 Case Report
Case Report

Affiliation:
1Department of Medicine, ESIC Postgraduate Institute of Medical Sciences and Research, New Delhi 110015, India
ORCID: https://orcid.org/0000-0001-6570-8577
Affiliation:
1Department of Medicine, ESIC Postgraduate Institute of Medical Sciences and Research, New Delhi 110015, India
Email: dramit_67@yahoo.com
ORCID: https://orcid.org/0000-0001-9348-7601
Affiliation:
2Scientist C, Indian Council of Medical Research, New Delhi 110029, India
ORCID: https://orcid.org/0000-0002-9129-4686
Affiliation:
1Department of Medicine, ESIC Postgraduate Institute of Medical Sciences and Research, New Delhi 110015, India
ORCID: https://orcid.org/0000-0003-1443-1680
Explor Endocr Metab Dis. 2024;1:77–82 DOI: https://doi.org/10.37349/eemd.2024.00009
Received: August 11, 2023 Accepted: September 22, 2023 Published: May 27, 2024
Academic Editor: Constantine A. Stratakis, Greece & ELPEN Research Institute, Greece
Hypoparathyroidism, deafness and renal dysplasia (HDR) syndrome is a rare genetic disorder caused by haploinsufficiency of the GATA3 gene. A very limited number of cases have been reported in the literature to date. Diagnosis is challenging as the phenotypic expression has wide heterogeneity due to variable penetrance of the underlying genetic mutation. Although the condition is inherited in an autosomal dominant pattern, sporadic cases do occur. This report presents a case of a 22-year-old female diagnosed with HDR syndrome, featuring bilateral cataract and bicornuate uterus. The GATA3 mutation detected in the patient was not identified in the family, suggesting it to be arising de novo. The present case report describes the rare phenotypic findings of bilateral cataract and bicornuate uterus associated with HDR, thus expanding the clinical spectrum of the syndrome.
Hypoparathyroidism, deafness and renal dysplasia (HDR) syndrome, first described by Barakat et al [1] in 1977, is a rare autosomal dominant disease. The disease may present at any age with varying combinations of these features. Some individuals may have some additional features also [2]. Transcription factor GATA3 mutation at chromosome 10 is responsible for the developmental anomalies in the parathyroid gland, auditory system, and kidneys [3, 4]. Due to the wide range of penetrance and expressivity of GATA3 mutations and the resultant phenotypic heterogeneity, the diagnosis of HDR syndrome is quite challenging. This report describes a case of a 22-year-old female with HDR syndrome with bilateral cataract and bicornuate uterus caused by a de novo mutation in the GATA3 gene.
A 22-year-old married female was admitted with complaints of bilateral hearing impairment since childhood, carpopedal spasm for six months, visual impairment in both eyes for six months, and two episodes of generalized tonic-clonic seizures in the preceding two weeks. There was no history of any trauma, chronic headache, ear discharge, neck surgery, bowel and bladder complaints, and long-term drug use. Her menstrual cycle had been regular and she reported a history of two abortions during the second trimester with no living issue. She was born through full-term normal vaginal delivery after an uneventful pregnancy from non-consanguineous parents. According to her parents, her prenatal and perinatal periods were uneventful. There was no history of any psychomotor abnormality or developmental delay. Her academic performance had been average. She had never reported any seizures as a child. There was no family history of deafness, renal disease, hypocalcemia, seizures, or mental retardation. She had two siblings and both were healthy. Clinical examination revealed an averagely built and nourished female having normal vital parameters. She had positive Trousseau’s and Chvostek’s signs. On central nervous system examination, her higher mental functions were normal with bilaterally diminished visual acuity. Ophthalmologic examination revealed the presence of bilateral cataract (Figure 1). The Rinnie’s, Weber’s test, and absolute bone conduction tests were suggestive of bilateral sensorineural deafness. Her sensory, motor, and cerebellar functions were normal. Other systemic examinations revealed no abnormality.
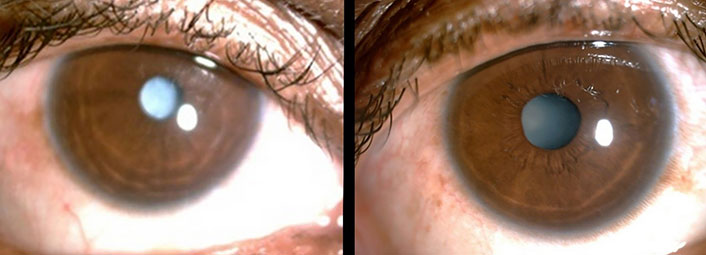
Bilateral cataract in the patient with hypoparathyroidism, deafness and renal dysplasia (HDR) syndrome
Laboratory investigations demonstrated persistently low corrected calcium and ionic calcium levels (5.2 mg/dL and 0.48 mmol/L respectively) with hyperphosphatemia (7.5 mg/dL), normal magnesium (1.90 mEq/L), normal serum alkaline phosphatase (217 IU/L), low intact parathyroid hormone (PTH) levels (9.74 ng/L) and normal calcitonin 4.74 ng/L. Serum Vitamin D3 level was decreased (24.3 ng/mL). The 24-h urinary calcium excretion was normal (150 mg/24 h). Her renal functions were mildly deranged with a blood urea level of 64 mg/dL and serum creatinine level of 1.4 mg/dL. Urinalysis showed no hematuria or proteinuria. Complete hemogram, liver function tests, serum electrolytes, blood sugar, and arterial blood gas analysis were within normal limits. Other hormonal assays including thyroid profile, luteinizing hormone (LH) and follicle-stimulating hormone (FSH), prolactin, and cortisol levels were within normal range. Electrocardiogram (ECG) showed prolongation of QT interval and her transthoracic echocardiography was normal. Chest X-ray revealed no abnormality. Abdominal ultrasonography (USG) demonstrated a small right kidney with multiple cysts and a left kidney with having large medullary cyst containing multiple concretions (Figure 2). USG pelvis revealed the presence of bicornuate uterus (Figure 3). USG of the neck was normal. Bilateral basal ganglia calcifications were seen on a brain computed tomography (CT) scan. Magnetic resonance imaging (MRI) of the brain showed hypointense lesions in the same regions (Figure 4). Pure tone audiometric examination showed bilateral moderately severe sensorineural deafness. On the basis of clinical features, and biochemical and radiological findings, there was a strong suspicion of HDR syndrome. Her family members were also evaluated for HDR, but nothing significant was found.
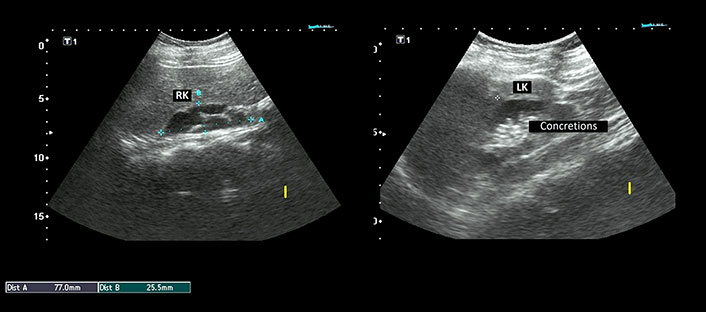
Ultrasonography (USG) abdomen showing right kidney (RK) measuring 77.0 mm × 25.5 mm with multiple cysts. The left kidney (LK) measured 87.7 mm × 42.3 mm and revealed a large medullary cyst with multiple concretions
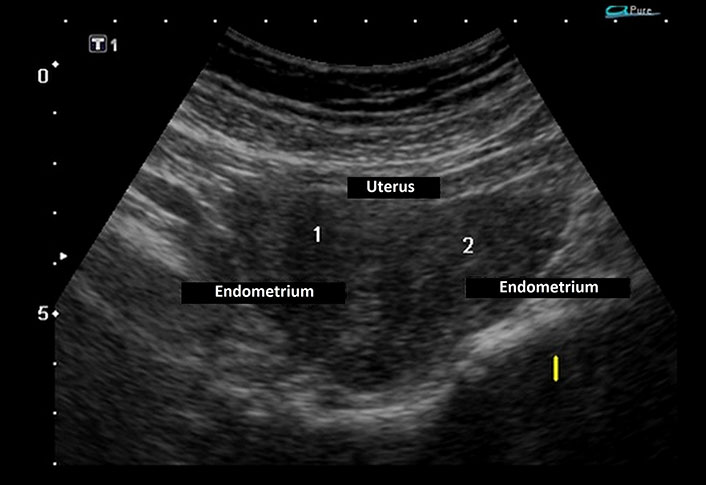
Ultrasonography (USG) pelvis revealed endometrium in the body of the uterus bifurcating cranially into two horns with intervening myometrium
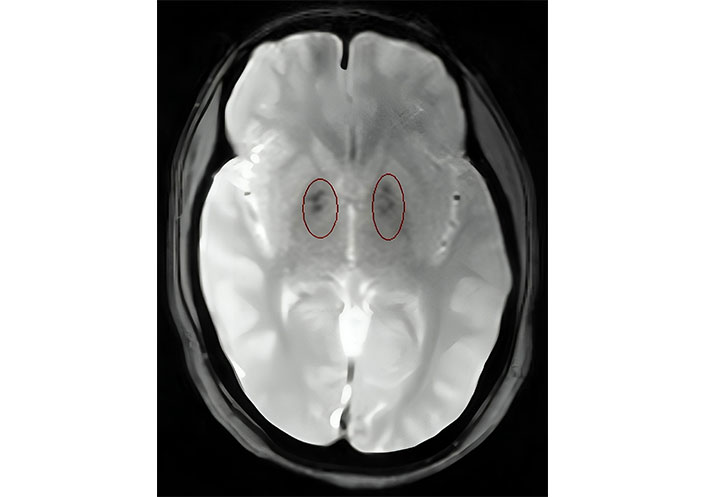
Axial T2 weighted magnetic resonance imaging (MRI) of the brain showing hypointensities in bilateral basal ganglia
The genetic analysis revealed a frame shift intragenic deletion (c.144delG) in exon 3 of the GATA3 gene causing a shift in the reading frame and generating a premature stop codon at codon 194. This mutation was absent in both parents and the siblings and hence found to be arising de novo. The karyotype of the patient was normal (46,XX).
The patient was initially treated with intravenous calcium and vitamin D, and showed marked improvement with a total calcium level of 8.6 mg/dL, phosphorus 6.5 mg/dL, and ionized calcium 1.09 mmol/L. After clinical stabilization, she was started on a high dose of oral calcium, and calcitriol along with dietary modification. Low-dose hydrochlorothiazide was administered to reduce hypercalciuria to prevent renal calculi formation. Later, she was operated on for cataracts and her visual acuity had improved significantly. On outpatient follow-up, she has been maintaining normal serum calcium and phosphorus levels.
Since the description by Bilous et al. [2], the unique dominantly inherited syndrome characterized by HDR has been established as a distinct clinical entity. HDR syndrome is primarily caused by the haploinsufficiency of the GATA3 gene on chromosome 10p15 [4]. GATA3 is a dual zinc finger transcription factor, critical for fetal development. It has two zinc finger domains, one acting to stabilize DNA binding affinity [zinc finger 1 (ZnF1)] and the other for DNA binding (ZnF2). The parathyroid glands, kidneys, breasts, inner ear, urinary bladder, rectum, and thymus are the most common sites of expression of GATA3 [5, 6]. The majority of the GATA3 mutations causing HDR syndrome reported to date are truncating mutations (i.e., nonsense, frameshift deletions, frameshift insertions, or involving a splice site) followed by gene deletions and missense mutations [7]. In our patient, a heterozygous G deletion at position 144 of the GATA3 gene located in exon 3 resulted in a frameshift and a premature termination at codon 194. This mutation is hypothesized to result in a truncated protein with loss of dual zinc fingers—ZnF1 and ZnF2.
The spectrum of clinical features is highly heterogenous with the classical triad of HDR being observed only in about two-thirds of patients [5]. The clinical manifestations of hypoparathyroidism in HDR syndrome have wide variation with some patients having symptoms ranging from myalgia, sensory symptoms, neuromuscular irritability, and tetany to frank seizures caused by hypocalcemia, while others remain asymptomatic for years despite having significant hypocalcemia. Although our patient had symptomatic hypocalcemia for six months, the presence of intracranial calcifications, premature cataracts, and hyperphosphatemia suggested a longer duration of the disease.
Early-onset deafness has been considered the most consistent feature of HDR syndrome, having varying degrees of bilateral, symmetric, and sensorineural hearing loss, predominantly affecting the higher frequencies. Malfunctioning of outer hair cells has been suggested as the underlying mechanism of hearing loss in HDR syndrome considering the absence of otoacoustic emissions and the loss of frequency selectivity [8, 9]. The kidney involvement in HDR syndrome includes both structural abnormalities such as renal dysplasia, hypoplasia or aplasia, pelvicalyceal deformity, vesicoureteral reflux, cystic kidney as well as functional abnormalities such as hematuria, proteinuria, nephrocalcinosis, renal tubular acidosis, and renal failure [10–14].
In addition to the classical clinical triad, our patient had bilateral cataracts and a bicornuate uterus. While some suggest chromosomal deletion involving other genes to be causative of additional manifestations such as genital tract malformation, unilateral megalencephaly, severe mental retardation, autism, basal ganglia calcification, and bilateral cataracts, others believe that the underlying genetic mechanism is the same that causes the syndrome as GATA3 expression can be found in developing central nervous system, foregut, liver, and the eyes. However, these organs seem to be less prone to the haploinsufficiency of GATA3 since they are not generally affected in patients with this syndrome [15–18].
Treatment is symptomatic based on the clinical abnormalities and disease severity. The treatment plan should be individualized to target the symptoms resulting from the distinct mutation. The severity of kidney disease generally defines the overall prognosis. Minor renal anomalies, such as renal cysts or small kidneys, require only routine monitoring to detect the onset of chronic kidney disease, which should be recognized and treated early to prevent the progression to end-stage renal disease [19].
In conclusion, rare genetic diseases such as HDR syndrome are often difficult to diagnose due to the heterogeneity of the clinical presentation. This case report tends to highlight that HDR syndrome should be considered in differential diagnosis while evaluating patients with persistent hypocalcaemia and sensorineural deafness, with or without renal involvement. Early detection of sensorineural deafness is critical to prevent neurodevelopmental disorders including cognitive and motor impairments, emphasizing the role of evoked acoustic potentials in infants and children with chronic hypocalcaemia of unknown etiology. As the HDR syndrome has not been fully understood to date, genetic studies are required in the future not only to expand the spectrum of disease-causing mutations but also to gain further insights into the role of other genetic mutations in the causation of the associated abnormalities.
HDR: hypoparathyroidism, deafness and renal dysplasia
USG: ultrasonography
ZnF1: zinc finger 1
RC: Conceptualization, Investigation, Writing—original draft, Writing—review & editing, Supervision. AK: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. ST and PR: Validation, Writing—review & editing. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
The Declaration of Helsinki was adequately addressed and the study was approved by the ethics committee of ESIC Postgraduate Institute of Medical Education and Research, New Delhi.
The informed consent to participate in the study was obtained from the participant.
The informed consent to publication was obtained from the participant.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 3927
Download: 31
Times Cited: 0
