
Abstract
Stress is a state of threatened or perceived as threatened homeostasis that can be induced by various external and internal stimuli such as psychosocial factors, inflammatory or injurious conditions, and infections. In order to restore body homeostasis, adrenal glands produce and secrete glucocorticoids (GCs) and catecholamines (CAs), which are the main stress hormones that support the survival and adaptation of the organisms to the new environment. In contrast to the rather beneficial impact of acute and short-lasting stress, chronic stress and related dysregulation of the stress system is implicated in the development of many non-communicable diseases, including cancer. Particularly, ever-increasing experimental and clinical evidence implicates the involvement of CAs and GCs as well as the overexpression of their receptors in the activation of the major pathways involved in tumour development, metastasis, and resistance to various therapies. More importantly, results of experimental and epidemiological studies revealed that overexposure to stress hormones during pre- and early postnatal life might induce life-long or even transgenerational dysregulation of the stress system and predispose it to the development of various tumours. Although the exact mechanisms involved in the latter process are not yet fully known, it has been demonstrated that GC-induced epigenetic modifications can change the expression of several key genes involved in the regulation of the stress system, tumour initiation, and epigenetic imprinting. When such alterations occur in stem/progenitor cells (SPCs), this might not only lead to long-term dysfunction of the stress system but might promote the generation of cancer stem cells (CSCs). This review article discusses a hypothesis that stress hormones-mediated epigenetic reprograming of various SPCs during sensitive developmental periods, might contribute to their dysfunction and increased sensitivity to malignant transformation, thereby promoting tumorigenesis.
Keywords
Foetal programming, stress system, tumorigenesis, chronic stress, glucocorticoid adjuvant therapy, cancer, the hypothalamic-pituitary-adrenal axis dysregulation, cancer stem cellsIntroduction
Stress is omnipresent in daily life. It is a state of threatened or perceived as threatened homeostasis in response to many external and internal stressors [1]. The human body reacts to these stressful stimuli by elucidating an individualized, short- or long-lasting adaptive stress response [2]. The latter event is orchestrated by the stress system consisting of the hypothalamic-pituitary-adrenal (HPA) axis and locus coeruleus (LC)-norepinephrine (NE)/autonomic nervous system (ANS). The ANS comprises the sympathetic-adrenal-medullary (SAM) system and the parasympathetic nervous system (PNS) [3]. The stress response is an evolutionary mechanism designed to help the organism properly respond, survive, and adapt to multiple threats in a constantly changing environment. Exposure to stressful stimuli such as immediate danger, injury, challenge, or panic initiates a fight, flight or freeze response, which enhances arousal, and focused attention, supports cardiac function, restrains immune response, and improves oxygenation and energy supplementation (glucose and free fatty acids) to the brain, heart and skeletal muscles [4]. Stress response is initiated and controlled by several major brain regions such as the hippocampus, amygdala, hypothalamus, and downstream parts of the reward system (i.e., striatum and prefrontal cortex) [5]. With the exception of the freeze response, which is mediated by the PNS and neurotransmitter acetylcholine, the fight-or-flight response is mostly executed by glucocorticoids (GCs) and catecholamines (CAs). These stress hormones are produced and secreted by the adrenal glands and in the case of NE additionally by neurons located in the LC (brain) and the sympathetic nervous system (SNS) (periphery). Stress-induced GCs and CAs secretion depend on coordinated action of multiple stress mediators including, among others, arginine vasopressin (AVP), corticotrophin-releasing hormone (CRH), adrenocorticotropic hormone (ACTH), dopamine (DA), serotonin, oxytocin, pituitary adenylate cyclase (AC)-activating polypeptide (PACAP), or inflammatory cytokines, especially interleukin (IL)-6 [4, 6–8]. Clinical and experimental studies have demonstrated that exposure to short-lasting stressors (spanning maximally a few hours) such as immediate threats, challenges, or a bout of physical activity has an overall positive effect on the organism. Particularly, a short-term stress response was found to enhance protective immunity, improve cognitive function, to stimulate neurogenesis thereby promoting learning processes and adaptation [9–11]. On the contrary, long-term exposure to stressful stimuli leads to dysregulation of the stress system and is associated with increased susceptibility to certain infectious, metabolic, cardiovascular, and neuropsychiatric diseases as well as cancer development [12]. In particular, many experimental, clinical, and epidemiological studies have demonstrated that various chronic psychosocial stress situations, for example, social isolation [13], depression [14, 15], denial, and hopelessness [16] positively correlate with enhanced cancer progression and lower overall postsurgical survival of patients with already-diagnosed cancers [17]. Although the mechanisms linking chronic stress-induced dysregulation of the stress system with tumorigenesis are not fully recognized, many in vitro and preclinical studies demonstrated that enhanced exposure to stress hormones and aberrant expression of their receptors promote several key processes involved in tumour initiation and progression. Those are among others, tumour angiogenesis, and neovascularization [18], immunosuppressive tumour microenvironment [19], promotion of epithelial-to-mesenchymal transition (EMT) and malignancy [20–22], and induction of resistance to chemo- and radiotherapy-induced cell death [23] as well as to immunotherapy [24].
There is increasing evidence that early life stress (ELS) or exposure to stress hormones during sensitive developmental stages or puberty may result in persisting dysregulation of the HPA axis and increased predisposition to chronic stress-related disorders [25]. In fact, multiple retrospective and observational studies performed in humans showed that maternal and early life stressors including exposure to war, malnutrition, psychosocial stress, depression/anxiety, or natural disasters are associated with hypersensitivity of the stress system, low-grade inflammation, and several metabolic complications such as type 1 diabetes [26–28]. Mechanisms involved in this process are complex and involve stress hormone-induced epigenetic changes including, DNA methylation (DNAm) and DNA demethylation, alterations in non-coding RNA (ncRNA) expression or the chromatin structure in the key regulatory brain centres and organs of the stress system [29]. During a late gestational age, rising concentrations of stress hormones are necessary for the preparation of the foetus for the external environment by promoting proper organ maturation and shaping of the stress response, a process known as developmental programming [30]. However, maternal stress or administration of synthetic GCs (sGCs) during the early prenatal stage was shown to hijack this process leading to improper development, maturation, or dysregulation of certain GC-sensitive organs e.g., the lungs, brain, and systems including the immune system or the stress system [31, 32]. More importantly, there is strong experimental evidence that GC and CAs-mediated maladaptive changes may not only permanently affect the adult life of the conceptus but they can also be passed to the next generations through germ cells [33, 34]. However, such long-lasting phenotypic changes observed in somatic cells can only be sustained when an epigenetic stabilization of gene transcription or programming occurs in cells that are capable of self-renewing, are multipotent, and are required for organ development and regeneration [33]. In fact, the ability of stress hormones to modulate various stem/progenitor cell (SPC) populations, including among others, mesenchymal stem cells (MSCs) [35], neural stem cells (NSCs) [9], hematopoietic progenitor cells (HPCs) [36] is increasingly recognized. Prolonged stimulation with sGCs was shown to induce senescence and cell death in NSCs, which might translate into suppressed neurogenesis and a higher predisposition to certain neurodevelopmental or neurodegenerative disorders observed in stressed individuals [37]. Similar effects were observed in the case of induced pluripotent stem (iPS) cells. Particularly, an exposure of these cells to GCs during their differentiation into mature microglia cells was shown to impair the function of the latter cells resulting in enhanced inflammation, cellular senescence, and reduced proliferation [38].
These modulatory effects of GCs on stem cell (SC) populations and the developmental programming process can additionally be exerted by other stress hormones including e.g., CAs [39] and oxytocin [40]. It is therefore conceivable, that ELS-induced epigenetic changes might ultimately alter the functioning of undifferentiated somatic SPCs and their differentiated progeny (daughter cells), which could contribute to the various chronic stress-associated disorders [41, 42].
In summary, the existing data support the involvement of dysregulated stress response including, an enhanced sensitization of the stress system, aberrant secretion of stress hormones, and differential expression of their receptors in tumorigenesis and cancer progression. Furthermore, overexposure of somatic SCs to stress hormones, especially during sensitive periods of human development may hypothetically induce epigenetic reprogramming in those cells thereby affecting their functionality and enhancing susceptibility to malignant transformation in adult life.
Activation and regulation of the stress response
Several internal and external conditions including, anxiety, challenges, hypoxia, malnutrition, injury, systemic infection, toxic chemicals, or physical activity are known to trigger the stress response (Figure 1). This response depends on the nature of stress (infections, physical or psychosocial), its duration (acute or long-term), and various individual factors such as resilience, age, or sex [43]. Depending on these variabilities, the stress response might involve coordinated action of various stress mediators, including e.g., neurotransmitters [epinephrine (E), NE, DA, acetylcholine, and serotonin], steroids (for example, cortisol in humans and corticosterone in rodents), peptides (CRH, ACTH, vasopressin, endorphins, PACAP, oxytocin, leptin, growth differentiation factor 15 or orexin) and inflammatory mediators including prostaglandins, nitric oxide or cytokines [44, 45].
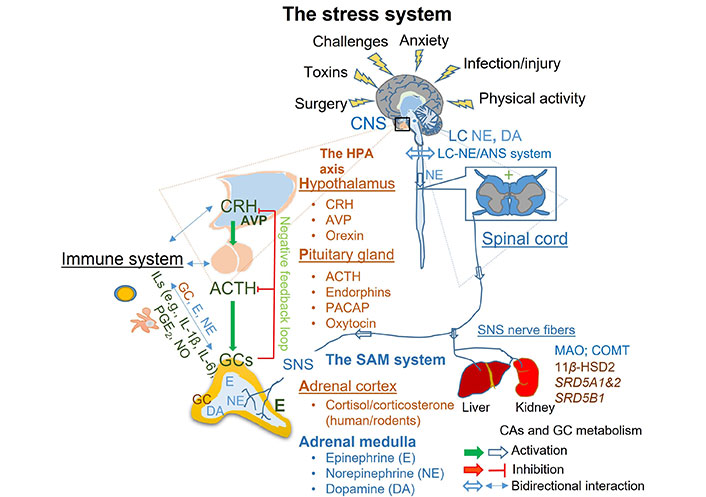
Activation, regulation, and termination of the stress system. Upon initial detection of the major threat by the central nervous system (CNS), the stress response is initiated and mediated through the coordinated action of the HPA and the LC-NE/ANS. The stress response results in rapid local and systemic release of CAs namely NE and E produced by activated LC in the brain stem, SNS, and adrenal medulla. This response is promptly followed by upregulation of CRH and AVP from the hypothalamus, ACTH from the pituitary, and GCs from the adrenal cortex. The stress response is terminated through negative feedback activation and degradation of stress hormones in the kidney and liver. HPA: hypothalamic-pituitary-adrenal; LC: locus coeruleus; NE: norepinephrine; DA: dopamine; ANS: autonomic nervous system; CRH: corticotrophin-releasing hormone; AVP: arginine vasopressin; ACTH: adrenocorticotropic hormone; PACAP: pituitary adenylate cyclase-activating polypeptide; GCs: glucocorticoids; ILs: interleukins; SNS: sympathetic nervous system; SAM: sympathetic-adrenal-medullary; COMT: catechol-O-methyltransferase; 11β-HSD2: 11β-hydroxysteroid dehydrogenase type 2; MAO: monoamine oxidase; PGE2: prostaglandin E2; SRD5A1&2: steroid 5α-reductase types 1 and 2; SRD5B1: steroid 5β-reductase; NO: nitric oxide; CAs: catecholamines
Due to the high complexity of the stress response and current limitations in existing literature, a further focus of this article is on the specific role of the two major mediators of stress namely, GCs and CAs.
Regulation of GC production and action during stress
Acute stress activates neurons located in the nucleus paraventricular of the hypothalamus to secrete CRH and AVP into the hypophyseal portal system, where they reach the anterior lobe of the pituitary gland (Figure 1). In the latter organ, CRH alone or in combination with AVP stimulates corticotropic cells to release proopiomelanocortin (POMC), which is an ACTH precursor. Upon POMC cleavage, ACTH is released into the general circulation through which it can reach the adrenal cortex and stimulate GC synthesis [46]. During various infectious and traumatic events, the systemic and intraadrenal upregulation of inflammatory cytokines and other mediators (e.g., nitric oxide or prostaglandins) can further boost adrenal GC production [47, 48]. In order to limit the adverse effects of prolonged exposure to those hormones and to restore homeostasis, further GC production is reduced in a process known as a negative feedback loop. In the latter process, elevated concentrations of GCs bind their receptors located in the hippocampus, the hypothalamus, and the pituitary gland and reduce further CRH and ACTH production, which limits the activity of the adrenal gland [49]. In addition, GCs inhibit their own signalling through upregulation of the FKBP5, which chaperon protein together with heat shock protein 90 (Hsp90) binds to GC receptor (GR) thereby preventing its nuclear translocation [50].
The peripheral action of GCs is regulated by differential expression and a combination of mineralocorticoid receptors (MRs) and GRs in a cell and tissue-specific manner [51]. Although MR is the main receptor for aldosterone and is expressed mostly in the epithelial tissues of the kidney or liver, it has a 10-fold higher affinity to cortisol (the major GC in humans) as compared to GRs [52]. The high affinity of MRs to GCs and their expression pattern in the brain including, the hippocampus, amygdala, and prefrontal cortex [53], indicates its involvement in the regulation of the stress response. Indeed, a recent study has demonstrated that MRs control the basal expression of the FKBP5 gene and therefore can determine tissue sensitivity to GCs [54]. To date, two main GR subtypes exist namely GR-α and GR-β, which are encoded by the GR gene (NR3C1). They are generated by the alternative splicing of the terminal exon nine, and each of them has at least 8 isoforms resulting from using different translation initiation sites [55]. Genomic actions of GCs are executed mostly by their binding to the cytoplasmic GR-α, which releases Hsp90 and associated FKBP5 protein-mediated inhibition of GR spontaneous nuclear translocation. Upon this process, GC-GR binds to many GC response elements (GREs) located in the promoter- and enhancer-regions of multiple GC-regulated genes [27]. Activation of nucleus-based GR-β and its binding to the GRE elements does not alter gene expression but instead regulates GR-α function [56]. In addition to above mentioned mechanism, GCs exert also non-genomic actions involving membrane-bound GRs [57]. For example, the regulation of the negative feedback loop [58] or suppression of lymphocyte function [59] were found to be regulated by GRs located under the plasma membrane [60], in a process involving GC-induced leucine zipper (GLIZ) proteins [61]. Besides repressing multiple inflammatory transcription factors, such as nuclear factor κB (NF-κB), and modulating the expression of thousands of genes in the nucleus, GCs can also regulate mitochondrial transcription through activation of mitochondrial GREs [62].
During stress, peripheral action and tissue sensitivity of those stress hormones depend largely on organ- and cell-specific GC signalling and the availability of GCs, which is regulated by corticosteroid binding globulins (CBGs) and 11β-HSD2 (11β-hydroxysteroid dehydrogenase type 2) [63, 64]. The latter enzyme is known to transform active cortisol into cortisone, an inactive metabolite, and is therefore highly expressed in the placenta [65], and in some aldosterone-sensitive tissues, like e.g., kidneys, where it limits adverse effects of GC-mediated MR activation. In addition, the bioavailability of GCs is also controlled by their metabolism in the kidney and the liver through the action of several GC degrading enzymes, including 5 α- and β-reductases [66].
Besides the aforementioned complex neuroendocrine interactions, a proper adrenal gland GC response during stress depends on the availability of cholesterol the major substrate for steroidogenesis [67], hereditary factors [68], and the crosstalk between multiple cell types coexisting in the adrenal microenvironment, such as adrenocortical, chromaffin, endothelial, neuronal, immune, and glial cells [69, 70].
Regulation of CA production and action during stress
Stress conditions induce rapid production and release of NE from noradrenergic neurons of the LC/NE brainstem nuclei in the brain, postganglionic sympathetic nerve terminals in various organs, and from the chromaffin cells of the adrenal medulla (Figure 1) [71, 72]. In addition to NE, the fight or flight response is characterized by a rapid release of E from the adrenal medulla into general circulation. E production is tightly regulated by the sympathetic-adreno-medullary system composed of the SNS- and the adrenal medulla. The exquisite production of E by the chromaffin cells in the adrenal medulla relates to their unique expression of phenylethanolamine N-methyltransferase (PNMT), which is a GC-regulated enzyme that catalyses E generation through methylation of NE [73]. During stress, the two abovementioned major arms of the stress system interact and support each other function at the level of the CNS [74] and the adrenal gland [75, 76]. CAs are known to induce prompt changes in the physiology of many major organs including the heart, lungs, and muscles. They also exert a major impact on the function of the neuronal system, immunity, and metabolism [77]. Cellular responses to those hormones depend largely on the differential expression of two main classes of adrenergic receptors (ADRs), namely α-ADR (ADRA) and β-ADR (ADRB) present in cellular membranes. These responses can be additionally differentiated in a cell-specific manner due to the presence of nine known subtypes of those receptors, α1 (1A, 1B, 1D), α2 (2A, 2B, 2C), and β (β 1–3) [78]. CAs bind to their respective ADRs, which as in the case of ADRB alters confirmation of these G protein-coupled receptors allowing their Gs-α subunit to activate AC. Upon activation, AC promotes the generation of a 3’, 5’-cyclic adenosine monophosphate (cAMP) a secondary messenger capable of binding to regulatory subunits (R2) of protein kinase A (PKA). This step leads to PKA activation and allows it to phosphorylate several target proteins including members of the mitogen-activated protein kinases (MAPKs) family. Consequently, activated MAPKs enhance the transcriptional activity of cAMP response element-binding protein (CREB), which transcription factor is responsible for differential gene expression and many post-translational protein modifications induced by CAs. After transducing signals, ADRs are phosphorylated by PKA or G protein-coupled receptor kinases and internalized, which temporarily leads to their desensitization [79]. During chronic stress, CAs are known to alter the expression of several gene sets including those associated with the organism’s defences, adaptation, and tissue tumorigenesis [19]. Besides altering the gene expression, CAs were also shown to introduce posttranslational protein modifications [19, 80]. Unlike activation of the HPA axis, stress-induced SAM activation and related CA production were found to be preserved or even enhanced following repeated exposures to the same stressor [81]. Biological functions of CAs can be promptly terminated by their reuptake into neurons, in which cells they can be either translocated back into the storage vesicles through the vesicular monoamine transporters (VMATs) or undergo degradation by MAO [82]. In non-neuronal cells, CA metabolism can also occur through COMT action. Finally, due to their short lifetime of approximately two and a half minutes, plasma CAs are rapidly degraded and cleared from circulation, thereby decreasing their systemic actions [83].
Dysregulation of the stress system
Chronic psycho-socioeconomic stress, early life adversity, depression, post-traumatic stress disorder, obesity, or prolonged use of sGCs are conditions usually associated with dysregulation of the HPA axis [4]. The latter condition is characterized either by hyper- or hyposecretion of GCs and alterations in peripheral and central expression and activity of their receptors [84–86]. In most cases however, chronic stress-induced dysregulation of the stress system is often manifested by the flattening of GC production pattern and complete loss of its normal daily circadian and ultradian rhythms [87]. During this condition, constantly elevated cortisol concentrations may continuously affect the expression of thousands of genes in various human cells, leading to stress-induced pathologies. For example, a persisting overexposure to GCs suppresses the expression of its own receptor (GR-α) in the HPA regulatory brain regions, and in immune cells, which contributes to dysregulation of negative feedback and low-grade inflammation, respectively [88, 89]. This can result in dysregulation of the stress response evidenced by attenuation of the awakening pick in cortisol concentrations, which might be associated with morning fatigue, pain, and inflammation [90].
Moreover, unlike the HPA axis response, stress-mediated CA secretion is enhanced during the encounter of the second stimuli. This can lead to steadily increasing activation of the SAM system. In fact, experimental studies have revealed that maternal stress or prenatal GC injections in rats induce the adrenal medulla hyperfunction, through mechanisms involving enhanced activation of the SNS [91] and related elevation of PNMT expression [92].
Mechanisms involved in the chronic- or ELS-induced dysregulation of the stress system are complex and depend on many additional factors including e.g., resilience, timing, type of stressor, or sex [93]. Nevertheless, an ever-increasing number of studies demonstrate the key involvement of the stress hormone-induced epigenetic alterations in cells constituting the stress system, and its regulatory brain centres may be involved in this process [94–96]. Furthermore, considering the life-long or even hereditary consequences of their actions, it is highly conceivable that SPCs might be the target cells [41, 42].
Epigenetic-based mechanisms involved in the HPA axis dysregulation
One of the most extensively studied examples of stress hormone-induced dysregulation of the stress system and related development of metabolic and psychological diseases is sGC supplementation in case of preterm deliveries. Unlike natural exposure of a foetus to GCs during late gestational age, which is necessary for the proper maturation of various organs including the lungs, brain, or kidneys, administration of sGCs [primarily betamethasone and dexamethasone (DEX)] [97, 98] to pregnant women with threatened preterm birth, might result in dysregulation of the stress system in the offspring [99]. Particularly, prenatal exposure to sGC resulted in age-, sex- and pregnancy stage-dependent increases or decreases in cortisol response to stress [100], and dysfunctional negative feedback in the offspring [101]. Moreover, those events were positively associated with psychological, metabolic, cardiovascular, and neuroendocrine dysfunction in adult life [102–105].
Further studies revealed that maternal chronic stress and synthetic GCs administration during prenatal and early postnatal life [104], two more sensitive times in child development, might induce a life-long or even transgenerational predisposition to the HPA axis dysfunction [106]. During that time, the genome shows high molecular plasticity, which allows epigenetic-based stabilization of transcriptional responses induced by environment-genome interactions [107] allowing adaptation to stress. During such a process, also known as developmental programing, prenatal GC administration might induce several maladaptive epigenetic changes in genes involved in the regulation of the stress system and GC signalling. Several epigenetic modifications that are known to persistently alter gene expression patterns leading to defined phenotypic changes are introduced by GCs. These include, among others, DNAm and DNA demethylation of various cytosine-guanine dinucleotides (CpG)-rich DNA regions situated close to GRE sites and global DNA demethylation steps [31, 85]. DNAm changes are mediated by GC-mediated induction or repression of several members of the ten eleven translocation (TET) family of 5-methylcytosine dioxygenases, which are actively involved in the demethylation process, as well as by altering the expression of DNA methyltransferases (DNMTs), such as DNMT1, DNMT3a and DNMT3b [108, 109]. Those tissue-specific [110] and transgenerational epigenetic actions of GCs are not limited to DNAm but were also found to include histone modifications (acetylation and deacetylation) [111], microRNAs (miRNAs) abundance [112] and chromatin remodelling [113].
Multiple clinical studies indirectly support the role of aforementioned epigenetic modifications in chronic stress-induced HPA axis dysregulation and foetal developmental programming. For example, differential DNAm of NR3C1, CRH, CRHBP, and FKBP5 genes that are involved in the activation and regulation of the stress response were found in mothers and children of war survivors [114]. This study indicated that high methylation of GRs in core blood and lower methylation of CRH in venous blood of traumatized persons could predict lower birth rates. Moreover, it also demonstrated enhanced methylation of the FKBP5 gene in the placentas of persons who experienced chronic stress and war [114].
Chronic stress, maternal depression, or perinatal stress were also found to enhance methylation of biologically relevant regions of the GR exon 1F promoter (NR3C1-1F), which was associated with increased cortisol stress response [115, 116]. In line with these data, a substantial correlation between different forms of prenatal stress and the GR-α1F methylation at CpG site 36 was found in a meta-analysis study involving in total of 977 individuals [117]. Furthermore, DNAm of GR-α1F in adults exposed to various grades of childhood trauma was positively correlated with laboratory stressor [Trier Social Stress Test (TSST)]-induced cortisol levels and could predict a specific direction of HPA axis dysregulation (hypo- vs. hyperactivity) [118]. Particularly, peak cortisol concentrations in response to the TSST were 62.3% higher in childhood trauma survivors who had increased NR3C1-1F DNAm as compared to those with low DNAm. However, the therapeutic usefulness of differential methylation of the NR3C1-1F promoter in peripheral blood cells in predicting an individual patient’s cortisol stress response may be difficult. It seems that GC stress responses might be in fact multifactorial and therefore results of various studies analysing this correlation should be more carefully interpreted [119]. For example, depending on the stress type, the number of CpG sites analysed, and the general quality of the study [94, 120], results and interpretations may differ. It has been shown that various early life stressors, including e.g., child trauma, depression, or anxiety, may be either associated with hypermethylation [115, 116] or hypomethylation [121] of NR3C1-1F promoter. On top of that, there are also studies showing no association between the abovementioned stress conditions and methylation of the NR3C1-1F promoter [119].
In addition to the GR-α1F, differential methylation of the FKBP5 gene, which is known to inhibit GR function [122], was found in various persons exposed to chronic stress. For example, in persons who experienced childhood trauma [123], or suffering from excessive GC secretion (Cushing’s syndrome), enhanced demethylation of specific CpG fragments of the FKBP5 gene [85] was reported. Demethylation of the FKBP5 gene found in immune cells in aged or psychosocially stressed individuals was later found to correlate with enhanced expression of this GR suppressor and low-grade inflammation [50].
Most of the evidence pointing to a link between epigenetic mutations induced by chronic- and early-life stress-dysregulated negative feedback mechanisms comes from experimental studies. In one such study social stress applied to pregnant rats enhanced the HPA responsiveness to stress in their offspring through increased hypothalamic CRH messenger ribonucleic acid (mRNA) and decreased hippocampal NR3C1 (GR-α) mRNA expression, which indicates a reduced GC negative feedback [124]. This observation is in line with other experimental studies showing similar pronounced GR-α downregulation in the hippocampus and other regulatory centres of the HPA axis [33]. Most convincingly, GR overexpression induced specifically in the hippocampal region [125] could ameliorate the negative consequences of stress-induced GR-α downregulation. Those results are also supported by the studies showing that deletion of GR-α (Nr3c1) specifically in the forebrain region including the hippocampus, can enhance GC production and impair the function of negative feedback [101]. However, enhanced global DNA hypomethylation in the hippocampus and frontal cortex [126] and region-specific hypermethylation [107] were also observed in other studies. Moreover, mechanisms involved in the negative regulation of the HPA axis by chronic stress might depend on the type and length of stress. For example, it has been demonstrated that an enhanced HPA activity (ACTH responsivity) rather than suppressed rapid GC negative feedback was involved in the HPA dysregulation in male rats undergoing chronic variable stress [92].
Furthermore, while most studies focused only on the key involvement of brain regulation, others investigated additionally the role of peripheral organs in the prenatal- and early postnatal stress-induced dysregulation of the stress system. For example, ELS-induced DNA hypomethylation permanently enhanced the expression of hypothalamic AVP [127] and pituitary POMC genes [128] resulting in sustained elevation of ACTH. Similarly, prenatal exposure to GCs was also shown to enhance the activity of the adrenal gland itself, through a mechanism involving DNA hypomethylation [129], impaired behavioural rhythms, and altered clock gene expression in the suprachiasmatic nucleus (SCN) [32]. Interestingly, dysregulation of the adrenal gland function was demonstrated to be age- and sex-dependent, as prenatal administration of GCs in male but not in female rats, resulted in an enhanced basal corticosterone production and in increased adrenal gland weight of 6 months-old rats, which effect was reversed at 12 months [130].
To sum up, the results of the abovementioned experimental and human studies strongly implicate the role of stress-hormone-induced epigenetic changes in the dysregulation of the stress system induced by chronic stress- and early-life stress.
Stress hormones-induced alteration in the function of SPCs and its possible role in stress system dysregulation
Chronic stress and exposure to sGCs during prenatal or early life can cause a lifelong or even transgenerational dysregulation of the stress system through mechanisms involving an epigenetically driven process known as developmental programming [104]. Such long-lasting phenotypic changes, however, can only be sustained when an epigenetic stabilization of gene transcription or programming occurs in the cell population that is capable of self-renewing, is multipotent, and is required in organ development and regeneration [41, 131].
The ability of stress hormones to modulate various stem/progenitor populations is increasingly recognized. In particular, GCs were found to promote the adipogenic differentiation pathway in MSCs through the induction of the master adipogenic regulator peroxisome proliferator-activated receptor γ (PPARγ) [35]. Besides the mesenchymal lineage, GCs can also alter the function and number of hepatic stellate cells (HSCs), which has been evidenced by the stress-induced expansion of embryonic [132] and adult HSCs [36].
Considering the aforementioned findings and high expression of GRs in the organs of the stress system, it is highly expected that acute and chronic exposure of the hippocampal, hypothalamic, pituitary, and adrenomedullary SPCs to stress hormones can induce such long-lasting changes thereby permanently altering their function [133].
One of the important brain regions that are involved in the regulation of GC-mediated negative feedback is the hippocampus. Due to its high content of CRH neurons and elevated expression of CRH-, MR-, and GRs, the hippocampus is particularly sensitive to the action of stress hormones [134]. More importantly, high plasticity accompanied by persistent neurogenesis, which is unique for this region, makes the hippocampus simultaneously one of the most sensitive regions for the adverse effects of chronic stress or ELS [135, 136]. Acute and especially chronic stress were found to differentially modulate neurogenesis, in a process that was highly dependent on a developmental stage, location, and nature of the exposure. Particularly, while short-term stress, such as a bout of physical activity, was shown to induce proliferation and survival of newly born neurons and to promote neurogenesis, chronic stress or exposure to GCs was shown to decrease NSCs number and impair generation of new neurons [9]. This is in line with the results of an elegant experimental study, which demonstrated that physiological but not stress-induced CRH concentrations were required for proper neurogenesis [137]. Recent in vitro and in vivo studies revealed that acute treatment with GCs could reduce proliferation and neuronal differentiation of various embryonic human and rat neuronal SPCs, in which process TET3-demethylase-mediated upregulation of Dickkopf 1 (DKK1) and downregulation of DNMT3a was shown to be involved [108, 138]. Most importantly, prenatal GC administration confirmed the heritable nature of this effect. Moreover, increased accumulation of the DKK1 in the hippocampus, which is a known WNT [the mammalian wingless-type mouse mammary tumour virus (MMTV) integration site] antagonist, was previously implicated in the age-related decrease in neurogenesis [139]. A similar GC-mediated effect has been also verified in another in vitro study, in which DEX treatment of embryonic rat cortical NSCs induced premature aging of those cells, which effect was transmitted to their daughter cells. In particular, DEX-stimulation of NSCs was shown to inhibit their proliferation and change the expression of genes involved in cellular senescence e.g., Cdkn2a (p16) and Cdkn1a (p21), and mitochondrial function e.g., mitochondrial nicotinamide adenine dinucleotide dehydrogenase 3 (mt-Nd3). These observations have been associated with DNA hypomethylation, reduced expression of DNMTs, enhanced susceptibility to oxidative stress and apoptosis [37]. Therefore, stress-relevant elevation of GC concentration in the brain especially during key developmental time may not only impair NSC-driven neurogenesis but also induce aging and decrease the number of those multipotent cells. In the latter process, enhanced autophagy of the [sex determining region Y (SRY)-box 2] (SOX2)-expressing hippocampal neuronal SPCs was implicated [140]. Furthermore, it has been shown that exposure of human HPC line to GCs can permanently change their DNAm and lead to vulnerability of those cells to secondary acute GC challenge [29]. The latter observation suggests that reprogramming of the hippocampal SPCs might be involved in dysfunctional stress response.
Activation of the HPA axis during stress depends additionally on the activity of CRH-expressing neurons located in the paraventricular nucleus (PVN) region of the hypothalamus. Using an in vivo optical recording of neuronal activity, it has elegantly demonstrated that while stress-induced rapid activation and adaptation of the hypothalamic CRH neurons to the same stressors, their function in stress-free conditions was reduced by GCs [141]. Suggesting that GCs may not only control the activation of these hypothalamic CRH neurons during stress but also affect their function during physiological conditions. The hypothalamus has been recognized as an additional centre of neurogenesis and the NSC niche in the brain [142]. In particular, lineage-tracing experiments provided evidence that specialized radial glial cells that can be found in the third ventricle of the arcuate nucleus, called tanycytes, represent the human NSC (hNSC) population [143]. Similarly to the hippocampal NSCs, the application of early stressors during the first days of life was found to decrease the proliferation and numbers of these SOX2+/Nestin+ adult NSCs [hypothalamic NSCs (htNSCs)] [144]. Furthermore, it has been demonstrated that those hypothalamic SPCs may directly respond to and adapt blood-born signals in response to various inflammatory, metabolic, and aging stress conditions. Interestingly, stress-induced loss of adult hypothalamic neuronal SPCs lowered the secretion of various exosomal miRNAs from those cells to the cerebrospinal fluid, which correlated with the incidence of dietary obesity, pre-diabetes, and aging [145]. Finally, the transcriptomic analysis of DEX-treated embryonically derived hypothalamic neural-progenitor/stem cells (NPSCs) demonstrated differential expression of many GR-regulated genes e.g., Fkbp5, cystic fibrosis transmembrane conductance regulator (Cftr), and family with sequence similarity 107 member A (Fam107a). The high induction (300-fold) of the Fam107a gene encoding for a discoidin domain receptor 1 (DDR1) [146] was additionally verified on the protein level in the hippocampus and the hypothalamus [147]. Overexpression of DDR1 was shown to enhance the activity of microglial cells and increase the expression of metalloproteases, which was linked with blood-brain-barrier disruption [148]. On the contrary, DDR1 inhibition was cytoprotective, as it prompted the autophagy-mediated removal of neurotoxins from the neuronal cells [149]. Those observations indirectly suggest that DDR1 upregulation may be involved in the neurodegeneration process, which is observed in the case of chronic stress.
In addition to the hypothalamus, stress can also affect the number and function of SPCs in the pituitary gland. For example, it has been revealed that while stress decreases the number of SOX2- and SOX9-expressing pituitary SPCs, it promotes also their mobilization and differentiation into major endocrine cell types [150, 151]. Furthermore, it has been found that perinatal administration of DEX to mice impaired the negative GC-feedback mechanism in 3-month-old animals by reducing the annexin A1 (ANXA1)-dependent inhibitory actions on ACTH release [152].
Chronic stress was also shown to influence the functioning of the SPCs in the adrenal gland, which is the main effector organ of the stress system. Prolonged ACTH stimulation of the adrenal cortex during chronic stress conditions, often results in an enhanced weight of the adrenal glands, hypertrophy of the cortisol-producing cortical region (the zona fasciculate), the medulla, and reduced cell size of the aldosterone-producing region (zona glomerulosa) [153]. Moreover, an enhanced proliferation in the subcapsular region in the outer zone of the adrenal cortex containing the Sonic hedgehog (Shh)-expressing cells and in the capsular region containing Gli1-positive adrenocortical SPC population was reported [154, 155]. Although the chronic- or ELS-stress-induced possible changes in the epigenome of the Gli1+ Wilms’ tumour 1 (WT1)+ adrenal capsular SPCs have not been directly addressed, the acute responses of those cells to severe stress were recognized [156]. Recently a hypothetic involvement of specific epigenetic changes and the contribution of different stem/progenitor pools was demonstrated in sex-specific differences in the regenerative capacity of the adrenal cortex [157, 158]. The latter observation suggests that stress-induced programming might also affect the functioning of adult adrenal SPCs. However, additional studies are required to further clarify the involvement of acute and chronic stress on the functionality of those cells.
Despite being considered a non-regenerative organ, the adrenal medulla was recently found to contain several embryonic and adult adrenomedullary SPCs, which can respond to various chronic stressors [159, 160]. Most of the CA-producing cells are derived from neural crest SPCs that after the acquisition of a Schwann cell precursor (SCP) fate and initiation of SOX10 expression migrate along the nerves to form the adrenal medulla [161]. The maintenance of the adrenal medulla in postnatal life was shown to be supported by Nestin-positive, self-renewing, and multipotent SPCs [159, 162]. Using cell fate tracing and a repetitive restraint stress model, stress-induced differentiation of those cells towards the endocrine, neural, and glial lineages at the cost of decreased cell number was observed [160].
Collectively and as summarized in Figure 2, chronic- or ELS-stress stress can influence multiple SPCs present in the stress system, promoting their recruitment, proliferation, differentiation, and cell death. Furthermore, through the accumulation of the epigenetic changes induced by stress hormones in those SPCs, various stable epigenetic modifications are introduced and stabilized that subsequently alter their response to the second challenge. Thus, chronic stress might alter the function of SPCs of organs constituting the stress system potentially contributing to its dysregulation.
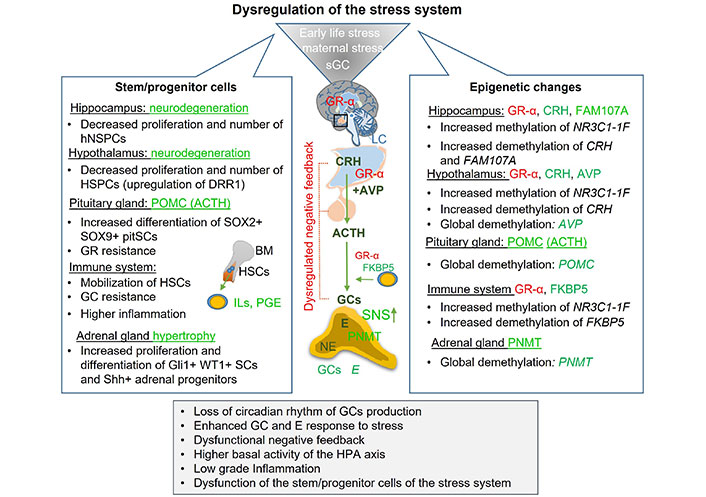
Mechanisms involved in the chronic- and early-life stress-induced dysregulation of the stress system. Overexposure to the stress hormones was shown to impair function and decrease proliferation and the number of various SPCs of the stress system. In addition, chronic pre- and postnatal stress or sGC therapy was shown to introduce several epigenetic modifications in genes involved in the activation, termination, and regulation of the stress system, leading to its dysregulation. Green symbolizes activation or upregulation, while red downregulation or inhibition. sGC: synthetic glucocorticoid; GR-α: glucocorticoid receptor-α; LC: locus coeruleus; CRH: corticotrophin-releasing hormone; AVP: arginine vasopressin; ACTH: adrenocorticotropic hormone; GCs: glucocorticoids; E: epinephrine; NE: norepinephrine; SNS: sympathetic nervous system; PNMT: phenylethanolamine N-methyltransferase; DRR1: discoidin domain receptor 1; SOX: sex determining region Y-box; pitSCs: pituitary stem cells; HSCs: hepatic stellate cells; WT1: Wilms’ tumour 1; SCs: stem cells; Shh: Sonic hedgehog; HPA: hypothalamic-pituitary-adrenal; FAM107A: family with sequence similarity 107 member A; NR3C1-1F: glucocorticoid receptor exon 1F promoter; POMC: proopiomelanocortin; BM: bone marrow; HSPCs: hypothalamic stem and progenitor cells; hNSPCs: hippocampal neural stem and progenitor cells; ILs: interleukins; PGE: prostaglandin E
Involvement of stress system dysregulation in tumorigenesis and cancer progression
An increasing number of studies demonstrate that maternal- or early-life stress-induced dysregulation of the stress system involving increased or decreased concentrations of stress hormones as well as an alteration in the expression of their receptors, might promote several key processes involved in tumour initiation and progression. Those are among others, tumour angiogenesis, and neovascularization [18], immunosuppressive tumour microenvironment [19], promotion of EMT and malignancy [20–22], and induction of resistance to chemo- and radiotherapy-induced cell death [23] as well as to immunotherapy [24]. Therefore, an enhanced predisposition to tumour development in adult life might result from a life-long dysregulation of the stress system. Indeed, there is evidence that ELS such as childhood adversity might increase a person’s cancer risk [163, 164]. This statement is further supported by other epidemiological studies showing the important role of psychosocial stress, such as depression, social isolation, distress, or trauma in the more rapid progression [14, 165] and development [166, 167] of various tumours. In particular, poverty, lack of education and social support, immigration status, and social isolation were found to correlate with the time of diagnosis, stage, and overall survival of patients with colorectal [167] and breast cancer [166]. Moreover, it has been shown that enhanced psychological distress, depression, or anxiety is associated with lower quality of life, worse treatment outcomes, and decreased survival of patients with prostate [168] or epithelial ovarian cancers [15].
Potential involvement of the HPA axis dysregulation in tumorigenesis
Chronic psycho-socioeconomic stress, depression, post-traumatic stress disorder, obesity, or prolonged use of sGCs were all found to be associated with dysregulation of the HPA axis, which is characterized either by hyper- or hyposecretion of GCs and enhanced peripheral expression and activity of their receptors [84–86]. Furthermore, in many experimental, pre-clinical, and clinical studies, the strong association between the HPA axis dysregulation and phenotypic hallmarks of cancer was documented. In one such study, high expression of stress hormone receptors in primary tumours was shown to be associated with their enhanced aggressiveness. Particularly, an analysis of 481 cases of human ovarian cancer tissues demonstrated an enhanced expression of GRs especially in high-grade and advanced-stage ovarian tumour tissues, which was associated with poor prognosis [169]. One of the possible explanations for this observation is that GR overexpression is required for tumour dissemination to other organs. Indeed, in the case of prostate cancer, enhanced expression of GRs was only evidenced in metastatic lesions [170] but not in primary cancer. In addition, elevated GR expression was positively correlated with increased mammary tumour cell invasion and lung metastasis in drug-resistant breast cancer patients [171].
Interestingly, the actions mentioned above of GR may be modulated by other nuclear receptors specific for certain endocrine tumours, such as androgen receptors (ARs), estrogen receptors (ERs), or progesterone receptors (PRs). In particular, high expression of GRs in primary breast tumours was correlated significantly with shorter relapse-free survival, but only in the early stage, ER-negative tumours. On the contrary, high GR expression in patients with ER-expressing tumours was beneficial for their survival [172]. A recent meta-analysis of twenty-five clinical studies verified these data showing that high GR expression was associated with enhanced disease progression in cases of endometrial cancer, ovarian carcinoma, and triple-negative breast cancer [173]. Using a human-relevant mouse model of metastasising breast cancer, in which experimental study, breast carcinoma cells are injected into pre-cleared mammary fat pads (orthotropic transplantation), activation of GRs at distant metastatic sites was found to be required for colonization and heterogeneity of metastases, as well for reduced survival of transplanted mice [22]. However, results obtained from those studies should always be carefully interpreted as injections of carcinoma cells to immune-deficient mice are devoid of natural immune-cancer interactions. In addition to breast cancer, another prime example of modulatory actions of other nuclear receptors is evidenced in the case of prostate tumours. Particularly, high expression of GRs was observed in human prostate cell lines treated with AR antagonists [170]. Elevated expression of GRs correlated with increased growth of prostate cancers especially in AR-negative tumours, by mimicking AR actions such as upregulation of AR-regulated genes and limiting the effectiveness of standard chemotherapeutic therapies. Based on these results, evaluation of GR expression was suggested to be considered in patients with prostate cancer receiving enzalutamide, an inhibitor of the AR. This statement is based on a study showing that AR blockade can enhance local cortisol concentrations through suppression of 11β-HSD2 and promote GR activation in human models of prostate cancer [174].
Multiple studies have implicated the GR expression and activation in the chemoresistance of various tumours. In the latter case, GR-related elevation of chemotherapy resistance-associated oncoproteins, such as lens epithelium-derived growth factor p75 (LEDGF/p75) and clusterin (CLU) were implicated in the potential mechanism of tumorigenic GR actions [175, 176]. A recent study has demonstrated that GR can directly bind to LEDGF/p75 promoter regions and enhance its expression. Consequently, downregulation or knockdown of GR in prostate carcinoma cell lines led to the downregulation of LEDGF/p75 expression, reduced clonogenicity and tumoursphere formation as well as enhanced their sensitivity to docetaxel-induced tumour cell death [177]. As for the possible mechanism, GR-induced CLU upregulation was shown to inhibit DNA damage-induced apoptosis through mechanisms involving e.g., stabilization of Ku70 (70 kDa subunit of Ku antigen) and Bcl-2-associated X protein (Bax) interaction, which prevents its translocation to the outer mitochondrial membrane and subsequent release of cytochrome c (cyto c) [178]. Despite its known proapoptotic actions in the case of lymphoid malignancies [179], GCs were demonstrated to reduce the efficiency of various chemotherapeutic and radiotherapeutics regimens. Particularly, a retrospective analysis of glioblastoma patient cohorts and results of studies using a disease-relevant mouse model of this highly aggressive brain cancer have demonstrated that GC adjuvant therapy inhibited chemotherapy and radiotherapy-induced cell death [180]. Similarly, administration of synthetic GCs such as mifepristone or CORT125134, reduced carboplatin/gemcitabine-induced cell death of intraperitoneally transplanted human ovarian carcinoma cells in mouse xenotransplantation model of metastasising ovarian carcinoma. In the latter case, GCs-mediated upregulation of pro-survival genes including serum and GC-regulated kinase 1 (SGK1) and dual-specificity protein phosphatase 1 (DUSP1) were found to play a key role [181] possibly. In particular, GC-induced SGK1 activation was shown to promote mouse double minute 2 homolog (MDM2)-mediated p53 degradation, thereby preventing its tumour-suppressing and proapoptotic actions [182].
In addition, chronic stress-induced excess of GC hormones was found to interfere with immune therapies by suppressing the presentation of various tumour antigens to dendritic cells (DCs), inhibiting T-cell activation, and antitumour activity [183]. Particularly, the elevation of endogenous GCs in response to repeated social defeat and acute restraint stress was associated with the suppressed effectiveness of the immunogenic chemotherapy (oxaliplatin) and immunotherapy [programmed cell death protein 1 (PD-1) blockade] in urethane-induced lung cancer and colitis-associated colorectal cancer models. In the latter process, stress-induced activation of the GC-inducible molecule, TSC22D3 [glucocorticoid-induced leucine zipper (GILZ)] in tumour-infiltrating DCs, which inhibited type I interferon (IFN) responses in those cells and IFN-γ+ T-cell activation, was suggested as a possible mechanism [24, 183]. Most interestingly, recent data suggest that multiple tumour types can actively produce active GCs through intratumoural reduction of cortisone to cortisol mediated by the 11β-HSD1 enzyme. Enhanced tumour cortisol production was shown to promote an anti-inflammatory environment by attraction and activation of regulatory T cells (Treg), which are known to suppress conventional T-cell activity [184]. For a more detailed description of the immunomodulating effects of chronic stress and stress hormones especially in the context of suppression of protective immunity and tumour immune microenvironment please refer to recently published comprehensive review articles [185, 186].
Chronic stress-enhanced GC concentrations and overexpression of their receptors [22, 187] were also shown to enhance tumour cell proliferation in vitro and pre-clinical mouse models. For example, DEX administration increased the severity of dextran sulphate sodium-induced ulcerative colitis and promoted associated cancer development through mammalian target of rapamycin (mTOR) activation and related modulation of immune cell recruitment [188]. In another experimental study, stress levels of corticosterone were found to promote azoxymethane- and dextran sulphate sodium-induced colorectal adenoma and carcinoma in mice [187]. In the latter process, corticosterone treatment was associated with NF-κB and cyclooxygenase-2 (COX-2) upregulation in these tumours [187]. However, such experimental results have to be carefully interpreted and they might depend on the experimental design. Especially, because GCs are well-known inhibitors of COX-2 and NF-κB expression [189]. Moreover, their enhanced accumulation in colorectal cancer following 11β-HSD2 inhibition was shown to reduce COX-2 expression and prevent tumour growth, and metastasis in mice. Finally, as demonstrated in the case of various colon cancer cell lines, the differential expression of GR-α protein and response to DEX treatment may be a limiting factor in the clinical translation of that data [190].
Involvement of CAs and ADRs in tumorigenesis
Some retrospective studies have also investigated the role of ADRs in tumour progression. In one such study, using 237 human distinct ovarian cancer tissues, an elevated expression of β2-ADR (ADRB2) was found in around 19% of investigated cases, and it positively correlated with high levels of anxiety symptoms and depression symptoms in those patients, but not with overall ovarian cancer mortality [191]. The more direct connection linking chronic stress-associated elevation of stress hormones and their signalling with tumorigenesis is based on experimental studies using animal models of human cancers (Figure 3).
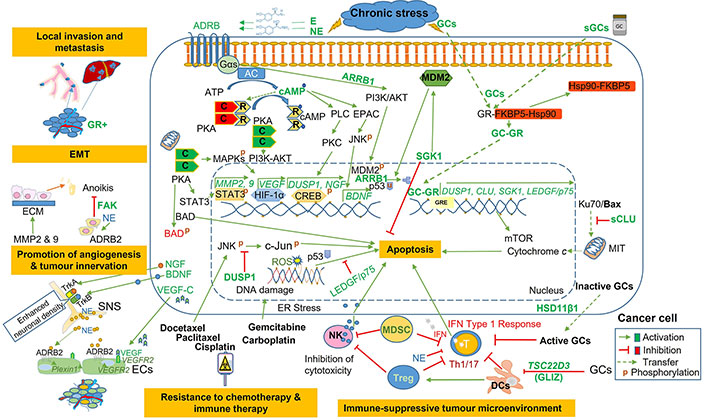
Pathways involved in chronic stress- and stress-hormones-induced tumorigenesis. Elevation of stress hormones (CAs and GCs) and upregulation of their receptors (ADRB2 and GR-α) promote several hallmarks of tumorigenesis. GR: glucocorticoid receptor; EMT: epithelial-to-mesenchymal transition; NE: norepinephrine; ADRB2: β2-adrenergic receptor; SNS: sympathetic nervous system; Treg: regulatory T cells; IFN: interferon; DCs: dendritic cells; GCs: glucocorticoids; GLIZ: glucocorticoid-induced leucine zipper; E: epinephrine; sGCs: synthetic glucocorticoids; AC: adenylate cyclase; PKA: protein kinase A; cAMP: 3’, 5’-cyclic adenosine monophosphate; MDM2: mouse double minute 2 homolog; SGK1: glucocorticoid-regulated kinase 1; Hsp90: heat shock protein 90; MAPKs: mitogen-activated protein kinases; ER: estrogen receptor; Ku70: 70 kDa subunit of Ku antigen; Bax: Bcl-2-associated X protein; DUSP1: dual-specificity protein phosphatase 1; CREB: cAMP response element-binding protein; CLU: clusterin; LEDGF/p75: lens epithelium-derived growth factor p75; GRE: glucocorticoid response element; mTOR: mammalian target of rapamycin; ROS: reactive oxygen species; NK: natural killer; T: T lymphocytes; MDSC: myeloid-derived suppressor cell; Th1/17: T helper type 1/17; ECs: endothelial cells; VEGF: vascular endothelial growth factor; NGF: nerve growth factor; BDNF: brain-derived neurotrophic factor; MMP: matrix metalloproteinase; MIT: mitochondria; C: catalytic; R: regulatory subunits of PKA; FAK: focal adhesion kinase; ECM: extracellular matrix; ARRB1: arrestin beta 1; BAD: Bcl-2 antagonist of cell death; PLC: phospholipase C; PKC: protein kinase C; TrkA: tropomyosin receptor kinase A; Gαs: Gs α subunit; PI3K: phosphatidylinositol-4,5-bisphosphate 3-kinase; AKT: protein kinase B; EPAC: exchange protein activated by cAMP; JNK: c-Jun N-terminal kinase; STAT3: signal transducer and activator of transcription 3; HSD11β1: 11β-hydroxysteroid dehydrogenase type 1; sCLU: secretory clusterin; HIF-1α: hypoxia-inducible factor-1α
The most frequently used mouse model of chronic stress is immobilisation (restraint), which enables studying the effect of prolonged elevation of stress hormones in the cancer progression or efficacy of anticancer therapy.
Using the restraint mouse model, CA elevation was shown to promote tumour progression through the inhibition of various forms of cell death. Particularly, enhanced levels of CAs and activation of the ADRB2 signalling evoked by restraint stress in mice with orthotropic transplantation of human ovarian cancer cells lowered ECM-withdrawal-induced cell death, known as anoikis, which promoted the growth of this type of tumour. The latter process was found to be mediated by the activation of FAK [192]. Using a similar chronic stress model, elevated E and the ADRB2 signalling was associated with decreased apoptosis incidence and tumour involution generated by subcutaneous (ectopic) injection of human prostate cancer cells through mechanisms involving ADRB2-PKA-Bcl-2 antagonist of cell death (BAD) interaction [193]. In addition to the aforementioned mechanisms, CAs were shown to inhibit tumour cell death induced by various therapies. For example, chronic immobilisation stress was found to protect intraperitoneally injected human ovarian cancer cells (HeyA8 and SKOV3ip1) from paclitaxel- and cisplatin-induced apoptosis through induction of DUSP1 [194]. Activation of this protein by NE was found to occur through the action of ADRB2-cAMP-PLC-PKC-CREB signalling [194]. In this study, activated DUSP1 was demonstrated to deactivate c-Jun N-terminal kinase (JNK) phosphorylation (activation) induced by taxels and thus inhibit c-Jun-induced apoptosis. Furthermore, in vitro and in vivo silencing of this P-glycoprotein involved in multidrug resistance was shown to enhance cell death induced by mentioned chemotherapeutic drugs [23]. The clinical relevance of those experimental studies is supported by the Cancer Genome Atlas data, which provide evidence that elevated DUSP1 expression positively correlates with decreased overall and progression-free patient survival [194]. Besides regulating multidrug resistance, CA hormones were also found to protect from cisplatin-induced DNA damage and associated cell death as shown in three human ovarian tumour cell lines [195]. In the latter process β-adrenergic signalling-induced p53 protein degradation, which acts as a tumour suppressor and DNA damage sensor, as well as a DNA double-strand break repair system may be involved [196]. In fact, chronic restraint stress-induced β-adrenergic signalling in mice was found to be involved in p53 ubiquitination and subsequent degradation through β-2 adrenergic receptors (β2ARs) and AKT-mediated activation of MDM2 [197].
Another mechanism involved in β-ADR signalling-enhanced cancer progression is the activation of the angiogenesis process [18, 198]. It has been shown that activation of the β2-adrenergic pathway presumably in tumour cells during restraint-stress promoted more intensive growth and tumour burden in ovarian cancer xenotransplantation mouse models through increased vascularization and angiogenesis [198]. However, a recent study using orthotropic co-transplantation of green fluorescent protein (GFP)-tagged either ADRB2-sufficient or deficient endothelial cells (ECs) together with PC-3 prostate carcinoma cells demonstrated elegantly a clear involvement of an EC-β-ADR signalling in the promotion of the angiogenic switch and related cancer progression [199]. Moreover, activation of the angiogenesis process in this tumour was found to rely mostly on adrenergic nerve-derived NE [199]. Similar dependence was also found in the study using orthotropic mouse models of breast cancer [200]. A recent study has investigated the potential mechanisms involved in the proangiogenic action of chronic stress. Those results revealed that stimulation of primary human umbilical vein ECs with ADRB2 agonist, isoprenaline, activates a proangiogenic pathway through upregulation of PlexinA1 and VEGFR2, which was associated with enhanced Janus kinase 2 (JAK2)-STAT3 phosphorylation [201]. Activation of the SNS during chronic stress has been strongly linked to pancreatic ductal adenocarcinoma (PDAC) development [202]. By using the restraint stress model, a stimulatory action of CAs on β2-adrenergic signalling, nerve infiltration, and tumorigenesis in mice with an early stage of pancreatic carcinoma was demonstrated [203]. Furthermore, this study clearly demonstrated that β2-adrenergic signalling might promote enhanced neuronal density and neurite outgrowth in PDAC through the upregulation of NGF by the tumour cells [203]. Similar results were demonstrated in the case of ovarian cancer, where activation of β3-adrenergic signalling by the restraint stress caused cAMP/EPAC-dependent induction of BDNF, which resulted in enhanced tumour innervation [204]. The dependence of tumour growth from sympathetic innervation has become increasingly clear in recent years and has been a major topic of some outstanding review articles [205, 206].
Besides its effect on tumour cell proliferation, chronic stress was also found to promote tumour progression through modulation of its microenvironment and inhibition of immune surveillance. For example, decreased NK cell cytotoxicity and T cell cytokine production along with increased numbers of immunosuppressive cells, such as MDSCs and Tregs were found in CA-rich tumours [207]. Furthermore, activation of the β-adrenergic signalling during chronic restraint stress was found to promote hepatocellular carcinoma (HCC) growth by mobilizing splenic myeloid cells to tumour tissues through alterations in C-X-C motif chemokine receptor (CXCR) 2-CXCL2/CXCL3 axis [208].
Accordingly, several stress-reducing therapies or pharmacological inhibition of adrenergic signalling were demonstrated to decrease the incidence and lower mortality of patients with diverse types of tumours including prostate cancer [209]. The latter results promoted launching and conducting the first clinical trials investigating the therapeutic effects of perioperative administration of ultra-short-acting beta-blockers in patients with lung cancer [210]. As demonstrated in the recent review article, the results of such clinical trials using beta-blockers are promising [211].
Collectively, chronic exposure to the stress hormones, overexpression of their receptors, and their enhanced signalling were found to correlate with cancer initiation, progression, degree of malignancy, decreased efficacy of the anti-cancer therapy, and shorter relapse-free survival. Based on these observations it is therefore highly plausible that chronic- and ELS or sGC adjuvant therapies-associated dysregulation of the stress system is involved in this process.
Hypothetical involvement of stress hormones-induced reprograming of adult SPCs in tumorigenesis
Several lines of evidence support the role of adult SPCs in the generation and progression of multiple tumours [212, 213]. Due to their indispensable contribution to tissue maintenance and regeneration, undifferentiated status, and longevity, SCs are very sensitive to various intrinsic and environmental stressors such as altered metabolism, accumulation of ROS, radiation, chronic inflammation, or toxins [214]. Therefore, generation, quiescence, and differentiation of SPCs are tightly controlled through epigenetic imprinting [215] and by additional tissue-specific factors derived from their close microenvironments known as a niche. In order to prevent potentially hazardous consequences of a life-long accumulation of multiple genetic and epigenetic mutations, SCs undergo premature senescence and/or apoptosis (Figure 4A) [213]. However, certain mutations leading to a dysfunctional DNA repair system can overcome this process and predispose those cells to malignant transformation (Figure 4B) [31, 131]. That process can be additionally promoted by cellular aging, which is associated with accumulating mutations in epigenetic machinery and dysregulation of cellular metabolism [216], or by certain therapeutic interventions [217].
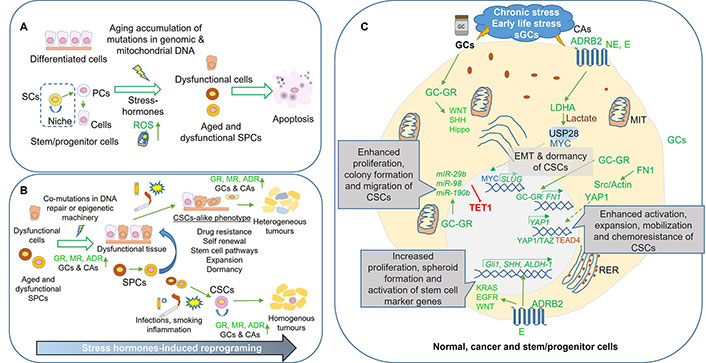
Stress hormone-induced epigenetic reprograming promotes tumorigenesis through the induction of SC-like features in differentiated and multipotent SPCs. Aging-associated accumulation of genetic and epigenetic mutations along with metabolism dysregulation in SPCs induce apoptosis. A. This prevents their negative influence on organ functioning and reduces the chance of malignant transformation; B. stress hormone-induced reprogramming in cells with active mutations in the DNA repair system protects those cells from cell death and may contribute to organ dysfunction and tumorigenesis. When exposed to mutagens (e.g., smoking) or infections, these cells may acquire SC-like characteristics, which will promote tumour formation or support the generation and maintenance of the cancer SCs (CSCs), giving rise to either heterogeneous or homogenous tumours, respectively; C. mechanisms involved in chronic stress-, ELS- or sGCs-associated tumorigenesis. Overexpression of stress hormone receptors and their increased intratumoural concentrations induce SC-like features, including expansion, mobilization, and chemoresistance in cancer cells as well as may contribute to the generation and maintenance of CSCs. Green color symbolizes activation or upregulation whereas red colour downregulation or inhibition. SCs: stem cells; ROS: reactive oxygen species; SPCs: stem/progenitor cells; GR: glucocorticoid receptor; MR: mineralocorticoid receptor; ADR: adrenergic receptor; GCs: glucocorticoids; CAs: catecholamines; CSCs: cancer stem cells; sGCs: synthetic glucocorticoids; NE: norepinephrine; E: epinephrine; WNT: the mammalian wingless-type mouse mammary tumour virus integration site; SHH: Sonic Hedgehog; TET1: ten eleven translocation 1; MIT: mitochondria; EMT: epithelial-to-mesenchymal transition; PCs: progenitor cells; KRAS: Kirsten rat sarcoma virus; EGFR: epidermal growth factor receptor; RER: rough endoplasmic reticulum; ALDH-1: aldehyde dehydrogenase-1; YAP1: Yes-associated protein 1; TAZ: transcriptional coactivator with PDZ-binding motif; TEAD4: TEA domain transcription factor 4; SLUG: snail family transcriptional repressor 2; FN1: fibronectin; USP28: ubiquitin-specific peptidase 28; LDHA: lactate dehydrogenase A
Stress hormones-induced the SC-like characteristic in normal and cancer cells and its role in the tumorigenesis
Chronic inflammation, dysregulation of the stress system [131], GC therapy, or smoking was demonstrated to contribute to cancer formation. Accordingly, it has been shown that GC hormones induce epigenetic changes, which not only can suppress the expression of certain tumour suppressor genes [31, 182] but also promote the activation of oncogenes [218–220]. Furthermore, dysregulated stress system and related stress hormones-induced epigenetic changes, or smoking may activate various SC pathways in both multipotent and differentiated cells, including, WNT, the Hippo, and SHH, thereby promoting certain stem-cell-like characteristics e.g., EMT, self-renewal or drug resistance (Figure 4C). Chronic exposure of bronchial epithelial cells to cigarette smoke was shown to cause an aberrant KRAS, WNT/β-catenin, and EGFR expression and reduced the number of mutations required for cancer initiation [221]. In addition, chronic inflammation and immune cell infiltration were shown to induce EMT and epigenetic changes in breast epithelial cells leading to activation of SC gene sets [222]. Further survival of those cells was found to be enhanced by CAs, for example, E-mediated induction of LDHA-dependent metabolic rewiring [223]. Moreover, stimulation of HSCs with NE enhanced expression and release of secreted frizzled-related protein 1 (sFRP1) from those cells, which through activation of the WNT pathway significantly promoted cell migration and invasion, epithelial-mesenchymal transition, and expression of cell proliferation-related genes and cancer stem cell (CSC) markers in HCC cells [224].
In addition to their effects on differentiated tissue cells, stress hormones may promote SC-like identity in existing cancer cells (Figure 4C). For example, DEX stimulation of three different human PDAC cell lines enhanced transforming growth factor β (TGF-β), NOTCH1, and SOX2 signalling and promoted expression of EMT markers in those cells. Those changes were then associated with gemcitabine resistance, self-renewal potential, induction of CSC signalling, and enhanced tumour spread observed in cancer xenograft mouse models [225]. Stress hormones were also found to exert their pro-tumorigenic actions through the modulation of several non-coding mRNAs [226, 227]. For example, GCs are known to enhance miR-29b expression, which factor was shown to increase proliferation, colony formation, and migration of CSC-like cells generated from various breast cancer cell lines. In the latter process, inhibition of TET1 a potent tumour suppressor was reported [226, 227]. Additional evidence comes also from studies investigating the role of GC-enhanced miR98 and 190b miRNA [228].
Stress hormones promote tumorigenesis through modulation of existing SPCs and the maintenance of CSCs
Unlike in both normal and cancer-differentiated cells [150], activating mutations in the WNT/ β-catenin pathway [229] or overexpression of mutated β-catenin in SOX2-expressing SPCs resulted in a homogenous cancer formation [230]. Moreover, aberrant activation of another key pathway controlling SPC proliferation and maintenance, the Hippo pathway, only in these SOX2-positive pituitary SCs, robustly induced their expansion and resulted in the formation of aggressive tumours [231]. These observations suggest that certain mutations in key SC pathways induced in differentiated cells by extrinsic factors, e.g., stress hormones, are strong enough to overcome cell-specificity [232], and may lead to a generation of heterogeneous tumours [233, 234]. However, they also indicate that when similar mutations occur in adult tissue SPCs, the resulting tumour might be more aggravated, more homogenous, and more aggressive.
During the latter process, a small number of tissue-specific SPCs may give rise to a limited pool of self-renewing and drug-resistant cancer cells, known as CSCs [198]. Currently, most of the studies isolating CSCs define these populations through an expression of cell surface SC markers (e.g., CD24, CD34, CD44, CD133, and CD166). Using this approach, several groups reported a successful identification and isolation of CSCs from brain, breast, liver, pancreas, prostate, or colon cancers [235–240]. However, as in the case of prostate CSCs, further studies demonstrated that multiple other proteins are required for their function as CSCs and their ability to sustain or renew cancer growth. Some examples of such proteins are α2β1 integrin, α6 integrin, multidrug resistance protein 1 (ABCB1), aldehyde dehydrogenase, CD117, CXCR4, E-cadherin, epithelial cell adhesion molecule (EpCAM), enhancer of zeste homolog 2 (EZH2) [241]. These proteins and signalling molecules are involved in the control of their self-renewal, proliferation, differentiation, migration, invasion, metastasis, sphere formation capacity, chemoresistance, SC maintenance, or clonogenicity. Other studies indicate the additional importance of the local microenvironment for the fate and maintenance of CSCs [242], especially those originating from mutated and transformed tissue-specific SPCs [243, 244].
Nevertheless, considering a high expression of GRs, MRs, ADRs, and responsiveness of various tumours to stress hormones, it is highly plausible that their sustained elevation is caused by dysregulated stress response or e.g., GC adjuvant therapy may be involved in the generation, maintenance, and therapy resistance of various organ-specific CSCs (Figure 4C). In a few studies, that evaluated the potential role of GCs on the latter mentioned cancer and SC hallmarks, a clear link between the administration of those stress hormones and enhanced tumorigenesis was observed. For example, in the case of the most aggressive type of brain cancer glioblastoma multiforme (GBM), stimulation of CD133 expressing glioblastoma CSCs with DEX enhanced their proliferation and self-renewal ability, which effect, however, required a presence of SC factors in the medium [245]. Furthermore, DEX protected glioblastoma CSCs from chemotherapy and nutrient deprivation-induced stress and promoted neurosphere formation [245]. It was suggested that in addition to the niche-derived factors, the formation and growth of CSCs are supported by stress hormones abundantly present in some tumours. This could in part explain the high prevalence of cancer metastases found in the adrenal glands [246]. In addition, it has been also found that stress hormones promote expansion and chemoresistance of existing breast CSCs through activation of the Hippo pathway [247]. In particular, GCs were found to enhance the expression and deposition of FN1 leading to the focal adhesion-Src pathway stimulation, cytoskeleton-dependent YAP activation and chemoresistance, and expansion of breast CSCs [247]. The latter finding was recently validated by an independent study showing that GCs mediate breast CSC maintenance, cell survival, metastasis, and chemoresistance both in vitro and in vivo through activation of key regulators of the Hippo pathway a TEAD4 [248].
More importantly, several studies demonstrated that stress hormones could promote tumour recurrence by influencing CSC functionality [249] and their reactivation from dormancy [250]. For example, E was found to enhance the number and spheroid formation of CSC-like cells from three nonsmall-cell lung carcinoma lines, which effect was dependent on cAMP signalling. This was additionally associated with enhanced expression of the SC markers in those cells, including, Shh, aldehyde dehydrogenase 1 (ALDH-1), and Gli1 [249]. Moreover, co-incubation of in vitro generated dormant CSCs with E-pre-treated neutrophils, but not other immune cells resulted in an exit from the dormancy and renewed tumour formation. The underlying mechanisms involved in E were proposed to involve enhanced S100A8/A9 (S100 calcium binding protein A8/S100 calcium binding protein A9) release from tumour-infiltrating polymorphonuclear cells, which subsequently induced the release of fibroblast growth factor from cancer cells [250].
To conclude, dysregulation of the stress system, cellular ageing, alteration in tissue microenvironments e.g., chronic inflammation, and extrinsic factors such as smoking or sGC therapy are associated with the generation and accumulation of several genetic and epigenetic mutations. Appearance of such mutations especially in the DNA repair systems or epigenetic regulators, were shown to predispose to tumour formation. Although such mutations may also induce CSC-like characteristics in various differentiated normal or tumour cells, their acquisition by the SPCs is associated with the formation of more aggressive and drug-resistant tumours, suggesting a critical involvement of the latter cells in stress-associated tumorigenesis. This observation is supported by clinical studies showing that chronic stress and dysregulation of the stress response were found to contribute to the maintenance and propagation of various CSCs in patients suffering from chronic stress, anxiety, depression, and suicidal behaviour [226, 251, 252].
Conclusions
The stress response is a highly dynamic and complex process, whose function is additionally influenced by the age, sex, type, and duration of the stressor and an individual ability to cope with it, known as resilience. Increasing experimental evidence indicates that maternal psychosocial stress, including, depression or trauma, and early life adversity can contribute to chronic-, life-long-, or even transgenerational- dysregulation of the stress system in the offspring. In the latter process, a key involvement of the stress hormone-mediated epigenetic-based modification of factors involved in the activation, termination (negative feedback), and regulation of the stress system, including among others AVP, CRH, POMC, NR3C1, FKBP5 genes were found. More importantly, it has been convincingly demonstrated that some of those epigenetic changes might induce differential and cell-specific expression of stress hormone receptors, enhanced sensitivity of the stress system, and aberrant concentrations of stress hormones.
However, in order to be maintained such epigenetic changes should be induced in the SPCs, which are undifferentiated, pluri- or multipotent cells capable of sustaining organ growth and its long-term regeneration. Therefore, it is highly plausible that those epigenetic alterations induced by the stress hormones during sensitive periods of human development such as pre- and postnatal periods, may not only change the function and fate of those SPCs but also ultimately cause a long-term dysregulation of the whole stress system. They are especially considering that those cells, which heavily rely on the epigenetic mechanism to sustain their multipotent and undifferentiated status, do express receptors for stress hormones and are highly dependent on their specific microenvironments known as niches. In this regard, several studies demonstrated that overexposure to stress hormones could alter the niche, metabolism, and functioning of the SPCs of the stress system, resulting in increased differentiation and reduced numbers of these cells.
Consequently, an increasing number of experimental studies demonstrated that chronic maternal- and ELS, or administration of synthetic GCs, results in low body weight at birth, development of mental and metabolic diseases, as well as increased predisposition to various tumours. In the latter case, it has been reported that overexposure to stress hormones (mainly CAs and GCs) and upregulation of their receptors can promote all major hallmarks of tumour development, including tumour angiogenesis [18], neovascularization [18], EMT [20], malignancy [20–22], or induction of resistance to chemotherapy [23]. Moreover, considering the evident actions of stress hormones on various SPCs, including modulation of dormancy, induction of developmental pathways, and alterations in DNAm, it is highly plausible that dysregulated stress system might also influence new and existing CSCs.
Available evidence summarized and discussed in the current review strongly indicates that stress hormones-mediated epigenetic reprograming of various SPCs may lead to dysregulation of the stress system and promote cancer development. However, many open questions remain and the results of various clinical trials testing the effectiveness of anti-stress therapies are inconclusive, which hinders a proper preclinical to clinical transition and their inclusion into current medical guidelines in oncology. For example, a large epidemiological study performed in Denmark with over 1 million children revealed that ELS was associated with 4.54 times higher all-cause mortality, including accidents, suicides, and cancer [253]. Similarly, another large epidemiological study revealed that especially in the women population, childhood adversity was associated with reduced risk of malignant melanoma and brain cancers, while simultaneously increasing predisposition to cervical and breast cancers [164]. However, contrary to those studies, clinical trials using e.g., GR and ADR inhibitors or various stress-lowering therapies reported mixed results so far [254]. The latter suggests that there are still many unknown mechanisms, which may be involved in stress-associated tumorigenesis.
Many factors could contribute to the failure of recent clinical trials such as lack of standardization of the study design and analysis, as well as using different definitions of previous and current mental conditions (based on self-reports). Furthermore, unlike in experiments using animals, clinical studies usually involve patients with heterogeneous backgrounds, different ages, sexes, and sensitivity to stress. Furthermore, an over-interpretation of the results of animal studies or the inability of the current mouse models to phenocopy the complex psychological condition of humans, which is shaped by individual stress resilience and life-long experience can contribute to the low reproducibility of such studies.
Longitudinal and multicentre studies using standardized protocols are necessary to provide valuable and more homogenous information regarding the effect of ELS and chronic stress on the dysregulation of the stress system and its relation to tumorigenesis. In this regard, a longitudinal analysis of the babies born during the recent coronavirus disease 2019 (COVID-19) pandemic and their mothers may prove useful. Most importantly, due to its severity and global spread, the COVID-19 pandemic involved an unprecedentedly large number of diverse societies, countries, and people of different ages and sex. Especially considering the fact that SARS-CoV-2, which is severe acute respiratory syndrome coronavirus type 2 causing COVID-19, was shown to infect the organs of the stress system [255], thereby contributing potentially to its dysregulation [41, 256, 257].
Moreover, despite rather strong evidence of the destructive influence of stress hormones on neuronal SPCs, less is known about their effects on the individual peripheral organs of the stress system. In addition, since most of the epigenetic studies have analysed the effects of GCs, it would be of great interest to evaluate the potential modulating effects of CAs. However, a general lack of such studies might also point to the different actions of those two major stress hormones. Therefore, additional studies exploring the long-term effects of GCs and CAs on the epigenome of adrenal, pituitary, and hypothalamic SPCs should be performed. In this regard, it is very interesting to investigate, whether SARS-CoV-2 can also infect SPCs of the stress system, and to analyse its long-term consequences, in addition to the effects of the epigenetic imprinting caused by temporal overproduction of the stress hormones.
Finally, since many studies demonstrated a clear modulatory role of stress hormones in the survival, propagation, and metastatic potential of existing CSCs, and considering rather frequent use of those hormones e.g., as adjuvant therapy, it is very important to better understand the mechanisms involved in their pro-tumorigenic processes and raise awareness about their potential side effects.
Abbreviations
| 11β-HSD2: | 11β-hydroxysteroid dehydrogenase type 2 |
| ACTH: | adrenocorticotropic hormone |
| ADRB2: | β2-adrenergic receptor |
| ADRs: | adrenergic receptors |
| ARs: | androgen receptors |
| AVP: | arginine vasopressin |
| cAMP: | 3’, 5’-cyclic adenosine monophosphate |
| CAs: | catecholamines |
| COVID-19: | coronavirus disease 2019 |
| COX-2: | cyclooxygenase-2 |
| CpG: | cytosine-guanine dinucleotides |
| CRH: | corticotrophin-releasing hormone |
| CSCs: | cancer stem cells |
| DDR1: | discoidin domain receptor 1 |
| DEX: | dexamethasone |
| DNAm: | DNA methylation |
| DNMTs: | DNA methyltransferases |
| DUSP1: | dual-specificity protein phosphatase 1 |
| E: | epinephrine |
| ECs: | endothelial cells |
| ELS: | early life stress |
| EMT: | epithelial-to-mesenchymal transition |
| ERs: | estrogen receptors |
| GCs: | glucocorticoids |
| GR: | glucocorticoid receptor |
| GREs: | glucocorticoid response elements |
| HPA: | hypothalamic-pituitary-adrenal |
| HSCs: | hepatic stellate cells |
| LC: | locus coeruleus |
| LEDGF/p75: | lens epithelium-derived growth factor p75 |
| miRNAs: | microRNAs |
| MRs: | mineralocorticoid receptors |
| NE: | norepinephrine |
| NF-κB: | nuclear factor κB |
| NR3C1: | glucocorticoid receptor gene |
| NSCs: | neural stem cells |
| PDAC: | pancreatic ductal adenocarcinoma |
| PKA: | protein kinase A |
| POMC: | proopiomelanocortin |
| SAM: | sympathetic-adrenal-medullary |
| SCs: | stem cells |
| sGCs: | synthetic glucocorticoids |
| Shh: | Sonic hedgehog |
| SNS: | sympathetic nervous system |
| SOX: | sex determining region Y-box |
| SPCs: | stem/progenitor cells |
| TET: | ten eleven translocation |
Declarations
Author contributions
WK: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. MS: Writing—original draft. AW and GPC: Writing—review & editing. All authors read and approved the submitted version.
Conflicts of interest
Prof. George P. Chrousos is the Editorial Board Member of Exploration of Endocrine and Metabolic Diseases, but he had no involvement in the decision-making or the review process of this manuscript. The other authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This study has been funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) Project [A01: 314061271-TRR 205] (to WK). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2024.