
Abstract
Apoptosis is a crucial process to maintain the correct balance between healthy cells and committed-to-death cells in every tissue. The internal (or mitochondrial) and external (or death receptor) pathways are responsible for driving a series of molecular events that lead to apoptosis by releasing pro-apoptotic proteins, such as B-cell lymphoma-2 (BCL-2) homology 3 (BH3)-only proteins and second mitochondria-derived activator of caspases/diablo inhibitor of apoptosis protein-binding mitochondrial protein (SMAC/DIABLO), that in turn activate the caspase family of proteases. By counterbalancing the apoptogenic machinery, anti-apoptotic BCL-2 family members turn off pro-apoptotic signalling, favouring cell survival, a circumstance that is particularly pronounced in tumour cells in which apoptosis is deranged. Therefore, targeting the defective apoptotic process has become a viable therapeutic option for the treatment of several cancers and much effort is being made in the research and development of effective compounds. This review discussed and updated the most promising therapeutic strategies that target deranged apoptosis process in cancer by mimicking the pro-apoptotic effects of BH3-only and SMAC/DIABLO proteins.
Keywords
Apoptosis, BH3-mimetics, SMAC/DIABLO, kinase inhibition, peptidomimetics, nanoparticlesIntroduction
In a multicellular organism, life requires death. Although this statement might seem a contradiction, in the biological context it represents a sine qua non requirement for the healthy survival of the organism itself. In fact, proliferation of newly generated cells via mitosis and death of damaged or unrequired cells that need to be cleared from the body must be kept strictly balanced to ensure normal physiology and correct tissue function. Conversely, disruption of this finely tuned homeostasis, either by uncontrolled cell proliferation or inadequate cell removal, may lead to the development of various diseases, such as cancer, neuro-degenerative disorders, and autoimmune diseases [1, 2].
Regulated cell death (RCD) is a complex process that comes in many forms, leading to the elimination of undesired or defective cells through a controlled and programmed series of biochemical and molecular events [3]. Based on morphological and structural details manifested in tissues or even in one single cell, different modalities of RCD have been identified so far, such as necroptosis, ferroptosis, pyroptosis, lysosome-dependent cell death, among many others [4, 5]. Interestingly, each of these cell death programs involves specific molecular events that can be targeted by pharmacological or genetic treatments [5]. The most known and well-characterized form of controlled cell death is termed apoptosis. Apoptotic cell death is a tightly regulated intracellular program that can be triggered by either external or internal stimuli, which in turn activate the extrinsic (death receptor mediated) or the intrinsic (mitochondria-mediated) pathways, respectively. Finally, both the apoptotic pathways converge into the activation of a family of cysteine-aspartic proteases, the caspases (CASPs), which degrade cellular components by forming apoptotic bodies and prepare dying cells for clearance avoiding any interference with the surrounding cells and tissues.
Extensive scientific literature has helped to consolidate the concept that dysregulated apoptosis underlies various diseases and, especially in cancer, it represents a hallmark of neoplastic transformation and development. For this reason, targeting apoptosis in various tumours has proven to be strategic and decisive in improving patient survival. Several strategies to re-activate defective apoptotic signalling in cancer tissues have been employed so far, including those that mimic the natural sequence of molecular events underpinning the apoptotic cascade, such as the inhibition of pro-survival signalling due to the sequestration of pro-apoptotic B-cell lymphoma-2 (BCL-2) family proteins by BCL-2 homology 3 (BH3)-mimetics or the quenching of the inhibitory activity of X-linked inhibitor of apoptosis protein (XIAP) on CASPs using second mitochondria-derived activator of CASPs/diablo IAP-binding mitochondrial protein (SMAC/DIABLO) peptidomimetics. While the death of tumour cells can lead to an attractive response to therapy, the selection, growth, and spread of resistant cells can ultimately be fatal and therefore should be prevented. It should be noted that only a limited number of Food and Drug Administration (FDA)-approved anticancer agents directly target apoptotic pathways, mainly small molecules specifically designed to counteract anti-apoptotic BCL-2 family members. Here, we summarized the recent development of specific bio-mimetic pharmacological agents that directly target the apoptotic regulatory machinery and the preclinical and clinical data that illustrate the contexts in which these treatments will be applicable. Finally, we described innovative strategies to overcome barriers to their successful clinical translation.
Intrinsic or mitochondria-mediated pathway
Key regulators of the molecular machinery for the intrinsic apoptosis pathway are proteins belonging to the BCL-2 family, bearing one or more BH domains (i.e. BH1–BH4) [6]. Noteworthy, the BCL-2 family comprises both pro-apoptotic and pro-survival/anti-apoptotic members, whose balanced modulation determines cell fate decisions between life and death. After apoptotic stimuli, such as cellular stress [7] or damage signals, including DNA damage [8], oxidative stress [9], and growth factor withdrawal [10], BCL-2 interacting mediator of cell death (BIM), BH3-interacting domain death agonist (BID), p53-upregulated modulator of apoptosis (PUMA), and potentially other BH3-only activator proteins are unleashed via upregulation or cleaveage [1, 2]. In addition, several genes have been related to apoptosis, either stimulating or inhibiting it [11]. For example, activation of the c-Myc and Bax proto-oncogenes as well as the E2F transcription factor induce apoptosis. Notably, normal and malignant cells are distinguished by fundamental differences in MYC-linked signalling pathways that can alternatively drive apoptosis or stimulate cell proliferation/transformation [12]. Conversely, the BCL-2, ABL, and RAS oncoproteins favour the inhibition of apoptotic cell death [13].
The BH3-only activators, if not promptly sequestered by pro-survival proteins such as BCL-2, BCL-extra large (BCL-XL), and myeloid cell leukaemia-1 (MCL-1), or whenever the binding capacity of the pro-survival network is overwhelmed, bind to and activate either or both the pro-apoptotic pore-forming proteins BCL-2 associated X protein (BAX), BCL-2 antagonist/killer 1 (BAK1), and possibly BCL-2 family apoptosis regulator (BOK). At the mitochondrial surface, activated BAX and BAK1 oligomerize and form pores, resulting in the mitochondrial outer membrane permeabilization (MOMP) with the consequent release into the cytosol of several apoptogenic molecules, including SMAC/DIABLO, reaper-related serine protease [human serine protease homologous to bacterial heat shock endoprotease A2 (HTRA2/OMI)], and cytochrome c (Cyt-c). In the cytosol, such apoptogenic molecules lead to the direct activation, as in the case of Cyt-c which forms activating apoptosome platform by binding the apoptotic protease-activating factor 1 (APAF1), or indirect activation, as for SMAC/DIABLO and HTRA2/OMI that interact with and counteract CASP-inhibitor proteins (e.g., XIAP) [14], of the initiator CASP9 which, in turn, triggers the executioner CASPs 3 and 7. At antagonizing this chain of pro-apoptotic events, the pro-survival/anti-apoptotic proteins of the BCL-2 family can sequester not only the BH3-only activator proteins but also the BH3-only activated BAX and BAK1 monomers [15], resulting in the block of apoptotic cascade (Figure 1).
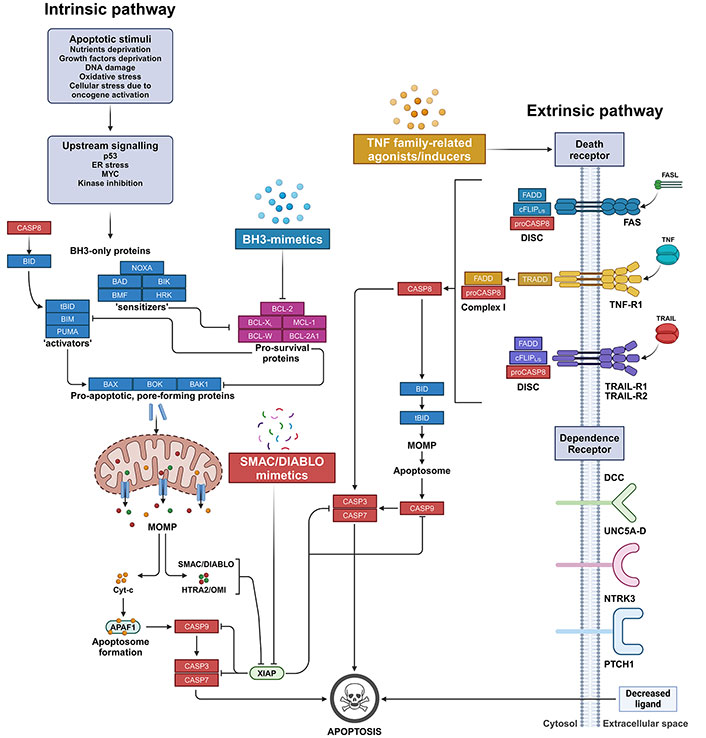
The apoptosis pathways. On the left, the intrinsic mitochondria-mediated apoptosis pathway initiates with cell stress or damage signals, which trigger the pathway leading to the upregulation, such as for BIM or PUMA, or cleaveage, as in the case of BID, which is activated into its truncated form (tBID), of pro-apoptotic BH3-only ‘activators’. BH3-only activator proteins can be sequestered by pro-survival BCL-2 family members that consequently block the apoptotic cascade. Conversely, when the pro-survival proteins network is absent or saturated by BH3-only ‘sensitizers’, BH3-only activators can bind and activate the pro-apoptotic pore-forming proteins BAX or BAK1, and possibly BOK. These, in turn, oligomerize and form pores resulting in the MOMP. Pore-forming proteins in their monomeric form can be equally sequestered by pro-survival BCL-2 family proteins that, therefore, switch off the downstream execution phase of apoptosis. Upon MOMP, apoptogenic SMAC/DIABLO, Reaper-related serine protease (HTRA2/OMI), and Cyt-c are released from the mitochondrial intermembrane space into the cytosol. In the cytosol, Cyt-c forms the apoptosome by recruiting and binding to APAF1. Apoptosome enables the direct activation of the initiator CASP9, which triggers the executioner CASP3 and CASP7, finally resulting in the degradation of nuclear constituents as well as of core and cytoskeletal proteins. The activation of the CASPs is eased by SMAC/DIABLO and HTRA2/OMI apoptogenic molecules, which sequester and/or degrade members of the IAPs, such as XIAP. BH3-mimetics are a class of small molecule compounds that sensitize cells to apoptosis by saturating the binding capacity of pro-survival BCL-2 family members, thus blocking their anti-apoptotic activity. On the other hand, SMAC/DIABLO mimetics belong to a class of peptidomimetics that prevent the inhibition of CASP activation by IAP proteins. On the right, the extrinsic DR-mediated apoptosis pathway originates from the binding of either FASL, TRAIL, or TNF to their corresponding DRs displayed on the extracellular surface of plasma membrane. The binding leads to the formation in the cytosol of either the DISC or the Complex I signalling complex for the transduction into the cell of the extracellular apoptotic signal. Finally, the assembly of these complexes promotes the activation of CASP8, which mediates either the direct proteolytic activation of executioner CASPs (i.e. CASP3 and CASP7) or their indirect triggering via the proteolytic activation of BID into tBID, which in turn promotes MOMP and the downstream events as in the intrinsic apoptotic pathway. TNF family-related agonists/inducers comprise a heterogeneous class of molecules that target either TNF-related ligands or their receptors, resulting in their direct or indirect upregulation and/or activation. In addition, in some cells, extrinsic apoptosis can also be induced by dependence receptors as a consequence of the transduction of lethal signals due to decreased concentrations of specific extracellular ligands. The therapeutic strategies described in this review are highlighted. APAF1: apoptotic protease-activating factor 1; BAD: B-cell lymphoma-2 antagonist of cell death; BAK1: B-cell lymphoma-2 antagonist/killer 1; BAX: B-cell lymphoma-2 associated X protein; BCL-2: B-cell lymphoma-2; BCL-2A1: B-cell lymphoma-2 related protein A1; BCL-XL: B-cell lymphoma-extra large; BH3: B-cell lymphoma-2 homology 3; BID: B-cell lymphoma-2 homology 3-interacting domain death agonist; BIK: B-cell lymphoma-2 interacting killer; BIM: B-cell lymphoma-2 interacting mediator of cell death; BMF: B-cell lymphoma-2 modifying factor; BOK: B-cell lymphoma-2 family apoptosis regulator; CASP8: caspase 8; Cyt-c: cytochrome c; DCC: deleted in colorectal cancer netrin receptor; DISC: death-inducing signalling complex; DR: death receptor; ER: endoplasmic reticulum; FADD: Fas-associated death domain; FASL: Fas ligand; HRK: B-cell lymphoma-2 family member Harakiri; HTRA2/OMI: human serine protease homologous to bacterial heat shock endoprotease A2; IAPs: inhibitor of apoptosis proteins; MCL-1: myeloid cell leukaemia-1; MOMP: mitochondrial outer membrane permeabilization; NTRK3: neurotrophic receptor tyrosine kinase 3; PTCH1: protein patched homolog 1; PUMA: p53-upregulated modulator of apoptosis; SMAC/DIABLO: second mitochondria-derived activator of caspases/diablo inhibitor of apoptosis protein-binding mitochondrial protein; tBID: truncated B-cell lymphoma-2 homology 3-interacting domain death agonist; TNF: tumour necrosis factor; TNF-R1: tumour necrosis factor receptor 1; TRADD: tumour necrosis factor receptor associated death domain; TRAIL: tumour necrosis factor-related apoptosis-inducing ligand; TRAIL-R1: tumour necrosis factor-related apoptosis-inducing ligand receptor 1; UNC5A-D: receptors of the Uncoordinated-5 family homologs A-D; XIAP: X-linked inhibitor of apoptosis protein. Created in BioRender. Venerando, A. (2024) BioRender.com/t26w727
Note that, although CASP activation stands for a crucial and ubiquitous downstream event in the apoptotic cell death, only MOMP represents the main point of engagement at which cell is doomed to die [16–19]. As a matter of fact, it has been clarified that prevention of CASP activation by using specific inhibitors can control the kinetics of apoptosis program, simply delaying the execution of an already committed cell death [2, 20, 21].
Extrinsic or death receptor-mediated pathway
On the other hand, the extrinsic pathway of apoptosis is triggered by activation or oligomerization of transmembrane proteins belonging to the superfamily of death receptors (DRs) or, alternatively, when specific extracellular factors reach a particular concentration threshold in the extracellular environment, inducing the transduction of lethal signals. DR1–8 can be grouped into two main categories, according to the adapter protein involved: i) Fas ligand (FASL) and tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) activated receptors [i.e. Fas, TRAIL receptor 1 (TRAIL-R1), and TRAIL-R2] that recruit the death-inducing signalling complex (DISC) for the transduction of apoptotic signals; and ii) TNF receptor 1 (TNF-R1), TRAMP, DR6, and EDAR, which conversely enrol TNF-R associated death domain (TRADD) as adapter protein to bind other factors and kinases, leading to the formation of a signalling complex (named Complex I) for the transduction of apoptosis or cell survival [4]. In both cases, once DR is triggered, initiator CASP8 is directly activated, as in the case of DISC formation, or indirectly activated by Complex I build-up, resulting in the switch-on of the downstream executioner CASPs. It should be pointed out that in the DR-mediated apoptotic pathway, mitochondria enhance CASP activation but, overall, they are non-essential components of the process [22] (Figure 1).
Execution phase
Activation of executioner CASPs in both the intrinsic and extrinsic pathways initiates the execution phase that represents the final step of the apoptotic cell death process. Indeed, executioner CASPs (i.e. CASP3, CASP6, and CASP7) in turn activate cytoplasmic endonucleases and proteases, which start to degrade nuclear constituents as well as core and cytoskeletal proteins, respectively [3, 23]. These events result in deep cytomorphologic changes, including cell shrinkage, chromatin condensation, and the formation of cytoplasmic apoptotic bodies, all signals that mark the cell as ready for clearance by the phagocytic system.
Targeting intrinsic pathway: the BH3-mimetic strategy
The BCL-2 regulated apoptotic pathway is activated in response to diverse cellular stress signals, resulting in the removal of damaged or superfluous cells. Such stimuli elicit the transcriptional and/or post-translational upregulation of the pro-apoptotic BH3-only proteins. So far, eight BH3-only proteins have been identified and validated to possess canonical pro-apoptotic activity, which displays different degrees of activation potential for pore-forming BAK1 and BAX, being BID, truncated BID (tBID), and PUMA the most potent activators [24]. The remaining BH3-only proteins are commonly referred to as ‘sensitizers’ and their principal role in the apoptotic mechanism is supposed to be the inhibition of pro-survival BCL-2 family protein counterparts [6, 25]. Indeed, the specific interactions between a hydrophobic groove formed by the BH1–BH3 domains on the anti-apoptotic proteins and the hydrophobic surface of the BH3 domain on the pro-apoptotic proteins (Figure 2A), conduct the block of apoptosis cascade by physically sequestering these latter transducers and tipping the balance in favour of cell survival [26]. On the contrary, when the binding capacity of the pro-survival system is saturated by the BH3-only sensitizers, apoptosis can proceed with the following downstream events. Interestingly, this molecular process represents the rationale at the basis of the BH3-mimetic anticancer strategy [27] (see below). In fact, evasion of apoptosis, which facilitates malignant transformation and cancer development, is one of the most representative and studied hallmarks of cancer [28]. The mechanisms by which cells elude apoptosis-controlled cell death vary widely between cancers and even within the same cancer type, including upregulation of pro-survival BCL-2 family members, downregulation or inactivation of pro-apoptotic proteins, and/or dysregulation of the pore-forming proteins BAX and BAK1 by means of mutations, post-translational or post-transcriptional modifications. Notably, cancer-specific roles have been ascribed to different BCL-2 family members. Indeed, it was shown that BCL-2 over-expression accelerates the development of MYC-induced mammary tumour development [29]. Similarly, the loss of genes encoding BAX, BIM, CASP2, or PUMA boosts tumour development in distinct models of breast cancer [30–32]. Further, in a mouse model of intestinal oncogenesis, PUMA deficiency worsened colorectal tumorigenesis as well [33]. In addition, BCL-XL expression resulted crucial for cell survival in cell lines derived from both colorectal [34] and non-small cell lung cancers [35]. Although most cancer cells display dysregulated apoptotic signalling, the heterogeneity in the expression of and dependence on BCL-2 family members is particularly evident in hematopoietic cancer cells [15, 36, 37]. As an example, over-expression of BCL-2, BCL-XL, or MCL-1 promoted the onset of leukaemia and lymphoma induced by over-expression of MYC proto-oncogene [38]. In addition, establishment of MYC-driven lymphoma and leukaemia is accelerated in mice lacking genes encoding BAX, BIM, BCL-2 antagonist of cell death (BAD), BCL-2 modifying factor (BMF), or PUMA [39–43], as single defect or in combination with p53 ablation [44].
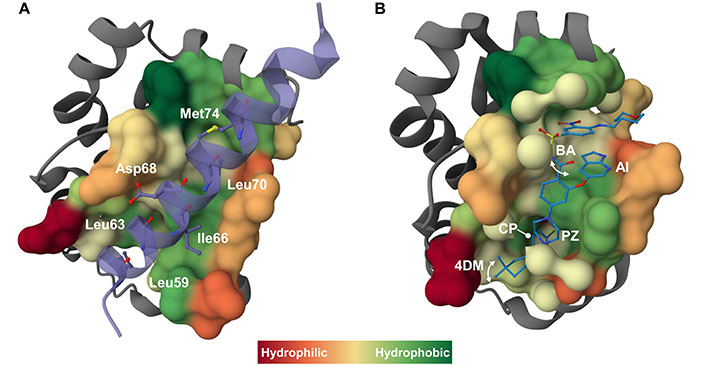
Mechanism of action of BH3-mimetics at a glance. A) The interaction of a pro-survival protein with a BH3 domain from a pro-apoptotic relative is exemplified by the crystallographic structure of BCL-2 in complex with a 31-mer BH3 BAX peptide (purple) (PDB ID 2XA0) [45]. The BAX peptide forms an amphipathic α-helix that binds to the BH3-binding groove of BCL-2, as commonly observed in all the reported crystallographic structures of anti-apoptotic BCL-2 proteins in complex with BH3 peptides [26]. Four conserved hydrophobic residues plus an additional non-conserved residue (i.e. Met74, in the BAX peptide) of the BH3 peptide represent the interacting consensus with the corresponding four hydrophobic pockets (namely P1–P4) within the BH3-binding groove on the pro-survival protein. Additional intermolecular ionic interactions occur involving conserved charged residues on the BH3 domains, such as the conserved Asp (Asp68 in the BAX peptide), and the corresponding oppositely charged residues on the pro-survival protein (e.g., conserved Arg residue). The key interacting residues in the BAX peptide are shown as sticks and balls and labeled. B) As an example of the BH3-mimetic interacting with pro-apoptotic BCL-2 family proteins, the crystallographic structure of BCL-2 in complex with venetoclax (ABT-199) (blue) (PDB ID 6O0K) [46] is presented. Venetoclax binds to BCL-2 by creating a deep pocket in the P2 region lined by a series of hydrophobic residues. Key chemical features of venetoclax are indicated, including the CP deeply buried in the BCL-2 P2 pocket, the PZ bridging the P2 and P4 pockets, and the AI bound in the BCL-2 P4 pocket. The 4DM ring and the BA can adopt two distinct conformations (schematized by a double arrow) above the BCL-2 P2 and the P4 pockets, respectively. Finally, interactions in the P4 pocket with Asp103 and Arg107 are thought to determine selectivity. In all images the molecular surface of the pro-survival BCL-2 protein surrounding the target molecule is colored based on hydrophobicity index. 3D images were created using Mol* program [47]. 4DM: 4–4-dimethylcyclohex-1-ene; AI: azaindole; BA: benzamide; BAX: B-cell lymphoma-2 associated X protein; BCL-2: B-cell lymphoma-2; BH3: B-cell lymphoma-2 homology 3; CP: 4-chlorophenyl; PZ: piperazine
It should be noted that in most cancer types the upregulation of anti-apoptotic BCL-2 family members is recognized as a specific tumour survival factor and is associated with a concomitant increase in pro-apoptotic proteins. Consequently, the enhanced binding capacity of anti-apoptotic proteins to pro-apoptotic BCL-2 members not only prevents apoptotic cascade but also makes cancer cells, compared to normal cells, more susceptible to cellular stress or damage stimuli [1]. This phenomenon is called “priming” and makes cancer cells readily to undergo MOMP even in the presence of weak pro-apoptotic signals [34]. As a matter of fact, in many cancers, escape from apoptosis during carcinogenesis does not necessarily yield an apoptosis-resistant cell, but rather it renders cancer cells sufficiently refractory to pro-apoptotic signalling during daily fluctuations in nutrients and environmental stressors. On the other hand, cancer cells, especially those of hematopoietic origin, are closer to the apoptosis threshold and therefore more prone to cytotoxic effect of chemotherapeutic agents and ionizing radiations [2, 48]. However, in too many patients, the impossibility of completely eradicating malignant cells fosters the selection and expansion of resistant cells to conventional anti-cancer therapies, making such therapeutic approaches ineffective over time with a high degree of relapse [1].
Several experimental evidences have underlined that defects in apoptosis signalling promote tumorigenesis and can render malignant cells resistant to various anti-cancer agents [49, 50]. In this context, BH3-mimetic drugs have been developed to directly activate the apoptosis machinery by binding to and inhibiting specific pro-survival BCL-2 family proteins in cancer cells [26, 27]. In other words, these novel small-molecule inhibitors mimic the hydrophobic interactions between pro-survival proteins and the BH3 domains of pro-apoptotic BCL-2 family proteins (Figure 2B), annihilating the anti-apoptotic capacity and therefore switching off the pro-survival escape of cancer cells [26].
BH3-mimetics are a complex and diverse group of molecules (Figure 3) that target major pro-survival BCL-2 family proteins (for example, the MCL-1-selective inhibitors S63845 [51] or AMG-176 [52], and the BCL-XL inhibitors A-1331852 [53] or A-1155463 [54]). Several BH3-mimetic drugs are currently under development or investigation in preclinical settings and some of them have already been evaluated or are currently in clinical trials for the treatment of various types of cancer (Table 1). It is important to note that these agents have on-target toxicities (e.g., thrombocytopenia due to platelet killing in response to BCL-XL inhibition [55]) and they should be used only in those patients in which tumour depends specifically on the level of the pro-survival protein being inhibited by the selective BH3-mimetic used, in order to avoid unnecessary toxicity and maximize efficiency of the treatment. For this reason, many efforts are being made to research and develop specific biomarkers to assist and guide the selection of the most suitable BH3-mimetic in each pathological setting [56–58].
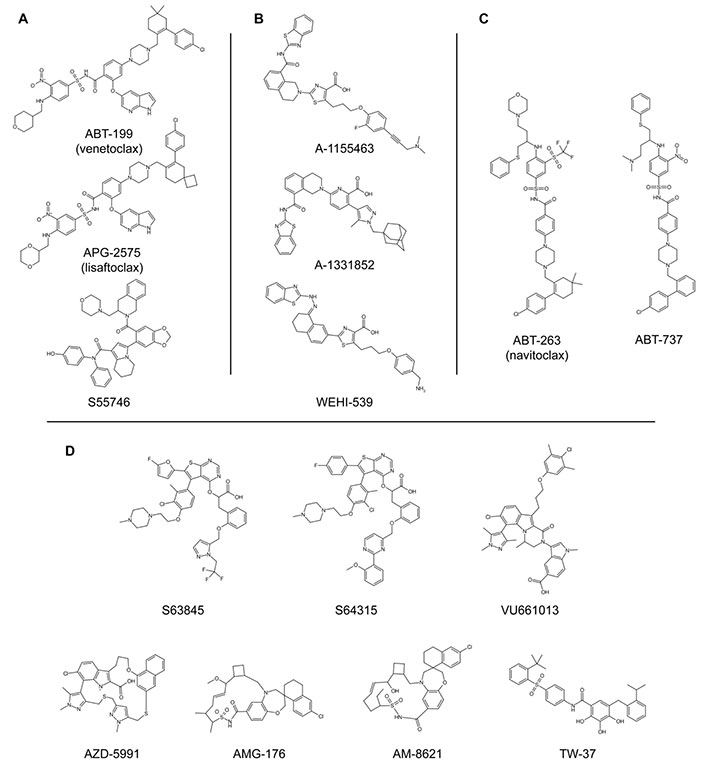
Chemical structures of BH3-mimetics. A) BCL-2-selective inhibitors; B) BCL-XL-selective inhibitors; C) promiscuous BH3-mimetic compounds that target BCL-2, BCL-XL, and BCL-W proteins; D) MCL-1-selective inhibitors. Refer to Table 1 for the bibliographic references of the discovery, the synthesis, and/or the patenting of each single compound [51, 52, 59–74]. BCL-2: B-cell lymphoma-2; BCL-XL: B-cell lymphoma-extra large; BH3: B-cell lymphoma-2 homology 3; MCL-1: myeloid cell leukaemia-1
BH3-mimetics in clinical trials for various tumour types. The chemical structures are shown in Figure 3. Note that only the completed clinical trials in which the compounds were used as single agents are reported. For ongoing clinical trials visit https://clinicaltrials.gov/. The bibliographic references refer to the discovery, the synthesis, and/or the patenting of the compounds
| Compound | Type of cancer | Clinical trials | References |
|---|---|---|---|
| ABT-199 (venetoclax) | Chronic lymphocytic leukaemia (CLL); acute myeloid leukaemia (AML); non-Hodgkin lymphoma (NHL); Waldenstrom macroglobulinemia |
| [59] |
| APG-2575 (lisaftoclax) | Waldenstrom macroglobulinemia |
| [60] |
| S55746 | CLL; B-Cell NHL; multiple myeloma (MM) |
| [61] |
| A-1155463 | Preclinical | - | [62] |
| A-1331852 | Preclinical | - | [63] |
| WEHI-539 | Preclinical | - | [64] |
| ABT-263 (navitoclax) | Ovarian cancer; small cell lung carcinoma; CLL; leukaemias; lymphomas; solid tumours; lymphoid malignancies; NHL |
| [65, 66] |
| ABT-737 | Ovarian cancer |
| [67] |
| S63845 | Preclinical | - | [51] |
| S64315 | MM; diffuse large B-cell lymphoma (DLBCL); AML; myelodysplastic syndrome (MDS) |
| [68, 69] |
| VU661013 | Preclinical | - | [70, 71] |
| AZD-5991 | Relapsed or refractory AML |
| [72] |
| AMG-176 | Higher risk MDS; Chronic myelomonocytic leukaemia; relapsed or refractory MM or AML |
| [52, 73] |
| AM-8621 | Preclinical | - | [52, 73] |
| TW-37 | Preclinical | - | [74] |
*: clinical trials in which the compound was used exclusively in combination regimen with other chemotherapeutic drugs; -: no data
To date, only the BCL-2 specific inhibitor venetoclax (ABT-199) [75] has been approved by the US FDA and by the European Medicine Agency for the clinical use to treat adults with chronic lymphocytic leukaemia (CLL) and acute myeloid leukaemia (AML) as monotherapy or, more effectively, in combination with other therapeutic agents [76]. In fact, except in a few specific hematologic diseases, such as CLL [77] or blastic plasmacytoid dendritic cell neoplasm [78], generally, BH3-mimetics as monotherapy have not produced high response rates [27]. Conversely, it has been demonstrated that such compounds do synergize with several anticancer agents, including chemotherapeutic drugs and oncogenic kinase inhibitors. As an example, the sequential combination of the microtubule-destabilizing agent vincristine and the MCL-1 selective inhibitor S63845 has been validated in rhabdomyosarcoma patient-derived xenograft model in which a reduction in tumour growth with a tendency to stabilize was observed [79]. Further, the combination of venetoclax and the BCL-XL inhibitor A‐1155463, in association with dinaciclib, a cyclin‐dependent kinase (CDK) inhibitor, which inhibits CDK1, 2, 5, and 9, showed potentiated cytotoxic effect both in vitro, in multiple myeloma (MM) cell lines, and ex vivo, in MM patient-derived plasma cells, suggesting a potential therapeutic option for MM patients overexpressing or with high dependence on MCL‐1 and displaying a poor prognosis [80]. Finally, targeting BCL-2 protein family by using the BH3-mimetic ABT-737 concomitantly with the inhibition of mammalian target of rapamycin (mTOR) pathway by the specific inhibitor CCI-779, enhanced drug-induced cytotoxicity in acute lymphoblastic leukaemia treatment [81].
Overall, the well-evidenced synergistic effect arising from the co-administration of BH3-mimetics in combination with different anti-cancer treatments can be explained by the general upregulation of BH3-only proteins, such as BIM, PUMA, and NOXA, that in turn inhibit the pro-survival BCL-2 family proteins, which are present in malignant cells but have escaped from the scavenger effect of the specific BH3-mimetic drug employed [31, 82, 83].
Despite the promising effects of BH3-mimetics in a wide range of malignancies, they are not curative by themselves, and over time relapses occur inevitably [26]. In addition, primary and secondary resistance to BH3-mimetics starts to emerge from clinical trial data, hampering their effective and widespread therapeutic applicability [26, 27]. The most complete data set on this issue comes from the venetoclax experience in patients with lymphoid malignancies. Indeed, in those patients, the poor response to the therapy despite high BCL-2 expression, has been attributed to high expression levels of multiple different pro-survival proteins [84] or to mutations within BCL-2 itself, which confer resistance to the drug [85]. Merging the analysis of patients receiving therapy with data from the preclinical studies investigating BH3-mimetics targeting also other different pro-survival BCL-2 family proteins has led to the general finding that resistance to BH3-mimetic drugs may arise either from loss of the targeted pro-survival protein, increased expression of non-targeted pro-apoptotic BCL-2 family members or decreased expression of critical pro-apoptotic proteins [2, 27].
Considering all these aspects and bearing in mind the experimental evidence from both the preclinical analysis and the clinical outcomes, it appears clear that combination therapies in which BH3-mimetics are co-administered with other compounds targeting collateral aspects of cancer cell survival and proliferation would represent the most effective anti-tumour strategies [2, 86]. In this respect, oncogenic kinases epitomize the ideal BH3-mimetic co-targets. In fact, frequently, oncogenic kinases inhibition leads to proliferation arrest in sensitive cancer cells, but not necessarily in their regression [87–91]. Consequently, combining BH3-mimetic drugs to drive apoptosis with inhibitors of oncogenic kinase pathways has been postulated to produce durable clinical responses with less collateral damage. Thus, several BH3-mimetics, such as AZD-5991 [92, 93] and AM-8621 [94] (MCL-1 specific inhibitors), ABT-737 [95] and ABT-263 [96] (both inhibiting BCL-2, BCL-XL, and BCL-W proteins), and the BCL-2, BCL-XL, and MCL-1 inhibitor TW-37 [97], have been demonstrated to synergistically improve the response of tumour cells to v-raf murine sarcoma viral oncogene homolog B1 (BRAF) and mitogen-activated protein kinase kinase (MEK) inhibitors.
Among the long list of the kinome members (i.e. the kinome is the protein kinase complement of the human genome, including over 500 different genes) that have been associated with human cancer initiation and progression [98, 99], and beyond the most targeted key oncogenic kinases [89], there is an interesting example of a protein kinase that promotes the growth and clonal expansion of cancer cells by antagonizing apoptosis cascade through a “non-oncogene” addiction mechanism and by sustaining key survival signalling pathways, such as those dependent by signal transducer and activator of transcription 3 (STAT3), p53, Wnt/β-catenin, AKT, and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [100, 101]. Indeed, the Ser/Thr acidophilic protein kinase CK2, a constitutively active and pleiotropic tetrameric enzyme composed of two catalytic (α or its isoform α’) and two regulatory (β) subunits, is ubiquitously present in all eukaryotic tissues of all organisms, where it plays fundamental roles in cell biology by phosphorylating hundreds of substrates [100–103]. Interestingly, CK2 has been found commonly overexpressed in both solid and blood tumours [104, 105]. Noteworthy, in mantle cell lymphoma, in which overexpressed CK2 sustains B-cell receptor (BCR) linked signalling and pro-survival BCL-2 family proteins, CK2 inhibition enhanced the cytotoxicity induced by the combination of ibrutinib, the first-in-class Bruton tyrosine kinase (BTK) inhibitor, which switches off BTK-dependent BCR signalling, and the BCL-2 specific inhibitor venetoclax, providing a potential strategy to overcome primary and secondary resistance to these drugs [106].
After a long coldness to consider it as a convenient drug target due to its ubiquity, protein kinase CK2 could represent a valid anticancer co-target considering its anti-apoptotic, pro-migration, and pro-proliferation functions [107]. It should be noted that, although over time several CK2-specific small-molecule inhibitors have been developed, CX-4945 (silmitasertib) [108] is the only CK2-specific inhibitor drug under clinical evaluation in patients with relapsed or refractory MM (clinical trial identifier NCT01199718). One of the causes that have slowed down the advancement of so many CK2-specific inhibitors, which conversely have proven to be very potent at least in vitro, can be traced to their low bioavailability and poor penetration of cell membranes. However, these problems could be overcome by the opportunities offered by nanotechnology. Indeed, the development of specific nanocarriers would have the potential to increase the delivery [109, 110] and targeting for those in vivo poorly effective CK2 inhibitors.
Targeting intrinsic pathway: SMAC/DIABLO-mimetics strategy
In the cancer cells, the final steps of apoptosis pathway (i.e. activation of executioner CASPs by CASP9) are usually modulated by the inhibiting effect of the IAPs, and can be activated by SMAC/DIABLO, a cytotoxic protein. This protein was discussed for the first time in 2000 by Du et al. [111] and Verhagen et al. [112] in two different studies. Both the research groups observed that SMAC/DIABLO is a functional homolog of Drosophila Reaper, Hid, and Grim, the function of which is countering the inhibitory activity of IAPs. They observed that SMAC is generally stored in the mitochondria and released into the cytosol upon apoptotic stimuli under the control of BCL-2 family of proteins. After release, SMAC binds IAPs in the cytosol and activates the apoptosis. IAPs are composed of three baculovirus IAP repeat (BIR) domains, namely BIR-1, BIR-2, and BIR-3, each of them able to inhibit the CASPs, both the activator CASP9 and the executioner ones, involved in the apoptosis cascade, therefore stopping the execution phase. In detail, BIR-2 inhibits CASP3 and CASP7, while the linker region between BIR-1 and BIR-2 is of crucial importance for the competitive inhibition of both. Instead, BIR-3 inhibits CASP9. Notably, BIR domains can also interact with SMAC/DIABLO and consequently SMAC/DIABLO binds different IAPs. SMAC/DIABLO is a 239 residues long protein but only a short portion formed by the sequence Ala-Val-Pro-Ile (AVPI) (see Figure 4), is able to interact with the region domains of BIR-2 or -3 (not simultaneously), inducing the activation of CASPs. On the contrary, SMAC/DIABLO is not able to bind BIR-1 [113]. From a structural point of view, the crystal structure and NMR studies [114, 115] conducted on AVPI peptide interacting with BIR-3 domain, demonstrated that the NH-amino Ala of AVPI interacts with Glu314 and Gln319 residues of BIR-3 due to strong hydrogen bonding, instead the carbonyl group of Ala forms an optimal hydrogen bond with indole NH of Trp323. Moreover, Val forms hydrogen bonds between its NH and carbonyl groups with the amino and carbonyl groups of Thr308, while the side chain of Val of AVPI peptide remains exposed to the solvent. Further, the five-member ring of Pro shows Van der Waals interactions with the aromatic ring of Trp323 and Tyr324. Finally, the NH group of Ile forms a hydrogen bond with the carbonyl group of Gly306, while its side chain is inserted into the pockets formed by the hydrophobic portion of the side chains of Lys297 and Lys299 and the side chains of Leu292 and Val298 of BIR-3 domain (Figure 4). In addition, an intensive structure-relative relationship study was conducted by Wist et al. [116]. Indeed, they observed a dissociation constant (Kd) = 480 nM when SMAC binds to XIAP’s BIR-3 domain, but when Ala of AVPI sequence is replaced by Gly or Ser, the binding affinity is 20-fold decreased. Instead, the replacement with 2-aminobutyric acid (an unnatural amino acid), the binding activity shows a slight improvement. Noteworthy, the replacement of Val by Gly, Asp, and Pro shows a significant loss in binding affinity. Finally, Ile substitution by other hydrophobic amino acids such as Val, Phe, Trp, or Leu is preferred, being Phe the best choice. However, in the presence of charged or polar amino acids in place of Ile, such as Lys, Arg, Glu, Asp, or His, there is a detrimental effect on the binding affinity. Moreover, N-methylation of amide bond between Ala and Val leads to loss a fundamental hydrogen bond for the activity, instead the methylation of amide bond between Pro and Ile is well tolerated. In addition, Oost et al. [117] evidenced that the substitution of methyl group with an ethyl group in the side chain of Ala maintains the binding affinity; on the contrary, while the N-alkylation of the free amino group of Ala is well tolerated, the N,N-alkylation instead leads to the loss of binding to XIAP resulting in the activation of CASP activity. Further, they observed that the replacement of the pyrrole ring of Pro with heterocycles consisting of four or six rings results in a 7-fold reduction of the inhibitory activity; conversely, the introduction of hydrophobic substituents on the Pro’s ring can slightly improve the binding affinity. Notably, the L-stereochemistry of the residues is essential for the activity.
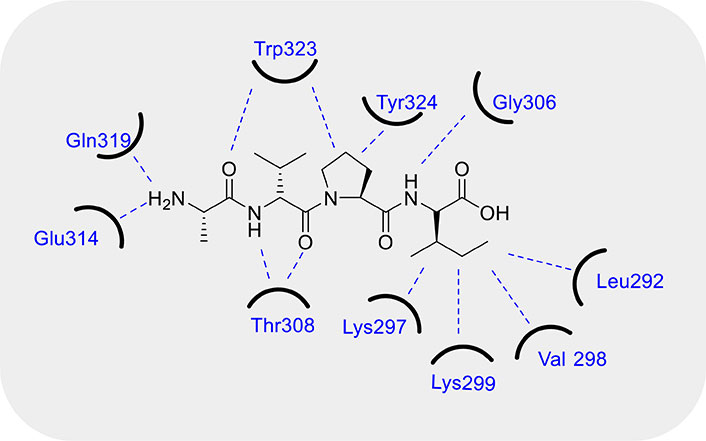
Fundamental interactions between SMAC/DIABLO and BIR-3 domain. Refer to the main text for an in depth description of the molecular interactions. BIR-3: baculovirus inhibitor of apoptosis protein repeat-3; SMAC/DIABLO: second mitochondria-derived activator of caspases/diablo inhibitor of apoptosis protein-binding mitochondrial protein
It should be noted that SMAC/DIABLO mimetics may represent a convenient strategy when used in combination with chemotherapeutic agents because they can help to overcome induced resistance to traditional anticancer therapies. In the following paragraphs, we will analyse SMAC/DIABLO mimetics that inhibit IAP and have the potential to combat chemoresistance when used in synergy with different anti-tumour therapies.
Monovalent SMAC/DIABLO mimetics
The first monovalent SMAC/DIABLO mimetic that entered phase I human clinical trial was GDC-0152, developed by Flygare et al. [118] (Figure 5). The chemical structure shows that the Pro at the position 3 is maintained, while the amino group of Ala at position 1 is converted into a secondary amine, which can interact with XIAP, and the side chain of Val is incorporated in a six-member ring. Finally, the last amino acid is an aromatic group able to enhance the interactions with the hydrophobic pockets of XIAP. The inhibitory constant (Ki) values obtained with GDC-0152 on the inhibition of XIAP, cellular IAP1 (cIAP1), and cIAP2 are 28, 17, and 43 nM, respectively. Interestingly, GDC-0152 is able to inhibit the growth of breast cancer after oral administration. GDC-0917, an SMAC mimetic of the second generation, differs from GDC-0152 in the presence of three aromatic groups in place of Ile (Figure 5). GDC-0917 was developed by Wong et al. [119] as a derivate of GDC-0152. Of note, GDC-0917, currently in clinical trials for the treatment of lymphoma, inhibits XIAP, cIAP1, and cIAP2 with Ki < 60 nM. GDC-0917 inhibits the tumour growth in the MDA-MB-231 xenograft model in a dose-dependent manner after oral administration.
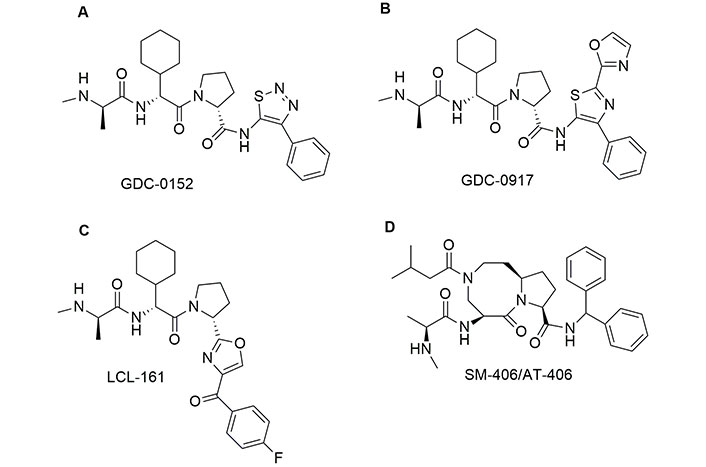
Chemical structure of monovalent second mitochondria-derived activator of caspases (SMAC) mimetics in clinical trials. See [118] for GDC-0152; [119] for GDC-0917; [120] for LCL-161 and [121] for AT-406 or visit https://clinicaltrials.gov/
The chemical structure of LCL-161, a potent inhibitor of IAP developed by Novartis [122] is reminiscent of both GDC-0152 and GDC-0917. However, in LCL-161 the Ile is replaced by a thiazole ring connected with 4-F-phenyl by a carbonyl bridge. Researchers observed that LCL-161 potentiates the anti-leukemic effect of nilotinib and PKC412 both in vitro and in vivo. Finally, LCL-161 reduces the intrinsic resistance to Tyr kinase inhibitors in leukaemia cells. In 2011, Cai et al. [121] developed a potent antagonist of IAPs named SM-406/AT-406, which is currently in clinical trials for acute myelogenous leukaemia, solid tumour, lymphoma, and head and neck carcinoma [123, 124]. This compound differs from the other SMAC/DIABLO mimetics in clinical trials, because the amino acids at positions 2 and 3 in the AVPI sequence are fused together. SM-406 binds XIAP, cIAP1, and cIAP2 with Ki = 66.4, 1.9, and 5.1 nM, respectively. This compound demonstrated good bioavailability, after oral administration, and showed a complete inhibition of the tumour growth in the MDA-MB-231 xenograft model. Monovalent SMAC mimetics are summarized in Table 2.
Summary of monovalent second mitochondria-derived activator of caspases (SMAC) mimetics. Monovalent SMAC mimetics in clinical trials and monovalent SMAC mimetics are presented in literature but not discussed in this review
| Compound | Type of cancer | Clinical trials | References |
|---|---|---|---|
| GDC-0152※ | Glioblastoma, leukaemia, and osteosarcoma | NCT00977067 | [125–127] |
| GDC-0917 | Lymphoma | NCT01226277 | [119] |
| LCL-161※ | Leukaemia | NCT03111992 | [128] |
| SM-406/AT-406※ | Solid tumours, lymphomas, and acute myelogenous leukaemia | NCT01078649 | [121] |
| LBW242 | Multiple myeloma | - | [129] |
| MV1 | Breast carcinoma | - | [130] |
| CS3 | Melanoma and breast carcinoma | - | [131] |
| Aza-phenylalanine 7 | Breast cancer | - | [132] |
| Compound 1 | Breast cancer | - | [117] |
| Compound 2 | Leukaemia | - | [133] |
| SM114 | Breast cancer | - | [134] |
| SM130 | Breast cancer | - | [134] |
※ Drugs in clinical trial for different cancer types; -: no data
Bivalent SMAC/DIABLO mimetics
The bivalent mimetics of SMAC/DIABLO are formed by two SMAC mimetics connected to each other via a linker [123]. Bivalent mimetics are more active than their monovalent counterparts, displaying greater binding affinity and potency in the activation of CASPs. Noteworthy, they can be administered intravenously. The bivalent SMAC/DIABLO mimetics are able to antagonize IAP in different ways: i) by degradation of TRAF2-associated cIAP1/cIAP2 and block of TNF-R1-mediated NF-κB activation [135]; ii) by inhibiting XIAP-dependent NF-κB activation via nucleotide-binding oligomerization domain-like (NOD) receptors [136]; and iii) by inducing degradation and ubiquitylation of ML-IAP [130].
Among different bivalent mimetics, birinapant and APG-1387 are the only two compounds that entered clinical trials (Figure 6). Birinapant (TL32711) was synthesized by TetraLogic Pharmaceuticals [137]. The chemical structure presents an Ala with a secondary amine and an ethyl group as side chain in the amino acid at position 2. Pro is replaced by a pyrrolidine-3-ol while Ile is replaced by a 6-F-indole. Note that the indole ring is used as a bridge to connect the two symmetric SMAC-mimetic moieties of the drug. Birinapant was tested on MDA-MB-231 cells where it induced CASP-dependent apoptosis after 72 h of treatment, as demonstrated by cleaveage of CASP3 substrate and cell death [135]. Birinapant is well tolerated by animals tested and induces tumour regression in MDA-MB-231 xenograft model [138]. Finally, it showed a good safety profile, pharmacokinetics, and pharmacodynamics [139].
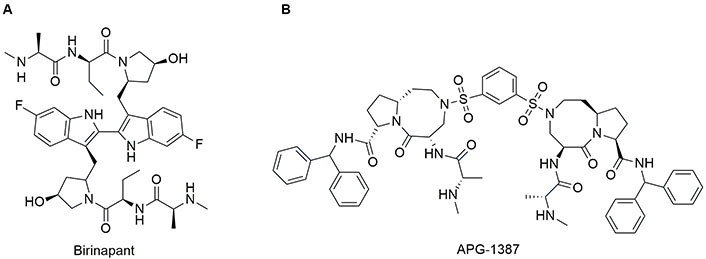
Chemical structure of bivalent second mitochondria-derived activator of caspases (SMAC)-mimetics in clinical trials. See [138] for birinapant and [140] for APG-1387 or visit https://clinicaltrials.gov/
APG-1387 possesses a chemical structure similar to SM406 with a 1,3-dihydrosulfonylbenzene as a linker to connect the two molecules. APG-1387 was developed by the group of Li et al. [140]. It has been demonstrated a synergic effect of APG-1387 in combination with TNF-α in an in vitro model of nasopharyngeal carcinoma in which the apoptosis was inhibited. Notably, in vivo, APG-1387 shows anti-cancer effects as single agent on nasopharyngeal carcinoma. Bivalent SMAC mimetics are summarized in Table 3.
Summary of bivalent second mitochondria-derived activator of caspases (SMAC) mimetics. Bivalent SMAC mimetics in clinical trials and bivalent SMAC mimetics are presented in literature but not discussed in this review
| Compound | Cancer type | Clinical trials | References |
|---|---|---|---|
| Birinapant※ | Acute myelogenous leukaemia | NCT01486784 | [128] |
| APG-1387※ | Nasopharyngeal carcinoma | NCT03386526 | [140] |
| Compound 9a | Breast cancer | - | [141] |
| Compound 3 | Leukaemia | - | [133] |
| BV6 | Rhabdomyosarcoma | - | [142] |
| SM-164 | Leukaemia | - | [143] |
| AEG40730 | Ovarian | - | [144] |
| Compound A | Ovarian | - | [145] |
| Compound 3 | Human glioblastoma | - | [146] |
| SMAC14-2X | Breast cancer | - | [147] |
| SMAC17-2X | Breast cancer | - | [147] |
| Compound 9a | Breast cancer | - | [148] |
※ Drugs in clinical trial for different cancer types; -: no data
Retro-inverse of SMAC/DIABLO
The cytotoxic peptide AVPI and the more active Ala-Val-Pro-Phe (AVPF) show poor stability in vivo with strong limitations in clinical applications [146]. To enhance the metabolic stability, a possible approach is the insertion of unnatural amino acids into the sequence [149]. The strategy developed by Hossbach et al. [150] in 2009 is represented by the insertion of D-amino acids to prepare partially retro-inverse, retro, and inverse SMAC-mimetics. The retro-inverse mimetics (Figure 7) were studied by molecular docking and in vitro on L1236 and L428 Hodgkin lymphoma cell lines, which display high levels of XIAP. Docking analysis suggested fPVA and avPf peptides as promising candidates with the best score obtained. The apoptosis assay was conducted with peptides at the concentration of 50 μM, confirming the docking data and showing that even AVPf, which obtained a low score, is able to inhibit XIAP. Furthermore, fPVA and avPf enhanced the sensitivity of L1236 cancer cells to the action of etoposide.
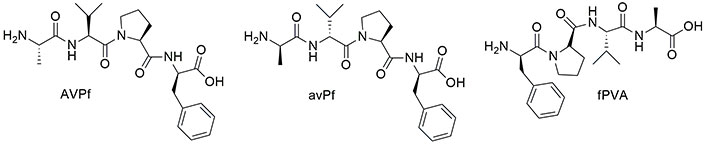
Chemical structure of retro-inverse second mitochondria-derived activator of caspases (SMAC) mimetics (see [150])
SMAC/DIABLO@nanoparticles mimetics
A different strategy to improve the bioavailability, the in vivo distribution, and the cellular uptake of SMAC/DIABLO mimetics toward the tumour site, is the use of nanoparticles (NPs) as nanocarriers functionalized with the cytotoxic peptide [151]. This approach was developed by Li et al. [152]. They used the N-terminal seven residues of the pro-apoptotic SMAC protein, called SMACN7, and connected it with a spacer to poly-Arg as cell penetrating peptide (CPP) (Figure 8A). Then the system was functionalized with a hydrophobic tail able to self-assemble into nanoscale micelles (SMACN7@NPs). The authors used MDA-MB-231 breast cancer cells and H460 lung cancer cells as in vitro models to test the system at the concentration of 20 μM for 2 h. SMACN7@NPs were distributed both into the cell membranes and cytoplasm. To evaluate the apoptosis, cells were exposed to SMACN7@NPs at the concentrations of 5 and 10 μM for 8 and 24 h. The total apoptotic cells increased from 10.4% at 5 μM to 67.1% at 10 μM. The late stage of apoptosis was enhanced from 0.8 to 5.9% at 5 μM and from 1.7% to 40.6% at 10 μM, demonstrating a dose- and time-dependent inhibition of XIAP. To improve the selectivity towards cancer cells, De Marco et al. [153] used the Arg-Gly-Asp (RGD) peptide, to recognize cancer cells specifically [154, 155]. SMAC/DIABLO and RGD peptides were connected directly to the silica NPs to form SMAC/RGD@NP (Figure 8A). The ability of the as-obtained system to activate CASP9 was evaluated on A549, U373, HeLa, Huvec, and fibroblast cells at the concentration of the nano-system of 1 μM for 6 h. They observed more than 40-fold improved activity in cancer cells, while in healthy fibroblasts it was improved only by 10-fold. Further, cellular uptake experiments demonstrated a strong internalization of the nano-system only in cancer cells. A dual-targeting peptide (DTP), in which the AVPI sequence was connected with the RGD peptide by Lys bridge and then directly loaded onto periodic mesoporous organosilica (PMO) NPs, was developed (DTP@PMO) (Figure 8A) [156]. The PMO NPs present an empty interior that can be filled with dyes or drugs. In this case, the authors introduced a fluorescent dye to follow the uptake of the nano-system by flow cytometry [156]. After treatment with DTP@NPs at the concentration of 0.02, 0.04, and 0.06 μg/mL for 48 h, HeLa cells displayed higher sensitivity than HT-29 cells. In addition, DTP@NPs were tested on cell models of acquired resistance to platinum compounds. Indeed, a synergic effect of 6 µM oxaliplatin in combination with 0.6 μg/mL DTP@NPs was observed in colorectal cancer cells after 96 h treatment with a strong decrease in the viability of cancer cells, thus suggesting a promising combined approach to treat colorectal cancers.
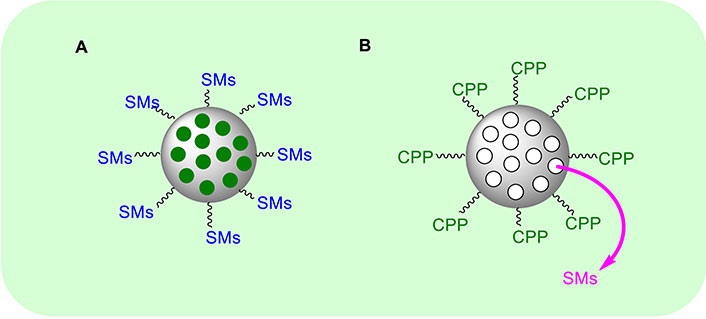
General approaches used to prepare SMAC/DIABLO@nanoparticle mimetics. CPP: cell penetrating peptide; SMAC/DIABLO: second mitochondria-derived activator of caspases/diablo inhibitor of apoptosis protein-binding mitochondrial protein; SMs: second mitochondria-derived activator of caspases mimetics
Further, an interesting approach to overcome the low bioavailability of AVPI peptides was developed by Nejabat et al. [157] who prepared SMAC@NPs mimetic where SMACN6 composed by AVPIAQ sequence as minimum element activating apoptosis via CASPs, was internalized into NPs (Figure 8B). The NPs were formed by PLGA (poly lactic-co-glycolic acid) and PEG (polyethylene glycol) with a di-amine functional group to form PLGA-PEG-NH2. To internalize the cytotoxic peptide, they used the solvent evaporation method. The outer shell of the NPs is functionalized by aptamers (Apts) to form the final system called Apt-NPs-SMACN6. Notably, Apts are able to interact specifically and with high affinity with their target, as in the case of mucin 1 (MUC1) transmembrane MUC glycoprotein that is overexpressed on the epithelial surface of cancer cells. The authors prepared a second system where doxorubicin (Dox) was used instead of SMACN6 peptide to form Apt-NPs-Dox. Both the systems were tested on 4T1, MCF-7, and C26 cell lines. The authors observed that Apt-NPs-SMACN6 improves the sensitivity of cancer cells to the action of Dox. Finally, Nikkhoo et al. [158], loaded chitosan NPs with the bivalent SMAC/DIABLO-mimetic BV6. The authors used carboxymethyl dextran conjugated with trimethyl chitosan (CMD-TMC) to enhance the cellular uptake [159]. The cytotoxic effects were evaluated on breast, colorectal, and melanoma cancer cell lines and an improvement in the apoptosis efficacy was shown when the nano-system was combined with siRNA [158].
Targeting the extrinsic pathway: the TNF family-related approach
From a different standpoint, interesting therapeutic approaches to re-activate apoptosis in cancer cells involve the use of agents that trigger the extrinsic apoptosis pathway.
In the extrinsic pathway, upon binding of appropriate ligands such as TNF, a regulatory cytokine and key signal transduction protein, and TRAIL, a pleiotropic cytokine, the pro-apoptotic DRs induce apoptosis either by using BID as an amplifier of the apoptotic signal (i.e. through the mitochondria involvement) or by the direct activation of the downstream CASPs [160]. However, DR signalling can be dampened by decoy receptors (DcRs) such as DcR3, which is overexpressed in several tumours [161]. Indeed, DcRs can sequester death ligands and compete for them on the cell surface, thus reducing the activation of pro-apoptotic DRs. Furthermore, CASP8 can also be actively degraded in tumour cells [162, 163], extinguishing the downstream apoptotic process.
Several small-molecule compounds targeting TNF-related ligands and their receptors have been developed so far, demonstrating promising activity in promoting cell apoptosis and suppressing the growth of cancer cells [164]. Some have even reached the clinical trial stage such as the TRAIL-inducing compound ONC201 [165], or imipramine (Figure 9), which induces apoptosis in glioblastoma cells by upregulating FASL and sustaining the activating cleaveage of CASP8 [166]. However, none of the TNF-related targeting compounds have been approved to date. It should be pointed out that TNF-based anticancer therapies stimulate systemic inflammatory responses due to TNF receptor-driven activation of NF-κB, an effect that restricts their use to locoregional treatments [167]. On the other hand, TRAIL-Rs present limited effects on NF-κB activation and for this reason, the development of TRAIL and TRAIL-Rs agonists has been favoured [160]. Nevertheless, the efficacy of TRAIL-targeting compounds against cancer is impaired by the widespread dysregulation of the extrinsic apoptosis pathway in such pathological conditions, including the downregulation of TRAIL-Rs and of the downstream signalling mediators [168]. TNF ligands are summarized in Table 4.
Chemical structure of some tumour necrosis factor (TNF) ligands in clinical trials. See [169] for ONC201 and [166] for imipramine or visit https://clinicaltrials.gov/
Summary of tumour necrosis factor (TNF) ligands. TNF ligands are presented in literature but not discussed in this review
| Compound | Cancer type | Clinical trials | References |
|---|---|---|---|
| Tetrandrine | Prostate cancer | - | [170] |
| Tannic acid | Embryonic carcinoma | - | [171] |
| Scutellarein | Hepatocellular carcinoma | - | [172] |
| CPT211 | Triple negative breast cancer | - | [173] |
| Apigetrin | Gastric cancer | - | [174] |
| Gentian violet | Cutaneous T-cell lymphoma | - | [175] |
| Compound 3 | Non-small cell lung cancer | - | [176] |
| Pemetrexed※ | Non-small cell lung cancer | NCT01769066 | [177] |
| ASTX660※ | Head and neck squamous cell carcinomas | NCT05245682 | [178] |
| Ruthenium complex 2c | Melanoma; breast cancer, lung cancer | - | [179] |
| MnIII complex | Triple negative breast cancer | - | [180] |
| Erinacine A | Colorectal cancer | - | [181] |
| C20E | Triple negative breast cancer | - | [182] |
| 1,3-diphenyl-2-benzyl-1,3-propanedione (DPBP) | Melanoma | - | [183] |
| Licochalcone B | Melanoma, squamous cell carcinoma | - | [184] |
| Erinacine S | Gastric cancer | - | [185] |
| Cedrol | Glioblastoma | - | [186] |
| 3β-Acetyl-nor-erythrophlamide (C5) | Lung cancer, lymphoma | - | [174] |
| Demethylzeylasteral (T-96) | Prostate cancer | - | [187] |
※ Drugs in clinical trial for different cancer types; -: no data
Conclusions
Apoptosis, one of the most studied RCD processes, is vital not only to remove defective or unwanted cells but also to foetal and embryonic development as well as to tissue remodelling. Despite the great clinical potential of apoptosis modulators/enhancers and the considerable research effort devoted to the development of effective apoptosis-targeting approaches, the success of such therapeutic interventions in the clinical setting is still limited. This paradoxical limitation is partly due to the on-target toxicity displayed by these compounds as well as by an emerging and still unresolved heterogeneity in the regulation of apoptosis (but also of other RCD) in the context of different pathophysiological conditions, at both different anatomical sites (possibly related to microenvironmental features) and even different stages of cellular differentiation. Notwithstanding, the employment of pro-apoptotic mimetics represents a transformative therapeutic approach in the never-ending fight against cancer. In this respect, a better understanding of the highly interconnected, diversified, and redundant apoptotic molecular machinery in different tumour environments, as well as the discovery of new and more reliable biomarkers to improve patient stratification will enable a rational patient-tailored selection of effective apoptosis modulator molecules, alone or in combination with other therapeutic targets, to enhance clinical outcomes in cancer treatment.
Abbreviations
| Apts: | aptamers |
| AVPI: | Ala-Val-Pro-Ile |
| BAK1: | B-cell lymphoma-2 antagonist/killer 1 |
| BAX: | B-cell lymphoma-2 associated X protein |
| BCL-2: | B-cell lymphoma-2 |
| BCL-XL: | B-cell lymphoma-extra large |
| BCR: | B-cell receptor |
| BH3: | B-cell lymphoma-2 homology 3 |
| BID: | B-cell lymphoma-2 homology 3-interacting domain death agonist |
| BIM: | B-cell lymphoma-2 interacting mediator of cell death |
| BIR: | baculovirus inhibitor of apoptosis protein repeat |
| BTK: | Bruton tyrosine kinase |
| CASPs: | caspases |
| CDK: | cyclin‐dependent kinase |
| cIAP1: | cellular inhibitor of apoptosis protein 1 |
| CLL: | chronic lymphocytic leukaemia |
| Cyt-c: | cytochrome c |
| DcRs: | decoy receptors |
| DISC: | death-inducing signalling complex |
| Dox: | doxorubicin |
| DRs: | death receptors |
| DTP: | dual-targeting peptide |
| FASL: | Fas ligand |
| FDA: | Food and Drug Administration |
| HTRA2/OMI: | human serine protease homologous to bacterial heat shock endoprotease A2 |
| Ki: | inhibitory constant |
| MCL-1: | myeloid cell leukaemia-1 |
| MM: | multiple myeloma |
| MOMP: | mitochondrial outer membrane permeabilization |
| MUC1: | mucin 1 |
| NF-κB: | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NPs: | nanoparticles |
| PEG: | polyethylene glycol |
| PLGA: | poly lactic-co-glycolic acid |
| PMO: | periodic mesoporous organosilica |
| PUMA: | p53-upregulated modulator of apoptosis |
| RCD: | regulated cell death |
| RGD: | Arg-Gly-Asp |
| SMAC/DIABLO: | second mitochondria-derived activator of caspases/diablo inhibitor of apoptosis protein-binding mitochondrial protein |
| TNF: | tumour necrosis factor |
| TNF-R1: | tumour necrosis factor receptor 1 |
| TRAIL: | tumour necrosis factor-related apoptosis-inducing ligand |
| TRAIL-R1: | tumour necrosis factor-related apoptosis-inducing ligand receptor 1 |
| XIAP: | X-linked inhibitor of apoptosis protein |
Declarations
Author contributions
AV and RDM: Conceptualization, Validation, Writing—original draft, Writing—review & editing, Supervision. DL: Validation, Writing—review & editing. All authors read and approved the submitted version.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2024.