaccines
While first-generation vaccines continue to be evaluated, recent research has prominently focused on developing second-generation vaccines. Second-generation vaccines encompass various candidates, including recombinant proteins produced through genetic engineering in cells like viruses and bacteria, purified fractions of native parasite antigens, synthetic peptides, and even genetically modified parasites. These second-generation vaccines offer several advantages, including enhanced feasibility for mass vaccination and their recombinant nature, which facilitates large-scale and cost-effective production [104].
Of particular interest are subunit vaccines, which involve the utilization of various subunits or recombinant candidates. Notable examples include LeIF, glycoprotein 63 (gp63), p36/LACK, A2, PSA-2/gp46/M-2, fucose-mannose ligand (FML), LCR1, ORFF, kinetoplastid membrane protein 11 (KMP-11), LmSTI1, TSA, hydrophilic acylated surface protein B1 (HASPB1), protein Q, CPB, and CPA [104, 105]. Many of these subunit vaccine candidates have demonstrated the ability to stimulate an effective protective immune response against Leishmania infection. One key advantage of subunit vaccines is their inherent safety, as they do not pose any risk of causing infection, making them suitable for immunocompromised individuals [105]. For instance, using a surface-expressed gp (gp63), a subunit protein, encapsulated within cationic liposomes, has been observed to augment the generation of IFN-γ-producing effector T cells. Furthermore, vaccination utilizing gp63 DNA has been demonstrated to induce robust immune responses and provide protective immunity. In several experiments evaluating their immunoprotective effects, viruses engineered to express the LACK (Leishmania homolog for receptors of activated C kinase) antigen, with or without adjuvants, exhibited substantial protective effects against Leishmania infection [106, 107]. Similarly, a virus expressing the promastigote protein surface of G46/M-2/PSA-2 proved efficacious in safeguarding against L. amazonensis infection [107].
Genetically modified parasite vaccines
Within Leishmania vaccine research, the development of live attenuated vaccines through genetic modifications is a prominent area of investigation. In this approach, the genes within the parasite responsible for its survival and/or virulence are subjected to modifications or deletions. Unlike their virulent counterparts, these genetically engineered parasites do not pose any risk of causing infection. However, they are designed to consistently elicit immune responses that closely emulate the protection achieved through natural infection processes [108].
Several live attenuated vaccine strains of various Leishmania species, such as L. major, L. mexicana, L. amazonensis, and L. donovani, have been developed through targeted modification or deletion of specific genes, these genetically engineered strains have demonstrated substantial efficacy in protecting both in susceptible mouse models CL and VL. Notable examples include the deletion of the biopterin transporter 1 (BT1) gene in L. donovani parasites, the A2-rel gene cluster in L. donovani, as well as the generation of null mutants for SIR2, HSP70-II, and KH1 in Leishmania infantum, all of which effectively conferred protection against virulent strains in mice [108]. Additionally, knockout of the p27 gene, responsible for encoding an amastigote-specific cytochrome C oxidase component, in L. donovani (gene Ldp27−/−) resulted in decreased parasitic burdens. It provided enduring protection against the development of both CL and VL associated with virulent strains [109].
The Leishmania centrin gene-1 represents an intriguing target for the development of potential mutant vaccine candidates, given its essential role in parasite growth and differentiation. Volpedo et al. [55] demonstrated that immunization with LdCen−/− parasites resulted in a notable augmentation of macrophages expressing MHC-II, leading to increased levels of IFN-γ-producing CD4+ Th1 cells and decreased levels of IL-10 and IL-4 secreting CD4+ Th2 cells. This immune response proved efficacious in providing protection against virulent parasites. In summary, the utilization of live, non-pathogenic/genetically engineered strains of Leishmania species presents itself as one of the most promising strategic alternatives in the fight against VL, as emphasized [109].
Third-generation (naked DNA) vaccines
Third-generation vaccines utilize DNA-based approaches, as outlined in reference [110]. In this relatively contemporary method, naked plasmid DNA or DNA encapsulated within a viral vector is administered either intradermally or intramuscularly. DNA vaccines are deemed safe due to the absence of pathogenic organisms that could potentially revert to virulent forms. They are also effective in stimulating IFN-γ production and activating dendritic cells, which are pivotal in providing protection against Leishmania infection [110].
DNA vaccines may be constructed to contain genes encoding individual antigens, such as gp63, LACK, or PSA-2, or they may encompass multiple genes encoding various antigens, including TSA, KMP-11, A-2, NH36, LmSTI1, CPs, and histones. To augment the immunogenicity of DNA vaccines, these vaccines may be primed by initially administering a boost with the expression of associated proteins using a recombinant virus-like modified vaccinia virus Ankara [111]. This strategic approach selectively stimulates diverse CD8+ T cells, specifically targeting Leishmania antigens.
New strategy vaccines
Progress in the scientific domain, alongside the previously mentioned approaches, has spurred exploration into novel methodologies for vaccine development. These include bioengineering strategies, such as advanced delivery systems and the development of chimeric vaccines. Additionally, non-pathogenic Leishmania species have emerged as a promising avenue of investigation in pursuing effective Leishmania vaccines [111, 112].
Vaccine antigen delivery systems
The field of drug delivery systems has been the subject of extensive investigation, particularly in treating cancer and infectious diseases. Among the various strategies explored, nanoparticles (NPs) have emerged as up-and-coming vaccine delivery platforms. NPs possess structural features that bestow several advantages upon vaccine delivery systems, including the controlled release of antigens, protection of vaccine antigens from degradation, site-specific delivery, and the potential to act as adjuvants [113]. These attributes of NP carriers collectively result in improved bioavailability of antigens, ultimately leading to the activation of the immune response.
In a specific study, the potential of cationic solid lipid NPs (cSLN) as carriers and delivery systems was demonstrated. Then injection of a fusion gene, pcDNA-A2-CPA-CPB-CTE, using cSLN was found to significantly enhance protective cell-mediated immunity [114]. This finding underscores the potential of NP-based drug delivery systems in vaccine development.
Chimeric vaccines
Lage et al. [115] introduced a novel in-silico synthetic recombinant vaccine, named ChimeraT. This innovative vaccine incorporates specific T-cell epitopes from Leishmania prohibitin, eukaryotic initiation factor 5a, and hypothetical LiHyp1 and LiHyp2 proteins. When administration with saponin as an adjuvant, ChimeraT elicited a Th1-type immune response and provided protection against L. infantum infection in BALB/c mice [116].
In a separate study, the F1F3 chimera protein, derived from the C-terminal domain of nucleoside hydrolase (NH, NH36), exhibited a significant decrease in the size of ear lesions caused by L. braziliensis. Moreover, it elicited robust CD4+ and CD8+ cytokine-secreting T cell responses, featuring a predominant presence of multifunctional CD4+ and CD8+ IL-2+TNF-α+IFN-γ+ T cells. As a result, chimeric proteins show potential as promising vaccine candidates for protecting against human diseases [117, 118].
Non-pathogen Leishmania spp. in vaccination
L. tarentolae, originally isolated from a reptilian host, represents a non-pathogenic Leishmania species in humans [119, 120]. Remarkably, this parasite plays a unique role in stimulating the maturation of dendritic cells, consequently leading to the production of IFN-γ and the induction of a Th1-type immune response. Moreover, it closely emulates the natural development of immunity, presenting a significant advantage over other strategies [121].
In an investigation by Breton et al. [122] it was observed that intraperitoneal administration of L. tarentolae induced protective immunity against L. donovani infection in BALB/c mice. Based on these findings, the authors proposed that L. tarentolae holds promise as a live vaccine candidate for combating Leishmania infections, without posing any risk of causing disease in humans.
mRNA vaccines
An efficacious vaccine should augment the inherent and adaptable immune responses and initiate an immunological memory reaction, affording prolonged safeguarding against infection. Recent investigations demonstrate that mRNA-based vaccines notably amplify these attributes [123]. Moreover, the mRNA platform facilitates the concurrent expression of diverse proteins, evoking immune responses against disparate epitopes stemming from distinct targets [124]. In a murine model, Duthie et al. [125] observed a substantial reduction in hepatic parasite load when they employed F2-RNA as an initial immunization, followed by a booster dose of recombinant LEISH-F2 protein. RNA vaccine technology can potentially provide a potent and feasible avenue for vaccine development. The creation of RNA vaccines necessitates solely the knowledge of the target gene sequence, obviating the need to cultivate pathogens or scale up recombinant protein production.
Novel compounds with therapeutic potential
In vitro studies
The effectiveness of leishmaniasis treatment depends on various factors related to the host, the environment, the administered drug, and the parasite [126]. The scientific community continually seeks novel therapeutic strategies and cost-effective, less toxic, and more potent compounds for leishmaniasis treatment. Consequently, numerous molecules that hinder Leishmania survival have been extensively investigated over the years. Initially, in vitro tests are employed in laboratory settings to assess the new drugs’ potential and their toxicity. These tests are valuable due to their cost-effectiveness, limited hazardous waste generation, and controlled experimental conditions [126–128]. This article focuses on newly discovered compounds exhibiting promising anti-leishmanial effects in vitro tests.
de Oliveira et al. [129] investigated the in vitro activity of five thiazole derivatives belonging to the thiazopyridines class (TP) and five from the thiazoacetylpyridines class (TAP) against L. infantum. Thiazoles constitute a class of compounds known for their diverse biological activities, including anti-tumor, antibacterial, and anti-inflammatory properties. In their investigation, all examined compounds displayed inhibitory effects on promastigote growth and demonstrated low cytotoxicity. Particularly noteworthy were TAP-01, TAP-04, and TAP-06, which exhibited enhanced efficacy against amastigotes, as evidenced by lower IC50 values (0.99 μM, 0.43 μM, and 0.59 μM, respectively). TAP-04 exhibited superior activity against both parasite forms, reduced cytotoxicity, and a notably high selectivity index (SI/amastigote = 137.37), approximately seven times higher than the minimum threshold for a promising drug. Additionally, the authors explored potential intracellular targets and the impact of the compounds on the parasite’s cell membrane. Promastigotes exposed to TAP-04 at half of its IC50 concentration exhibited notable alterations, including swollen mitochondria, disorganized mitochondrial ridges, changes in cell body morphology, and a rounded appearance, indicative of apoptosis-mediated cell death (Figure 3) [129].
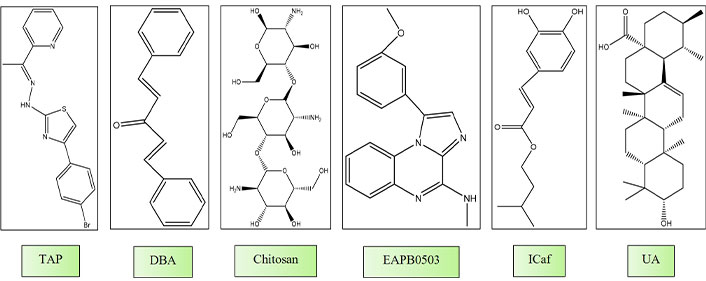
Chemical structures of molecules gone under in vitro and in vivo studies. TAP: thiazoacetylpyridines class; DBA: dibenzalacetone; ICaf: isopentyl caffeate; UA: ursolic acid
Chauhan et al. [130] investigated the leishmanicidal activity of trans-dibenzalacetone (DBA), a cost-effective and easily synthesizable monoketone analog of curcumin. They found IC50 values of 7.43 ± 1.88 μg/mL and 17.80 ± 1.42 μg/mL for amastigotes and promastigotes, respectively. Treatment of promastigotes with DBA-induced morphological changes, including cell reduction, rounding, and extensive cytoplasmic vacuolization after 24 hours. Furthermore, features associated with apoptosis, such as cell arrest in the G0/G1 phase, mitochondrial membrane potential depolarization, increased cytosolic Ca2+ levels, and reduced levels of GSH, were observed. Kinetoplastids like Leishmania rely on a peculiar form of GSH (trypanothione), making it a vital target for novel drug development. The unique mode of DBA, which induces apoptosis in the parasite, underscores its potential as a leishmaniasis treatment.
Chitosan is a biodegradable cationic polysaccharide renowned for its demonstrated antimicrobial, antileishmanial, and immunostimulatory properties [130]. Prior investigations have showcased its in vitro antileishmanial efficacy, with IC50 values spanning from 70 μg/mL to 240 μg/mL against promastigotes and amastigotes of L. infantum, L. amazonensis, and Leishmania chagasi [131]. A recent investigation assessed the in vitro antileishmanial efficacy of various forms of chitosan and its derivatives against extracellular promastigotes and intracellular amastigotes of L. major and L. mexicana. The researchers observed a pH-dependent antileishmanial effect of chitosan and its derivatives. Notably, chitosan and its water-soluble derivatives displayed minimal cytotoxicity against human squamous carcinoma cells (KB cells) at pH levels of 7.5 or 6.5. Obviously, chitosan and its derivatives exhibited approximately 7 to 20 times higher activity at pH 6.5, potentially attributed to enhanced ionization. The positively charged nature of chitosan might augment its antimicrobial efficacy by interacting with the negatively charged microbial membrane. The authors inferred that chitosan and its derivatives demonstrate pH-dependent antileishmanial effects and proposed that their mechanism of action involves the direct uptake of chitosan into the parasitophore vacuole via pinocytosis [132, 133].
El Hajj et al. [134] investigated the effects of imiquimod, an immunomodulatory drug, on L. tropica and L. major, and an imiquimod analog, EAPB0503, against both strains. Both compounds suppressed the replication of amastigotes, with EAPB0503 displaying enhanced potency, notably against L. tropica amastigotes. Imiquimod predominantly upregulated TLR7, while EAPB0503 exhibited a lesser extent of TLR7 modulation. Both compounds activated the canonical NF-кB pathway, inducing an immune response and upregulating i-NOS in infected macrophages. The authors proposed that imiquimod and EAPB0503 are promising candidates for treating L. tropica. Our research team has been investigating isopentyl caffeate (ICaf) and its anti-leishmanial properties. ICaf, which is derived from caffeic acid, has demonstrated significant efficacy against both the promastigote and amastigote forms of cutaneous (L. amazonensis) and visceral (L. chagasi) leishmaniasis [134, 135]. Nevertheless, the hydrophobic characteristics of ICaf pose limitations to its bioavailability. To address this constraint, an inclusion complex of ICaf within β-cyclodextrin (β-CD) was formulated, resulting in enhanced solubility while preserving its potent in vitro anti-leishmanial activity (Figure 3).
In vivo studies
The safety evaluation of novel molecules necessitates preliminary animal experimentation before they progress into clinical trials. These in vivo investigations are designed to elucidate various facets of the new molecule’s behavior within a biological system before its human testing [136]. The critical initial step in this process is the carefully selecting an appropriate animal model. This choice is paramount, as it dictates how much the animal model corresponds to human trials. Disparities in gastrointestinal physiology, enzyme activity, circulatory systems, and other characteristics render certain models more suited for specific research objectives. For instance, variations in metabolic processes between two species significantly impact the substance’s toxicity and efficacy. Additionally, it is worth emphasizing that animal models of human diseases facilitate the selection of compounds and the early assessment of their mechanisms of action and enhance our comprehension of the therapeutic index. This, in turn, leads to more informed decisions regarding clinical dosage selection for human trials [137, 138].
Ursolic acid (UA) is a compound that exhibits considerable potential in the context of anti-leishmanial activity. UA has been demonstrated to effectively combat both acute and chronic models of L. infantum (VL) and CL. UA treatment resulted in noteworthy reductions in parasitic burdens and elicited a modulation of the Th1 immune response, culminating in parasite eradication and reduced inflammatory responses. Notably, no signs of toxicity were observed, underscoring the well-tolerated nature of UA. Consequently, it is a promising candidate for treating visceral and CL [139].
Bilbao-Ramos et al. [140] conducted an assessment of the effectiveness of UA, a multifunctional triterpenoid known for its established antitumor, antioxidant, antimicrobial, and antifungal properties. Their study encompassed in vivo evaluations of UA activity in acute and chronic VL models. Furthermore, they developed topical formulations and assessed their efficacy in a chronic CL model.
In their investigation, the authors initially reported in vitro antileishmanial activity UAs against amastigotes of L. amazonensis and L. infantum. Notably, this activity was six times greater for L. amazonensis and three times greater for L. infantum amastigotes when compared to promastigotes. The SI of UA was found to be higher, ranging from three to eight fold greater, depending on the parasite strain, compared to miltefosine. The authors explained the heightened efficacy of UA against amastigotes in comparison to promastigotes by its dual mechanism of action. UA exhibited activity against the parasite itself and stimulated the host cell’s immunological response, particularly by increasing the production of nitric oxide (NO) in macrophages [141].
In the acute and chronic models of L. infantum (VL), UA was administered intraperitoneally at a daily dose of 5 mg/kg for seven consecutive days. Substantial reductions in parasitic loads were observed, with a 99.83% reduction in the spleen and liver in the acute model, and a 58% and 79% reduction in the spleen and liver in the chronic model.
In the chronic model of CL, the progression of lesions decreased by approximately 42% and 50% (at weeks 10 and weeks 15) during the topical administration of UA ointment (0.2%) for 28 days. However, it is noteworthy that the parasites were not eliminated after topical administration, as evidenced by increased inflammation at week 15. UA was found to modulate the Th1 immune response in the host, leading to parasite death through the increase of IFN-γ and the decrease of IL-4.
Notably, the research revealed no apparent signs of toxicity in the animals, underscoring the well-tolerated nature of the administered dose of UA. This supports the notion that UA holds promise as a candidate for treating both visceral and CL [141–143].
Current study involving heterocyclic compounds having leishmanial activity
Bekhit et al. [144] reported an article that contains “New heterocyclic hybrids of pyrazole and its bioisosteres: design, synthesis and biological evaluation as dual acting antimalarial-antileishmanial agents”. A novel set of pyrazole derivatives was synthesized by integrating five-membered heterocyclic elements, including thiazoles, thiazolidinones, 1,3,4-thiadiazoles, and pyrazolines. These compounds underwent an assessment of their in vivo antimalarial efficacy against Plasmodium berghei-infected mice. Subsequently, the most potent derivatives were subjected to further investigation to evaluate their in vitro antimalarial activity against the chloroquine-resistant (RKL9) strain of Plasmodium falciparum (Figure 4).
Quintal et al. [145] reported an article titled “Synthesis, structural characterization and leishmanicidal activity evaluation of ferrocenyl N-heterocyclic compounds”. Organometallic compounds possessing a tricomponent structure comprising ferrocene, an aromatic ligand, and a biofissionable link were systematically designed as potential leishmanicidal agents. These compounds demonstrated substantial activity against L. infantum visceral strain amastigotes, with an IC50 value of approximately 5 µM. Moreover, their therapeutic indexes reached 96 in U-937 macrophages, significantly surpassing the reference drug miltefosine.
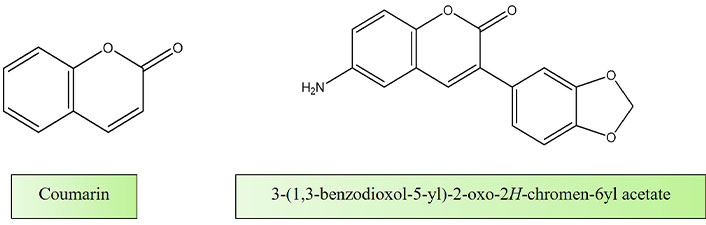
Mandlik et al. [146] published a research article titled “Biological Activity of Coumarin Derivatives as Anti-Leishmanial Agents” in which they delve into the investigation of natural compounds, specifically coumarins, as potential supplementary therapeutic agents to complement existing treatment modalities. Their study encompassed an in-depth analysis through in-silico screening of coumarin derivatives, followed by a comprehensive evaluation of their anti-leishmanial properties through a combination of in vitro and in vivo experiments (Figure 5).
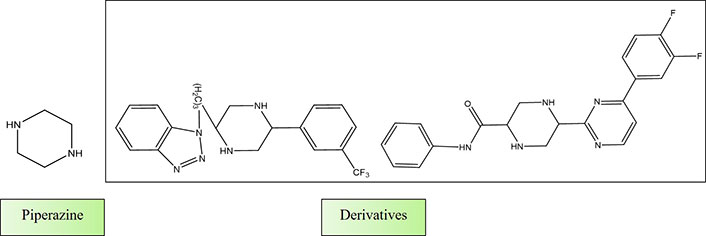
Shaquiquzzaman et al. [147] authored a review article titled “Piperazine scaffold: A remarkable tool in generation of diverse pharmacological agents”. In this comprehensive review, they elucidated the pivotal role of piperazine as a highly sought-after heterocyclic compound in the quest for new drug candidates. Piperazine’s presence was noted in numerous well-established and commercially available drugs, underscoring its significance. The broad spectrum of pharmacological activities exhibited by piperazine derivatives has positioned them as indispensable building blocks for the development of innovative therapeutic agents. The review emphasizes the piperazine derivatives’ therapeutic relevance and importance in drug discovery (Figure 6).
Al Nasr et al. [148] published a research article titled “New pyrano-4H-benzo[g]chromene-5,10-diones with antiparasitic and antioxidant activities”. This study, synthesized and examined a series of new pyrano-naphthoquinone derivatives for their efficacy against Trypanosoma brucei, L. major, and Toxoplasma gondii parasites.
The penta-fluorophenyl derivative exhibited notable effectiveness against T. brucei, with EC50 values in the single-digit micromolar range, and against T. gondii with sub-micromolar values. Furthermore, the 3-chloro-4,5-dimethoxyphenyl derivative demonstrated activity against amastigotes of L. major parasites comparable to AmB. Additionally, the study observed antioxidant activities in the bromophenyl derivatives, and their redox behavior was investigated using cyclic voltammetry. Notably, the antiparasitic and antioxidative activities of the new naphthoquinone derivatives appeared to be independent of one another (Figure 7).
Structures of derivatives synthesized
Note. Adapted from “New pyrano-4H-benzo[g]chromene-5,10-diones with antiparasitic and antioxidant activities” by Al Nasr IS, Jentzsch J, Shaikh A, Singh Shuveksh P, Koko WS, Khan TA, et al. Chem Biodivers. 2021;18:e2000839 (https://onlinelibrary.wiley.com/doi/10.1002/cbdv.202000839). CC BY.
Conclusions
The review article provides a comprehensive overview of the multifaceted landscape of leishmanial disease, encompassing its etiology, treatment strategies, and promising advancements in drug design. We have explored a broad spectrum of therapeutic approaches, including chemotherapy, combination therapy, thermo and cryotherapy, therapeutic vaccines, and immunotherapy, collectively contributing to the arsenal against this formidable disease. Chemotherapy remains a pivotal pillar in the treatment of leishmanial disease, with a myriad of anti-leishmanial drugs at our disposal. The rationale behind combination therapy, aiming to enhance treatment efficacy while minimizing drug resistance, has been discussed, shedding light on potential synergistic interactions between anti-leishmanial agents. Thermo and cryotherapy strategies present alternative modalities with the potential to complement existing treatment options, especially for CL.
The evolving frontier of therapeutic vaccines and immunotherapy offers the promise of long-term immunity and improved patient outcomes, and we have examined recent advancements in this domain. Furthermore, our exploration into structured-based and LBDD strategies underscores the importance of rational drug design in the quest for more effective, selective, and less toxic anti-leishmanial agents.
In addition, the review has highlighted the utilization of new heterocyclic compounds, a promising avenue of research that has gained prominence in recent years. These novel compounds represent a reservoir of potential candidates for drug development, offering fresh avenues for therapeutic intervention.
Abbreviations
| AmB: | amphotericin B |
| BCG: | Bacillus Calmette Guerin |
| CL: | cutaneous leishmaniasis |
| CPs: | cysteine proteases |
| DBA: | dibenzalacetone |
| gp63: | glycoprotein 63 |
| GSH: | glutathione |
| HIV: | human immunodeficiency virus |
| ICaf: | isopentyl caffeate |
| IFNs: | interferons |
| LBDD: | ligand-based drug design |
| LdTop1: | Leishmania donovani topoisomerase 1 |
| LST: | leishmanin skin test |
| LZ: | leishmanization |
| MCL: | mucocutaneous leishmaniasis |
| MT: | miltefosine transporter |
| NH: | nucleoside hydrolase |
| NPs: | nanoparticles |
| PKDL: | post-kala-azar dermal leishmaniasis |
| RF: | radiofrequency |
| SBDD: | structure-based drug design |
| Sb3+: | trivalent antimony |
| SbV: | pentavalent antimony |
| TAP: | thiazoacetylpyridines class |
| TLR: | toll-like receptor |
| UA: | ursolic acid |
| VL: | visceral leishmaniasis |
Declarations
Acknowledgments
I acknowledge the esteemed faculty and researchers at Bharati Vidyapeeth (Deemed to be University) Poona College of Pharmacy, Pune for their valuable contributions and support in the development of this review article. Their expertise and guidance have been instrumental in shaping the content and insights presented in this work. I extend my sincere gratitude to the institution for providing a conducive academic environment that fosters research excellence and innovation.
Author contributions
TM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing—original draft, Writing—review & editing. DK and DB: Formal analysis, Project administration, Supervision, Visualization, Writing—review & editing.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2024.