
 A
Correction to this article was published on 20 December 2024
A
Correction to this article was published on 20 December 2024
Abstract
Pheochromocytomas (PCCs) and paragangliomas (PGLs; together PPGLs) are uncommon neuroendocrine tumors arising from adrenal medullary chromaffin cells and sympathetic/parasympathetic paraganglia. Though PPGLs predominate in adult populations, pediatric cases of PPGLs represent more aggressive disease outcomes with 12% being diagnosed as metastatic. Metastatic disease (spread to bone, lung, lymph nodes, or liver) occurs in a subset of PPGLs, ranging from 15% to 17% depending on the underlying pathogenic variants. Historically, pulmonary metastases present clinically as multiple small lesions; however, cases of PPGLs with innumerable small metastases (a miliary pattern) overwhelming lung parenchyma define a novel yet exceptionally challenging disease presentation. This pattern of pulmonary lesions upon treatment and/or cellular lysis may lead to both respiratory decompensation as well as prolific catecholamine release, incurring significant morbidity and mortality if not appropriately managed. Of the 2,649 PPGL patients enrolled in our protocol from January 1, 2000, to April 30, 2023, 500 had metastatic disease, 122 were children/adolescents, and 3 of the 122 children/adolescents had extensive pulmonary metastatic disease. All three adolescent patients with extensive pulmonary metastases had cluster 1 PPGLs and suffered hypoxemia (due to pulmonary metastases) leading to overactive hypoxia signaling and catecholamine-induced signs and symptoms [among them hypertension and/or tachyarrhythmia(s)]. Interventions including surgery, chemotherapy, and radiotherapy were pursued. Two patients achieved disease stability, while one patient succumbed to disease. Ultimately these divergent outcomes emphasize the importance of recognizing poor prognostic factors and aggressive disease early, to select appropriate treatments. Optimal management of these patients must consider complications of catecholamine excess and the profound influence of hypoxia. Herein, we describe three adolescent cases of extensive pulmonary metastatic PPGL and the unique clinical challenges faced in treating these tumors alongside relevant literature to provide guidance on appropriate interventions (ClinicalTrials.gov identifier: NCT00004847).
Keywords
Pheochromocytoma, paraganglioma, adolescent, pulmonary, metastasesIntroduction
Pheochromocytomas (PCCs) and paragangliomas (PGLs; together PPGLs) are rare neuroendocrine tumors that arise from the medulla of the adrenal gland or extra-adrenal chromaffin tissue, respectively. Those arising from sympathetic paraganglia are often characterized by catecholamine production and secretion—often in excess—and catecholamine-induced signs and symptoms [1–3]. In contrast, head and neck PGLs (HNPGLs) found along the parasympathetic chains rarely secrete catecholamines [4, 5]. Although most PPGLs will not develop metastatic lesions, approximately 20% behave aggressively and ultimately develop metastatic disease influenced by the underlying pathogenic variant(s), tumor location, and size, as well as secretory status [6–11]. Metastatic lesions are commonly found in bones (64%), lungs (47%), lymph nodes (36%), and the liver (32%) [6, 12–17]. Lung lesions are usually found as solitary masses ranging from a few millimeters to several centimeters and are often surgically resected. When multiple pulmonary lesions are present, surgical resection cannot be attempted, but must be treated via systemic therapy.
Nonetheless, various therapeutic options must be considered when treating patients with extensive pulmonary metastatic disease as some treatments may be contraindicated. The presence of innumerable lung lesions can result in hypoxia, which can drive the progression of cluster 1 PPGLs. Within cluster 1 PPGLs the hypoxia signaling pathway is already overactive (see Figure 1) while also being a stimulus for further catecholamine release, resulting in adverse cardiovascular events and compensatory polycythemia [18–20]. Thus, treatment should be initiated in a monitored environment as rapid tumor cell lysis and consequent pulmonary edema can lead to catecholamine surges or respiratory decompensation respectively [21, 22]. Patients should also be serially monitored as treatment may suppress compensatory polycythemia/hematopoiesis. Therefore, chemotherapies or systemic radiotherapies must be rationally selected in these patients [23].
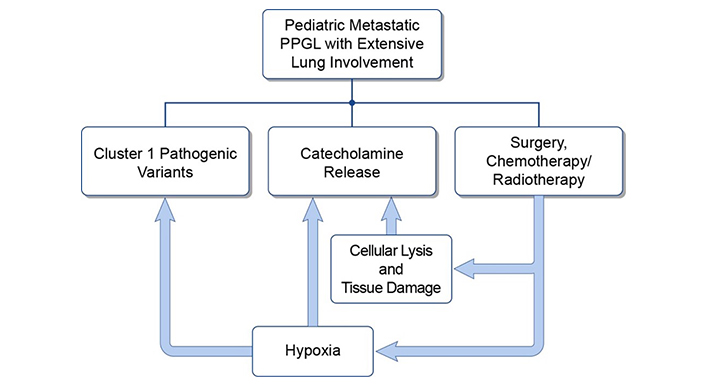
The interplay between hypoxia, catecholamine excess, and pulmonary tumor burden. PPGL: pheochromocytomas and paragangliomas
Catecholamine excess heightens morbidity and mortality. Nearly all PPGL patients experience hypertension (92%) while 10–20% suffer life-threatening complications (e.g., myocardial infarction, stroke, multiple organ failure) with adverse cardiovascular events being the main cause of death (up to 71%) [20, 24]. Therefore, treating both pediatric (< 18 years old) and adolescent (10 to 19 years old) metastatic PPGL with extensive pulmonary metastases presents clinical challenges (such as higher incidence of symptoms) which are discussed alongside demonstrative cases and relevant literature [9, 25, 26].
Case report
The case presentations for all patients are summarized below. For further information pertaining to biochemical laboratory data and the extent of tumor/metastatic involvement refer to Tables 1, 2 and 3, and Figures 2, 3 and 4, respectively. Alternatively, the patient cases are generally summarized in Figure 5 (timeline).
Patient 1: serial biochemistries
| Times the URL | 3/12/2020 plasma | 5/28/2021 plasma | 9/24/2021 plasma | Normal values* pg/mL [nmol/L] |
|---|---|---|---|---|
| Normetanephrine | 15.6 (1,743) | 0.6 [0.52] | 0.6 [0.55] | 18–112 [< 0.90] |
| Metanephrine | 1.5 (91) | U | U | 12–61 [< 0.50] |
| Norepinephrine | 0.8 [563] | 0.5 [400] | 112–750 | |
| Dopamine | U | 0.9 [25] | 0–29 | |
| Epinephrine | U | U | 0–50 | |
| Chromogranin A | 4.9 (453) | 0.9 [80] | < 93 |
Elevated values are bolded. * Note fractionated plasma values (e.g., fractionated metanephrines) are provided in picograms per milliliter in parenthesis (pg/mL) while total values (e.g., total metanephrines) are provided in nanomoles per liter in brackets [nmol/L]. Blank cells indicate an absence of laboratory data for the respective time point. URL: upper reference limit; U: undetectable/below the lower limit of the reference range
Patient 2: serial biochemistries
| Times the URL | 7/23/2019 urine | 7/23/2019 plasma | 9/13/2019 plasma | Normal values* pg/mL [μg/24h] |
|---|---|---|---|---|
| Normetanephrine | 1.8 [683] | 0.2 (25) | 0.5 (58) | 18–112 [103–390] |
| Metanephrine | 1.1 [204] | 0.4 (22) | 0.3 (18) | 12–61 [35–180] |
| Norepinephrine | 0.7 [56] | 0.3 (191) | 0.3 (251) | 112–750 [15–80] |
| Dopamine | 1.2 [462] | U | U | 0–29 [65–400] |
| Epinephrine | 0.1 [1.7] | U | U | 0–50 [< 21] |
| Chromogranin A | 1.0 (92) | [< 93] |
Elevated values are bolded. * Note fractionated plasma values (e.g., fractionated metanephrines) are provided in picograms per milliliter in parenthesis (pg/mL) while 24-hour urine values are provided in micrograms over 24 hours in brackets [μg/24h]. Blank cells indicate an absence of laboratory data for the respective time point. URL: upper reference limit; U: undetectable/below the lower limit of the reference range
Patient 3: serial biochemistries
| Times the URL | 08/11/2016 plasma | 2/9/2017 plasma | 02/10/2017 urine | 3/23/2018 plasma | Normal values* pg/mL [μg/24h] |
|---|---|---|---|---|---|
| Normetanephrine | 37.6 (4,209) | 43.6 (4,883) | 31.4 [12,238] | 4.8 (538) | 18–112 [103–390] |
| Metanephrine | U | U | U | U | 12–61 [35–180] |
| Norepinephrine | 9.9 (7,434) | 13.2 (9,928) | 25.9 [2,068] | 1.6 (1,200) | 112–750 [15–80] |
| Dopamine | U | 1.1 (33) | 0.2 [96] | U | 0–29 [65–400] |
| Epinephrine | U | 0.6 (29) | 0.2 [4.1] | 0.5 (23) | 0–50 [< 21] |
| Chromogranin A | 6.5 (605) | 5.6 (520) | 2.8 (263) | [< 93] |
Elevated values are bolded. * Note fractionated plasma values (e.g., fractionated metanephrines) are provided in picograms per milliliter in parenthesis (pg/mL) while 24-hour urine values are provided in micrograms over 24 hours in brackets [μg/24h]. Blank cell indicates an absence of laboratory data for the respective time point. URL: upper reference limit; U: undetectable/below the lower limit of the reference range
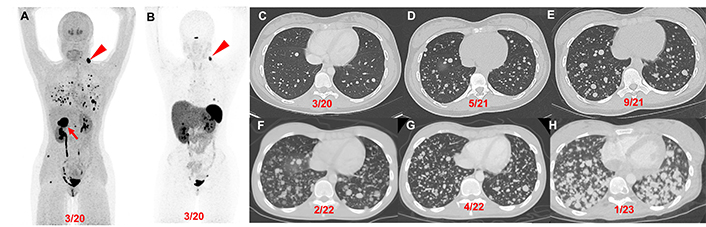
Patient 1 (14-year-old female, VHL pathogenic variant), serial images throughout the course of treatment: Anterior maximum intensity projection images of 18F-FDOPA PET/CT (A) and 68Ga-DOTATATE PET/CT (B) demonstrate a 4.4 cm right PCC (red arrow), left parapharyngeal neck mass (red arrowhead), and widespread metastatic lesions in the lungs and bones. Axial contrast CT images (C–H) in the same plane at various time points (03/2020, 05/2021, 09/2021, 02/2022, 04/2022, and 01/2023) show the patient’s progression (increase in number of lung lesions and tumor volume) despite therapy (3 cycles of CVD between 12/2021 and 2/2022; 3 cycles of axitinib between 3/2022 and 10/2022; and pembrolizumab and axitinib between 10/2022 and 02/2023). CVD: cyclophosphamide, vincristine, and dacarbazine; PCC: pheochromocytoma
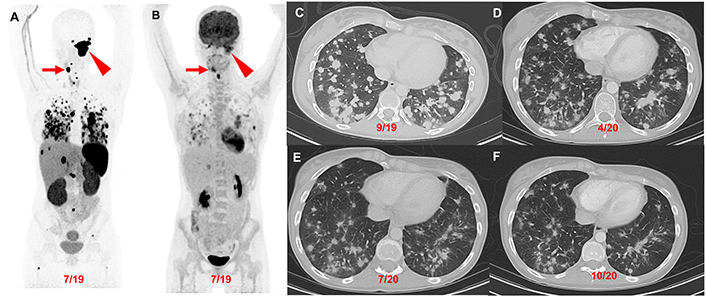
Patient 2 (23-year-old female, SDHD pathogenic variant), serial images throughout the course of treatment: Anterior maximum intensity projection images of 68Ga-DOTATATE PET/CT (A) and 18F-FDG PET/CT (B) demonstrate a 7.1 cm complex left jugular mass (red arrowhead) and a 2.5 cm right carotid body paraganglioma (red arrow) with widespread metastatic lesions in the lungs, mediastinum, liver, and bones. The axial contrast CT images (C–F) in the same plane at various time points (09/2019, 04/2020, 07/2020, and 10/2020) show the patient’s response (decrease in number of lung lesions and tumor volume) after 4 cycles of CVD therapy (between 10/2019 and 03/2020) with resolution of hypoxemia. CVD: cyclophosphamide, vincristine, and dacarbazine
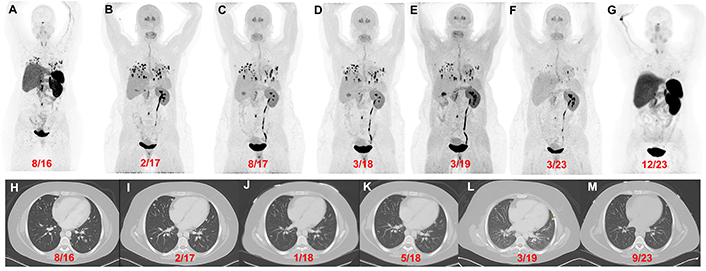
Patient 3 (26-year-old female, VHL pathogenic variant), serial images throughout the course of treatment: Anterior maximum intensity projection images of 68Ga-DOTATATE PET/CT (A and G) and 18F-FDOPA PET/CT (B–F) with a history of a right adrenalectomy (4.2 cm), right nephrectomy, and left periaortic- and aortocaval mass resection in 2016 [resection(s) prior to imaging] demonstrate widespread metastatic lesions in the lungs, retroperitoneal adenopathy, and a few bone metastases. 18F-FDOPA PET/CT images before (B), during (C), and after (D) treatment with CAPTEM. 18F-FDOPA PET/CT (E and F) imaging demonstrate disease stability with temozolomide monotherapy. 18F-FDOPA PET/CT on 03/2023 (F) shows a significant decrease in the number of lung lesions and a reduction in tumor volume compared to prior imaging. The axial contrast CT images (H–M) in the same plane at various time points (08/2016, 02/2017, 01/2018, 05/2018, 03/2019, and 09/2023) show progression of disease prior to chemotherapy (H and I) followed by stable disease (J–L) and a marginal decrease in tumor size(s) on 09/2023 (compared to 03/2019). However, a more robust response was appreciated on 18F-FDOPA PET/CT [02/2017 (B) vs 03/2023 (F)] and 68Ga-DOTATATE PET/CT [08/2016 (A) vs 12/2023 (G)]
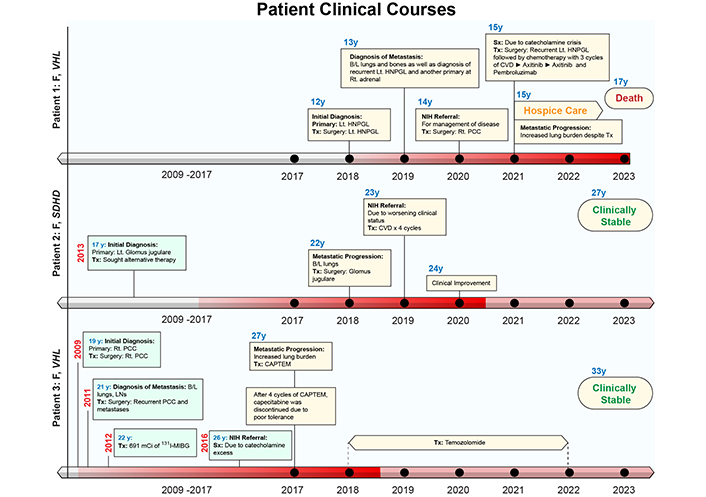
Patient clinical courses. The intensity of red color in the timelines on the x-axis represents the disease status. The darker shade represents worse disease status. B/L: bilateral; CAPTEM: capecitabine and temozolomide; CVD: cyclophosphamide, vincristine, dacarbazine; HNPGL: head and neck paraganglioma; 131I-MIBG: 131I-metaiodobenzylguanidine; LNs: lymph nodes; Lt: left; PCC: pheochromocytoma; PPGL: pheochromocytoma and paraganglioma; Rt: right; Sx: symptoms; Tx: treatment
Patient 1: At the age of 12 (2018) the patient was found to have a solitary left neck PGL measuring 7.5 cm. At the age of 14 (2020), the patient had elevated plasma normetanephrine, chromogranin A, and metanephrine (15.6, 4.9, and 1.5 times the upper reference limit, URL, respectively; Table 1) with a recurrent left neck mass, right PCC, and significant pulmonary metastases (miliary pattern) on imaging. Following tumor resection of the 4.4 cm right PCC subsequent immunohistochemical (IHC) staining of the primary tumor was positive for synaptophysin, chromogranin, and S100 in surrounding sustentacular cells with a Ki-67 proliferation rate of 10% consistent with PGL. Germline genetic testing confirmed a VHL pathogenic variant (VHL c.250G>T, p.Val84Leu).
The patient’s biochemistries normalized post-operatively, but at the age of 15 (2021), she developed hypoxemia requiring (2L) of supplemental oxygen and was found to have a progression of extensive multifocal lung metastases (Table 1, Figure 2). She was then treated with 3 cycles of cyclophosphamide, vincristine, and dacarbazine (CVD), complicated by multiple cytopenias and a 5-kg weight loss with progression of disease on imaging. Her disease progressed despite being transitioned to axitinib (3 mg, twice daily for 7 months) and then pembrolizumab (25 mg, twice daily for 5 months) (Figure 2). She was transitioned to hospice care and succumbed to disease at the age of 17 (2023).
Patient 2: At the age of 17 (2013) the patient was diagnosed with a left glomus jugulare PGL. After 5 years of pursuing alternative treatment, she presented to an outside institution at the age of 22 (2018) with visible outgrowth from her left ear with bleeding and was found to have an extension of the primary tumor, a right carotid body tumor, and extensive pulmonary metastases (miliary pattern) (Figure 3). She then underwent partial resection/debulking of the left glomus jugulare PGL (2018). IHC staining of the tumor was positive for synaptophysin, chromogranin, and S100 in surrounding sustentacular cells consistent with PGL. Genetic testing of the primary tumor revealed a pathogenic variant in SDHD c.124_127delinsATA (SDHD p. Glu42Ilefs*44).
The patient presented to our institution at the age of 23 (2019) and was found to have elevated urine normetanephrine, metanephrine, and dopamine (1.8, 1.1, and 1.2 times the URL, respectively; Table 2) and soon developed hypoxemia requiring (1L) supplemental oxygen. She responded to treatment with 4 cycles of CVD with reduced pulmonary tumor burden and resolution of hypoxemia (Figure 3). She then discontinued treatment and gave birth to two children at the age of 26 and is clinically stable.
Patient 3: At the age of 19 (2009) the patient was found to have a right 4.2 cm PCC upon evaluation for hypertension and headaches. Following resection, IHC staining was positive for synaptophysin, chromogranin, and S100 in surrounding sustentacular cells consistent with PCC. Germline genetic testing of the primary tumor (PCC) confirmed a VHL pathogenic variant (VHL c.439C>G, p. Leu147Val). At the age of 21 (2011), with elevated plasma biochemistries and metastatic PGL noted on 123I-metaiodobenzylguanidine (123I-mIBG) scintigraphy she underwent tumor resection (left periaortic tumor resection, right nephrectomy, and aortocaval mass resection) followed by 131I-mIBG radiotherapy (691 mCi total in 1 year).
The patient was referred to our institution at the age of 26 (2016) with signs and symptoms of catecholamine excess and hypoxemia requiring (2–3L) supplemental oxygen and was found to have elevated plasma norepinephrine and normetanephrine (37.6 and 9.9 times the URL respectively; Table 3) with extensive pulmonary metastases on imaging metastases (miliary pattern) (Figure 4). She was then treated with 4 cycles of capecitabine and temozolomide (CAPTEM, in 2017) then temozolomide (180 mg twice daily PO for 5 days every 28 days) for 6 years until the age of 32 (2022). At age 33 (2023) she was clinically stable and had a slight reduction in tumor burden (Figure 4).
Discussion
This study describes three female adolescent patients with metastatic PPGL and extensive lung involvement. Each patient had catecholamine excess and hypoxemia with common complications (e.g., hypertension) [3, 20, 24]. In this small series, surgical intervention of isolated PPGLs was the first-line treatment in all patients followed by adjuvant chemotherapy and/or systemic radiotherapy. Two patients had a favorable outcome, while one succumbed to metastatic disease. Thus, early intervention that considers the consequences of tumor progression associated with severe hypoxia and catecholamine excess is important in these cases. This is especially true when considering how these conditions are interrelated (Figure 1), as hypoxia worsens catecholamine release and drives the progression of PPGLs (or other cancers) with pseudohypoxia-associated pathogenic variants (like VHL and SDHD) [27]. As such, any systemic therapy must be carefully selected to avoid further complications when a risk of rapid tumor lysis is present (leading to pulmonary edema and respiratory failure and/or concurrent catecholamine surges) and/or to avoid treatment-induced pulmonary sequelae (like lung fibrosis) [23, 27, 28]. Therefore, all patients with metastatic disease, including patients with extensive pulmonary metastases, should have guideline-compliant and frequent re-evaluation consisting of (1) measurement of plasma or urine metanephrines and (2) anatomic (e.g., CT) as well as functional imaging [e.g., most often 68Ga-DOTA(0)-Tyr(3)-octreotate (68Ga-DOTATATE PET/CT)] [22, 29–31].
Pediatric hereditary PPGL
Pediatric cases of PPGL are exceptionally rare with an incidence of 0.45 cases per million in childhood/adolescence as opposed to 4.6 cases per million in adults which is a 10-fold greater incidence [2, 32, 33]. The most common cause of PPGLs in children and adolescents are pathogenic variants in cluster 1 genes: VHL (27–51%) and SDHD (8–10%). These PPGL variants are quite prevalent and on average present at the age of 9 (for VHL-associated tumors) and 10 (for SDHD-associated tumors), respectively [32–38]. In children, metastases may be synchronous (present at the time of diagnosis), but are more likely to develop over time, further emphasizing the need for lifetime surveillance so that providers can identify the signs and symptoms of progressive disease [32]. Recognition of these indicators allows for appropriate selection of treatment; the risk of metastasis in children (due to disease going undetected) is higher than in adults and may have catastrophic implications if stored catecholamines are suddenly released (by surgery, induction of anesthesia, and or interfering medications) [33]. Poor prognostic indicators include extra-adrenal tumor location (PGL compared to PCC), size (≥ 5 cm) at presentation, familial inheritance (in these cases often cluster 1A PPGLs), non-secretory status (as tumors are considered less differentiated), metastasis (especially metastases to the lung and liver), and metastatic interval (patients with a shorter metastatic interval are younger and more likely to have soft tissue metastases) [13, 16, 33, 38–40]. In contrast, favorable prognostic indicators include cluster 2 PPGLs (e.g., well-differentiated and often lower grade) and small tumors (preferably found in the adrenal gland) [13, 16, 32, 38–40].
Evaluation and screening
Evaluation for PPGL in pediatric patients tends to be prompted by symptoms (hypertension: 64–93%, headache: 39–95%, diaphoresis: 90%, palpitations: 53%) in 90% of patients or by detection of an incidentally noted mass [2, 13]. Subsequent biochemical analysis may be performed—taking care to avoid interfering agents—with plasma metanephrines, which are the most sensitive, or urinary metanephrines which are less sensitive but avoid the difficulties of venipuncture in children [3, 29, 41, 42]. Beyond metanephrine and normetanephrine, plasma 3-methoxytyramine (3-MTY, the metabolite of dopamine) or chromogranin A may reveal clinically silent/non-secretory tumors [3, 41, 43–46].
Whole-body imaging (e.g., CT and/or MRI) should be obtained when PPGL(s) are suspected with CT providing superior diagnostic sensitivity in identifying metastatic pulmonary lesions [13, 47]. Alternatively, MRI avoids radiation exposure but often requires sedation and endotracheal intubation while gadolinium (if used) has been shown to accumulate in the CNS with unclear significance [35]. The imaging modality (CT vs MRI) with the best benefit and least risk should be chosen on a case-by-case basis.
In general, anatomic imaging usually precedes functional imaging; but is lacking in its ability to characterize small nodules or bony lesions [47]. Functional imaging in PPGLs utilizes radiopharmaceuticals that target functional receptors/proteins for precise localization of these tumors within the body [13, 23, 47–49]. Radiopharmaceuticals such as 68Ga-DOTATATE, 18F-flurodopa (18F-FDOPA), 18F-fluorodopamine (18F-FDA), and 18F-fluorodeoxyglucose (18F-FDG) are used for PPGL imaging. However, 68Ga-DOTATATE, along with the non-specific radiopharmaceutical 18F-FDG are used most often to screen for PPGL [47, 49, 50]. Particularly, 68Ga-DOTATATE PET/CT has excellent sensitivity in cluster 1A (e.g., SDHx) PPGLs including in pediatric patients, HNPGLs, and metastatic lesions [10, 17, 49–60]. In comparison, 18F-FDOPA PET/CT has excellent sensitivity for tumors in cluster 1B (VHL, EPAS1/HIF2A) and cluster 2 (RET, MAX, NF1, etc.) PPGLs and apparently sporadic PCC [61–67]. 123I-mIBG scintigraphy is mainly performed along with 68Ga-DOTATATE to establish whether treatment with 131I-mIBG or 177Lu/225Ac-DOTATATE (respectively) would be suitable [23, 48–52, 68].
PET imaging is often used as a surrogate for invasive procedures in patients with metastatic or progressive disease. Where surgery or biopsy is avoided for fear of inducing the release of stored catecholamines resulting in catecholamine-induced hypertensive crisis, especially in children [24, 33]. Patients who can undergo resection or biopsy qualify for histopathological analysis to verify molecular pathogenesis active in tissues through IHC staining (with biochemical markers such as chromgogranin, synatophyosin, and Ki-67). A component of this analysis may include the Grading Systems for Adrenal PCC and PGL (GAPP) and/or the PCC of the Adrenal Gland Scales Score (PASS) validation systems which assist in the classification of metastatic PPGL. However, both systems have shown a lack of utility in the clinical environment displaying limited predictive ability [8].
Treatment
Treatment consists of operative and non-operative interventions. Notably, adrenoceptor blockade is advisable in secretory tumors before any intervention (operative or non-operative) that may cause tumor disruption. α-Adrenoceptor blocking agents followed by β-adrenoceptor blocking agents should be administered to avoid unbalanced adrenoceptor blockade which may precipitate (or worsen) hypertension or tachyarrhythmias and lead to PPGL crisis [1].
In operable patients with PPGL, resection is the first line of treatment for solitary lesions and is sometimes indicated for lesions causing mass effect or to decrease catecholamine excess associated with various symptoms/signs. Therefore, surgery (in those that qualify) collectively increases overall survival by 24% [69].
In inoperable patients with PPGL (due to poor performance status or location, for example, HNPGLs abutting eloquent neurologic structures, thus precluding surgery) systemic and/or local radiotherapies are the treatments of choice [33, 34].
Rate-of-growth (fast versus slow growing) in progressive disease, guides subsequent management. In more rapidly progressive inoperable disease, patients are most often treated with CVD chemotherapy, followed by tyrosine kinase inhibitors (TKIs), and finally, in some selected patients, immunotherapy [22, 69]. Immunotherapy using pembrolizumab, a humanized anti-PD-1 receptor antibody, is being investigated in clinical trials evaluating patients with cluster 1 and 2 tumors with PD-L1 expression but must be further reviewed [69–71]. Chemotherapy can elicit a response (defined as decreased or normalized blood pressure/decreased number and dosage of antihypertensive medications and/or reduced tumor size) in a few weeks as opposed to radionuclide therapies that may require 6–12 months [69]. CVD chemotherapy is usually the first line of treatment and is especially effective in patients with cluster 1 tumors (complete response: 11%, partial response 44%) [6, 69]. Chemotherapy with temozolomide (TMZ) is a common second line of treatment, due to its efficacy as a short-term treatment, or in conjunction with capecitabine (CAPTEM), which has been shown to prolong survival in advanced neuroendocrine tumors, but requires further study in PPGLs [70, 71]. TKIs are also being studied in the treatment of PPGL and may be an alternative if CVD and/or CAPTEM are ineffective or implemented as a first-line therapy [72–74]. Current TKIs under ongoing investigation include: sunitinib, pazopanib, cabozantinib, axitinib, and anlotinib which have been shown to elicit very promising responses in various cohorts overall [73, 75, 76]. Notably, both CVD and systemic radiotherapy may lead to cell lysis, consequent catecholamine release, and associated complications; therefore, close monitoring upon initiation of treatment is advisable.
In more indolent inoperable disease, patients are treated with targeted peptide receptor radionuclide therapies (131I-mIBG: low-specific-activity or high-specific-activity) or 177Lu-DOTATATE (Lutathera©) if their corresponding functional imaging (123I-mIBG or 68Ga-DOTATATE) demonstrates radiotracer uptake within their tumors. Previously, some treatment options included either high-specific-activity 131I-mIBG or 177Lu-DOTATATE (Lutathera©). Patients with sufficient bone marrow reserve, high catecholamine burden, and avidity on 123I-mIBG scintigraphy qualify for the FDA-approved high-specific-activity 131I-mIBG therapy (Azedra©), however, its production has been discontinued [23, 68, 77]. So, in such cases, low-specific-activity 131I-mIBG therapy is a suitable alternative [23, 68]. Lutathera© (177Lu-DOTATATE) is best suited for patients who are avid on 68Ga-DOTATATE or 64Cu-DOTATATE [23, 58, 68]. It is often well-tolerated with a disease control rate of up to 90% and can be given to patients greater than 65 years of age or patients with compromised bone marrow reserve [23, 68]. Thus, in hypoxic patients with disease that is avid on both studies (123I-mIBG and 68Ga-DOTATATE), perhaps Lutathera© may be preferable as it imposes less of a risk in suppressing erythropoiesis (an essential compensation in hypoxic patients), especially if high 131I-mIBG doses are considered for treatment [23]. However, endocrinopathies may be seen with Lutathera© [78, 79]. Other options for inoperable patients with 68Ga-DOTATATE avid disease include somatostatin analogs, such as lanreotide or octreotide, often in those with cluster 1 tumors, especially SDHx PPGLs [80–82]. Currently, a phase II trial evaluating lanreotide in inoperable/metastatic PPGL is ongoing (LAMPARA, NCT03946527) and its results would be pivotal in determining its efficacy in this cohort.
Therefore, in short, these imaging modalities have a two-fold benefit as they bind to and highlight somatostatin receptors upon tumors, allowing them to be imaged, while simultaneously demonstrating that therapy directed toward somatostatin receptors is effective [7, 82–84]. Alternatively, if patients progress on chemotherapy and radionuclide therapy, or cannot qualify for radionuclide therapy (because the disease is not avid on 123I-mIBG scintigraphy or 68Ga-DOTATATE PET), these patients may be treated with TKIs or chemotherapy with CVD or TMZ, as described above.
Avidity on various imaging modalities is a significant indicator of treatment viability and certain pathogenic variants have a higher incidence of positive response on certain therapies. CVD therapy has been shown to elicit continued tumor reduction in SDHB-mutated tumors. This positive response with CVD can also be extended to subsequent treatment with TMZ which has higher susceptibility due to increased methylation in the O6-methylguanine-DNA methyltransferase (MGMT) promoter region in SDHB-mutated tumors [6]. As such, a similar response is elicited in SDHx pathogenic variants due to similar pseudohypoxia-driven increases in succinate and DNA hypermethylation [6]. For those patients with fast-growing metastatic disease SDHx pathogenic variants that demonstrate sensitivity to 68Ga-DOTATATE PET/CT usually have a positive response to somatostatin analogs [23]. A trial at the NIH evaluating patients that underwent treatment with Lutathera© demonstrated a 6-month progression-free survival of 19.1 months (22.7 in sporadic cohorts, and 15.4 in SDHx cohorts) further demonstrating that differences in treatment efficacy can be observed with different pathogenic variants [23, 85]. If these, more therapeutic, treatment options do not suffice, experimental options (as discussed below) can be pursued.
On the horizon
Currently, promising therapies include antagonists of the hypoxia signaling pathway (such as belzutifan), which is overactive in cluster 1 pathogenic tumors, PRRT with somatostatin antagonist, and α-emitter based targeted radiotherapy [86–89]. Clinical trials assessing the utility of belzutifan (NCT04924075), a HIF-2α inhibitor currently approved for the treatment of tumors harboring a VHL pathogenic variant, are ongoing [86–88]. Belzutifan could be a promising treatment modality carefully selected for patients with lung metastases given its efficacy, tolerability, and mechanism as it opposes the overactive hypoxia signaling pathway. Nevertheless, caution must be exercised in patients with hypoxemia as belzutifan decreases erythropoietin production resulting in anemia and potentially worsening tissue hypoxia [87, 88, 90, 91]. Providers that encounter non-hypoxic pediatric patients with lung metastases from cluster 1 tumors and suitable bone marrow reserve should consider enrollment on a clinical trial if available. Alternatively, an initial pilot study with 225Ac-DOTATATE suggests that α-emitter therapy could control metastatic disease and improve the quality of life in PPGL patients [23, 89].
Conclusions
Pediatric patients with PPGL are predisposed to more aggressive disease presentations (multifocal/metastatic, extra-adrenal, and recurrent) over their lifespan compared to adult patients. If PPGLs metastasize to the lungs the risk of mortality may increase, especially when lesions are innumerable and secretory, as represented by patient 3 presented in this report. Despite these clinical challenges, by rationally considering the context of hypoxia, catecholamine excess, genetic background, and age, early intervention can minimalize morbidity and offer a favorable prognosis.
Abbreviations
| CAPTEM: | capecitabine and temozolomide |
| CVD: | cyclophosphamide, vincristine, and dacarbazine |
| HNPGLs: | head and neck paragangliomas |
| IHC: | immunohistochemical |
| PCC: | pheochromocytoma |
| PGL: | paraganglioma |
| PPGL: | pheochromocytoma and paraganglioma |
| TKIs: | tyrosine kinase inhibitors |
| TMZ: | chemotherapy with temozolomide |
| URL: | upper reference limit |
Declarations
Author contributions
KMC and KP: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. MAN: Conceptualization, Investigation, Writing—original draft, Writing—review & editing, Supervision. AJ, MP, AL, and TP: Investigation, Data curation. ST: Writing—review & editing. MJMK: Investigation. ASA, AD, AC, JG, and JDR: Investigation, Writing—review & editing. All authors read and approved the submitted version.
Conflicts of interest
Prof. Karel Pacak is the Editor-in-Chief of Exploration of Endocrine and Metabolic Diseases, but he was not involved in the decision-making or review process of this manuscript.
Ethical approval
This study was part of a protocol (NCT00004847) approved by the National Institutes of Health Institutional Review Board (Protocol: 00-CH-0093).
Consent to participate
Informed consent to participate in the study was obtained from the parents (and or guardians) of the participants.
Consent to publication
Informed consent to publish this article was obtained from the parents (and or guardians) of the participants.
Availability of data and materials
The datasets that support the findings of this study are available from the corresponding author upon reasonable request.
Funding
This study was funded by the National Institutes of Health [Z1AHD008735]. This work was supported by the Intramural Research Program of the National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2024.