
Abstract
In recent years, many societies have expressed increasing apprehension regarding the potential negative impacts of food additives, pesticides, and environmental contaminants on human health. Environmental or occupational exposure to these compounds can cause significant adverse effects on human health by causing temporary or permanent changes in the immune system. There is supporting evidence linking pesticides/food ingredients/contaminants-induced immune alterations to the prevalence of diseases associated with changes in immune responses. Hence, it is essential to comprehensively understand the key mechanisms contributing to immune dysregulation induced by these substances, including direct immunotoxicity, endocrine disruption, and antigenicity. The impact of pesticides/food ingredients and contaminants on the human body ranges from mild to severe, depending on their affinity for blood components. These compounds form complexes with blood serum proteins, influencing their metabolism, transport, absorption, and overall toxicity. Numerous studies in the literature have explored the interactions between serum proteins and various molecules, including pesticides, drugs, and food dyes. These investigations employed a range of techniques, including spectroscopy, electrochemical and chromatographic methods as well as molecular modeling and molecular dynamics simulations analyses. This recent review, spanning from 2020 to the present, has been employed to investigate the binding characteristics, mechanisms, and attributes of different food additives, pesticides, and contaminants with serum proteins by using various techniques such as steady-state fluorescence, circular dichroism and ultra-violet spectroscopies, and computational docking methods. The review provides insights into these compounds’ positions and affinities to proteins and possible effects on human health through detailed research studies.
Keywords
Analytical methods, food additives, interaction, pesticides, serum proteinIntroduction
Nowadays, a substantial growth in population and the swift advancement of global industrialization are associated with a rise in the release of various hazardous and harmful substances. Hazardous pollutants like dyes, heavy metals, fertilizers, pesticides, personal care products, food additives, nitrophenols, and medications are upsetting the balance of nature and generating environmental toxins at a disturbing pace. Therefore, due to significant ecological risks and adverse effects on human health, the pervasive existence and capacity of hazardous pollutants to infiltrate the environment have garnered widespread public attention in recent years. A comprehensive categorization and impact of these hazardous pollutants is shown below (Figure 1) [1].
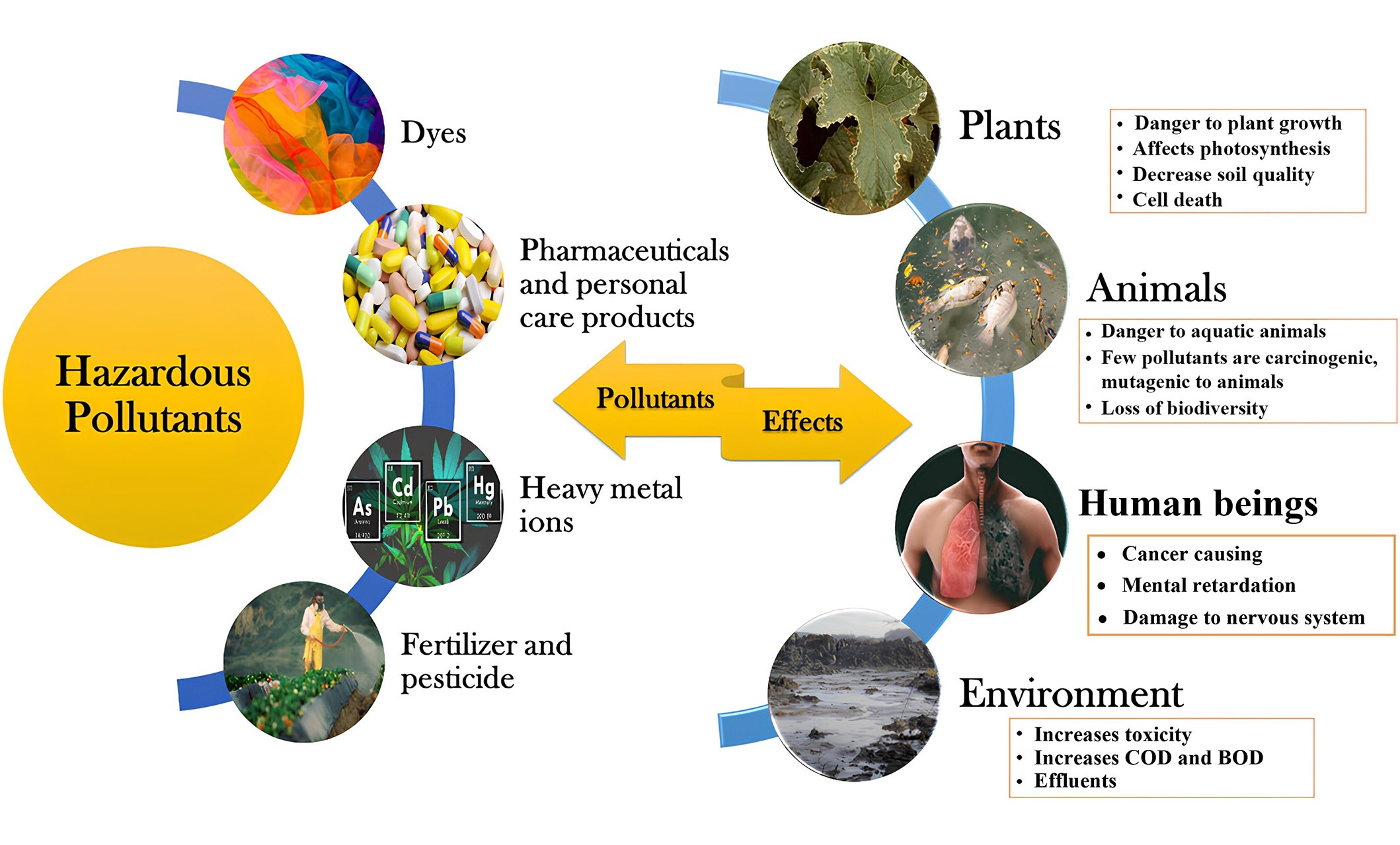
Broad classification and effects of hazardous pollutants. COD: chemical oxygen demand; BOD: biochemical oxygen demand
Note. Adapted from “Critical review on hazardous pollutants in water environment: occurrence, monitoring, fate, removal technologies and risk assessment,” by Rathi BS, Kumar PS, Vo DN. Sci Total Environ. 2021;797:149134 (https://doi.org/10.1016/j.scitotenv.2021.149134). © 2021 Elsevier B.V.
Because certain non-biodegradable organic pollutants persist in the environment for extended periods, the accumulation of these contaminants in living organisms ultimately infiltrates the food chain, significantly impacting the ecosystem. Specifically, organic contaminants like nitrophenols find extensive use in the production of pharmaceuticals, dyes, a variety of pesticides, and explosives. Due to their high acidity, water solubility, non-biodegradability, and carcinogenic properties, the unregulated utilization and release of these substances into the environment, without proper treatment, are leading to significant health concerns [2–5]. Furthermore, several neurotoxic and deadly pesticides are being widely employed to reduce crop losses by eradicating weeds and eliminating pests. In addition to impacting the reproductive and immune systems, the persistent consumption of food and water contaminated with pesticides is giving rise to various metabolic diseases [1–6]. While the primary purpose behind the utilization of food additives, whether natural or chemically synthesized, was to prolong shelf life and enhance the color, fragrance, taste, and nutritional value of food, their excessive use has posed a threat to human life. Natural food additives are predominantly produced by refining components extracted from plants or animals, while chemical food additives necessitate the use of chemical raw materials for their production. The food industry relies significantly on the application of chemical additives, but their overuse is a factor contributing to a range of gastrointestinal, neurological, and immunological diseases. The impacts of various contaminants and food additives on mental disorders are explored in the following sections and summarized in Figure 2 [7–10].
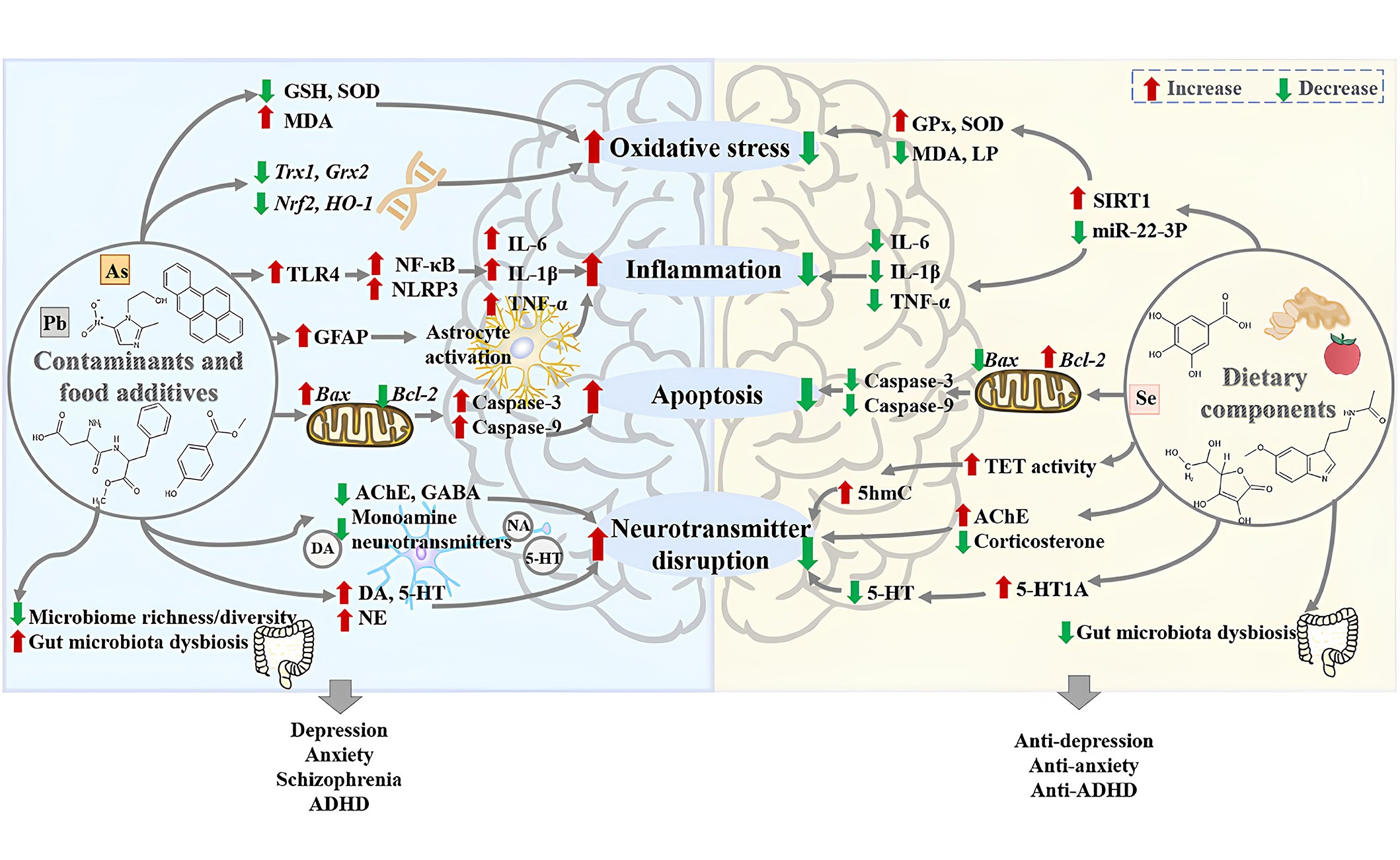
The harmful effects of contaminants and food additives on mental disorders as well as the protective effects of foods and dietary components against them, which is retrieved from the literature. The potential mechanisms of the harmful effects of contaminants and food additives on mental disorders include: (1) the contaminants and food additives could increase oxidative stress in the brain through increasing the level of malondialdehyde (MDA), decreasing levels of glutathione (GSH) and superoxide dismutase (SOD), and inhibiting the expression of antioxidant genes thioredoxin 1 (Trx1) and glutaredoxin 2 (Grx2), as well as the messenger RNA (mRNA) expression of nuclear factor erythroid 2 related factor 2 (Nrf2) and heme oxygenase-1 (HO-1); (2) the contaminants and food additives could induce neuroinflammation by activating astrocytes, enhancing the protein expression of Toll-like receptor 4 (TLR4), nuclear factor-kappa B (NF-κB), and nucleotide-binding domain and leucine-rich repeat related (NLR) family, pyrin domain containing 3 (NLRP3), and increasing the levels of pro-inflammatory cytokines interleukin 6 (IL-6), IL-1β, and tumor necrosis factor-alpha (TNF-α); (3) the contaminants and food additives could induce neuronal apoptosis by down-regulating the expression of B cell lymphoma-2 (Bcl-2), up-regulating the expression of Bcl-2-associated X protein (Bax), as well as the protein levels of caspase-3 and caspase-9; (4) the contaminants and food additives could disrupt neurotransmitters, especially monoamine neurotransmitters; (5) the contaminants and food additives could decrease the gut microbiome richness and diversity, as well as cause gut microbiota dysbiosis. Moreover, the potential mechanisms of the protective effects of dietary components against mental disorders caused by contaminants and food additives include: (1) dietary components could decrease oxidative stress in the brain by decreasing the levels of MDA and lipid peroxidation (LP), increasing the activities of SOD and glutathioneperoxidase (GPx) via the microRNA-22-3p (miR-22-3p)/sirtuin 1 (SIRT1) signaling pathway; (2) dietary components could reduce neuroinflammation through decreasing the levels of pro-inflammatory cytokines IL-1β, IL-6, and TNF-α; (3) dietary components could reduce neuronal apoptosis by down-regulating pro-apoptotic proteins caspase-3, caspase-9, and Bax, as well as up-regulating the expression of Bcl-2; (4) dietary components could regulate neurotransmitters by increasing activities of ten-eleven translocation (TET) and 5-hydroxymethylcytosine (5hmC) in the fetal brain, decreasing levels of corticosterone and 5-hydroxytryptamine (5-HT, serotonin); (5) dietary components could reverse gut microbiota dysbiosis caused by contaminants and food additives. AchE: acetylcholinesterase; ADHD: attention deficit hyperactivity disorder; GABA: γ-aminobutyric acid; GFAP: glial fibrillary acidic protein; NA: noradrenaline; NE: norepinephrine; DA: dopamine
Note. Reprinted from “New insights into the protection of dietary components on anxiety, depression, and other mental disorders caused by contaminants and food additives,” by Xiong RG, Li J, Cheng J, Wu SX, Huang SY, Zhou DD, et al. Trends Food Sci Technol. 2023;138:44–56 (https://doi.org/10.1016/j.tifs.2023.06.004). © 2023 Elsevier Ltd.
To safeguard consumers from contaminated foods, numerous national and international organizations, such as the European Food Safety Authority (EFSA), World Health Organization (WHO), US Environmental Protection Agency (EPA), and US Food and Drug Administration (FDA), have instituted regulations and guidelines to mitigate exposure to these chemicals. Owing to the efficacy of these regulations and legal frameworks, emissions of dioxins, dioxin-like compounds, and certain hazardous pesticides have notably decreased in recent years. The United Nations Environment Programme (UNEP) global monitoring plan for persistent organic pollutants has resulted in a consistent reduction in the levels of polychlorinated dibenzo-p-dioxins, polychlorinated dibenzofurans, polychlorinated biphenyls, and organochlorine pesticides in human milk. Nevertheless, the implementation of international codes and standards remains challenging, particularly in the face of the ongoing rise in the global population, projected to reach nine billion by 2050 [11–14].
Generally, investigating the binding properties, mechanisms, and characteristics of different food additives, pesticides, and contaminants with serum proteins is important for several reasons summarized below. Understanding the interaction of these substances with serum proteins helps determine their bioavailability. The binding capacity influences how these compounds are transported in the bloodstream and delivered to their target tissues. Additionally, it provides insights into potential toxicity and its impact on overall health [15, 16]. Besides, this information is vital for assessing the potential risks associated with exposure to these compounds. In cases where individuals are simultaneously exposed to pharmaceuticals and contaminants, understanding the binding interactions with serum proteins becomes essential. Because, drug interactions can alter the pharmacokinetics of therapeutic agents, affecting their efficacy and safety. Studying the interactions of contaminants with serum proteins is relevant not only for human health but also for understanding the environmental impact. The fate and transport of contaminants in ecosystems can be influenced by their interactions with proteins, affecting wildlife and ecosystems. In the context of food additives, studying their interactions with serum proteins is crucial for assessing food safety. These researches help to evaluate the potential health risks associated with the consumption of certain food products [17–22].
Therefore, exploring the interactions between these chemicals and human serum proteins is crucial for understanding the distribution, metabolism, and toxicity mechanisms of pollutants at the molecular level. While current research on pollutant toxicity predominantly emphasizes the dose-response relationship and cytotoxic effects on organisms, it is imperative to recognize that the essence of pollutant toxicity lies in the interaction between pollutants and biomolecules. Monitoring these interactions reveals variations in the chemical diversity of pollutant molecular structures, binding modes, binding free energy between pollutants and biomolecules, and significant distinctions in biomolecule conformation [23]. As a pioneering study, this review aims to assemble and combine the existing knowledge from diverse scientific studies, shedding light on the methodologies utilized to elucidate the binding properties, mechanisms, and characteristics of these compounds with serum proteins. By critically examining the analytical techniques, such as spectroscopy, molecular docking, and chromatographic methods, the review seeks to provide a nuanced understanding of molecular interactions. Moreover, the overarching goal is to contribute to the advancement of analytical approaches in this field, fostering a deeper comprehension of the implications of these interactions for human health and the environment. By consolidating the current state of research, the review aims to identify gaps in knowledge, potential areas for future investigation, and applications of these findings in areas such as risk assessment, pharmacokinetics, and the development of therapeutic interventions. In light of these considerations, this review delves into the interaction mechanisms of various food additives, pesticides, and contaminants with different serum proteins. The examination encompasses studies conducted from 2020 to 2023, providing a summary of the analytical and computational techniques employed in these investigations.
Methods employed to study the binding of food additives, pesticides, and contaminants with serum proteins
Basic information about serum proteins
Serum albumin is the predominant protein in blood plasma and is frequently employed as a representative protein in studies investigating molecule/ligand interactions. Human serum albumin (HSA) serves as the primary transport protein in blood plasma, playing a vital role in transporting both endogenous and exogenous ligands, including drugs and metabolites. The extent to which a molecule binds to HSA influences its in vivo delivery, distribution, effectiveness, and potential for toxicity [24, 25].
Bovine serum albumin (BSA) stands out as a versatile and widely used protein for different scientific purposes. BSA obtained from bovine blood can be used as a substitute for HSA in many experimental studies due to its structural similarity to HSA and its low cost. BSA was found wide application in protein quantitative analysis, in preserving the solubility and activity of enzymes and other biomolecules, and in immunoassays due to its inert nature as an ideal blocking agent. Additionally, BSA is a common component in cell culture media and provides essential nutrients and proteins to support cell growth due to its consistent and well-characterized composition [26, 27].
Human hemoglobin (Hb) stands out as a vividly red tetrameric protein, equipped with four oxygen-binding sites. Its primary role involves the transportation of oxygen, electrons, H+, CO2, and 2,3-bisphosphoglycerate within the bloodstream. While the structural and functional aspects of Hb have undergone comprehensive scrutiny, investigations into its interactions with various molecules have been largely confined to a subset of small compounds. Examining the interplay between Hb, a key physiologically active protein, and clinically relevant small molecules presents an opportunity to glean insights into Hb-mediated drug targeting. The exploration of bimolecular interactions involving proteins akin to Hb and specific small compounds holds significant promise within the field of biology, offering the potential to uncover novel perspectives on therapeutic strategies and drug design [28, 29].
Another serum protein, lysozyme, although not a dominant protein in the blood system, exhibits unique properties and plays an important role in maintaining homeostasis. The presence of lysozyme in blood, which is typically found in higher concentrations in body fluids such as tears and saliva, indicates the potential importance of this protein in immune defense. Owing to its antimicrobial properties, this protein may contribute to the innate immune response by disrupting bacterial cell walls. Although its concentration in the blood is lower compared to other blood proteins, the enzymatic activity of lysozyme makes it an effective protein in fighting infections and supporting general immune function in the bloodstream [30, 31].
Studying the interactions of all the above-mentioned proteins with different molecules provides information in terms of understanding potential or undesirable side effects, general kinetics of the compounds in the body, optimizing new molecular formulations, and distribution strategies in the body. For this reason, many different analytical techniques are frequently used to elucidate the interactions of serum proteins with molecules.
Basic information about analytical methods
Ultraviolet (UV)-visible (UV-Vis) absorption spectroscopy stands as a crucial and basic technique for discerning the interaction and complex formation of small molecules with serum proteins. Examining alterations in the absorbance profile of protein using UV-Vis absorption spectroscopy provides a comprehensive understanding of molecular interactions. Moreover, utilizing the Benesi-Hildebrand equation, the binding constant or association constant is calculated from UV-Vis absorption data, providing quantitative insights into molecule/ligand interactions with serum proteins [32, 33].
Fluorescence spectroscopy emerges as a highly sensitive and selective method for investigating the interactions between proteins and molecules. The intrinsic fluorescence spectra of proteins, mainly derived from aromatic amino acids such as tryptophan (Trp) and tyrosine residues, serve as a valuable indicator of structural changes induced by varying molecule concentrations. Quenching or enhancement mechanisms, generally categorized as static, dynamic, or mixed, can be easily interpreted from fluorescence data. Stern-Volmer and double logarithmic plots along with relevent equations provide quantitative data such as the Stern-Volmer constant, bimolecular quenching rate constant, binding constant, and the number of binding sites for the quenching or enhancement process. Thermodynamic parameters, such as entropy change, and enthalpy change offer insights into the non-covalent forces governing serum protein interactions. Calculated values and signs using the van’t Hoff equation, these parameters help identify the predominant stabilizing forces, whether electrostatic interactions, hydrophobic interactions, or hydrogen bonding and van der Waals interactions. Therefore, fluorescence spectroscopy emerges as a powerful tool not only for understanding molecular interactions but also for unraveling the thermodynamics behind these intricate binding processes. Furthermore, electron emission matrix spectroscopy, commonly referred to as three-dimensional fluorescence spectroscopy, has gained prominence as a scientific tool for scrutinizing the interaction mechanisms between molecules and serum proteins. This technique has emerged as a valuable resource in recent years, facilitating a comprehensive analysis of the microenvironment and conformational alterations within the structure of serum proteins induced by the presence of molecules. Its application provides a three-dimensional perspective, enhancing insights into complex molecular dynamics (MD) simulations involved in the interaction between serum proteins and molecules [34, 35].
The formation of complexes between serum proteins and molecules gives rise to energy transfer phenomena, involving the exchange of energy between serum proteins and interacting molecules. This energy transfer efficiency is elucidated through Förster (fluorescence) resonance energy transfer (FRET), a mechanism that entails the non-radiative transfer of energy from a donor fluorophore to an acceptor molecule. The success of FRET relies on specific conditions: proximity between the excited fluorophore (donor) and the drug/ligand (acceptor), overlap of the donor’s fluorescence emission spectrum with the acceptor’s excitation wavelength, correct orientation of the dipole moments of donor and acceptor molecules, and a high quantum yield of the donor [36, 37].
As another analytical tool, circular dichroism (CD) spectroscopy stands as a widely utilized method for examining alterations in the secondary and tertiary structures of serum protein induced by the presence of molecules. Specifically, changes in the secondary structure can be observed in the far-UV region of the CD spectra, where the serum protein CD spectra typically exhibit bands at different regions, indicative of α-helical content. These bands serve as highly sensitive markers for the binding of molecules. Increases or decreases in the intensity of the peaks of these bands provide information about the dynamic structural changes caused by the interaction. Moreover, the CD data can be further represented as mean residue ellipticity (MRE), offering a quantitative measure of the CD signal [32, 38].
Isothermal titration calorimetry (ITC) represents a widely employed technique for investigating the interactions between a protein and a molecule, providing insights into the energetics of complex formation. Through the measurement of energy changes during the process, ITC enables the determination of various thermodynamic parameters crucial for understanding the interaction between the molecules and serum proteins. Calculations involving changes in enthalpy, entropy, and the number of binding sites offer valuable information about the nature of the forces driving the complex formation [32, 39].
Fourier transform infrared (FTIR) spectroscopy serves as a crucial technique for revealing information about the functional groups within a molecule, producing a spectrum that acts as a unique molecular fingerprint. Specifically, FTIR proves valuable in characterizing secondary structural changes in proteins, both before and after binding with a molecule. FTIR not only facilitates qualitative observation but also enables quantitative analysis of changes in the secondary structure, including the amide I (C=O stretching) and amide II (C-N stretching coupled with N-H bending mode) bands, following the binding of a molecule [40, 41].
Nowadays, molecular docking stands as a crucial tool for unraveling the intricate interactions between small molecules and macromolecules, such as proteins and DNA, playing a pivotal role in rational drug/ligand design and development based on molecular structures. These studies offer predictive insight into the binding modes of drugs/ligands with macromolecules, allowing for informed decisions before embarking on experimental studies. Molecular docking not only validates findings obtained in the laboratory but also contributes to a deeper understanding of the binding orientation. Automated software facilitates these studies, predicting binding modes, and energetically favorable conformations between molecules and serum proteins. The process of molecular docking commences with acquiring the three-dimensional Protein Data Bank (PDB) structure of the macromolecule from databases like research collaboratory for structural bioinformatics or others, while molecule structures can be obtained from repositories like PubChem or ChemSpider. Multiple software options are available for docking studies, including AutoDock 4.0, AutoDock 4.2, AutoDock Vina, Hex 8.0, BSP SLIM online, and more. AutoDock, in particular, is widely employed for its reliability in predicting energetically favorable conformations. The selection of the energetically favorable conformer, typically the run with the lowest binding energy, allows for flexibility in drug/ligand docking, incorporating detailed molecular mechanics to calculate energy within the assumed active site [32, 42, 43].
MD simulations have proven to be a powerful tool for comprehending the intricate relationships between macromolecular structure and function. This computational approach calculates the time-dependent behavior of molecular systems, offering crucial insights for molecule design. MD simulation studies provide a detailed understanding of variations, stability, thermodynamics, and conformational changes within the structure of macromolecules in the presence of molecules. The binding of a molecule to a protein induces measurable conformational changes, quantified through root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), and formed hydrogen bonds [32, 42, 44].
Potential findings and latest applications of serum protein binding
Currently, numerous studies focus on residue detection and degradation methods of pesticides, environmental pollutants, and food products or their degradation metabolites. However, there is a notable scarcity of research on the toxicity mechanisms and transport properties of these compounds and metabolites in organisms. Therefore, in recent years, efforts have been made to investigate the binding interaction mechanisms of these substances with human serum proteins, especially in humans. In 2023, in their research, Cui et al. [45] explored the binding interaction mechanisms between HSA and the main degradation metabolites of pyrethroid insecticides, namely, 3-phenoxybenzoic acid (3-PBA) and 4-fluoro-3-PBA (4-F-3-PBA), through a combination of theoretical simulations and experimental validation. In this study, for fluorescence spectrum experiments, encompassing both fluorescence quenching and competitive binding experiments, HSA solutions were blended with two distinct small molecule solutions, maintaining an HSA concentration of 2 μmol/L and varying the small molecule concentrations from 0 μmol/L to 6 μmol/L. Following a 30 min water bath at temperatures of 298 K, 303 K, and 310 K, the fluorescence emission spectrum within the 290–450 nm range was scanned at a speed of 2,000 nm/min with an excitation wavelength of 280 nm. Steady-state fluorescence spectra revealed a static fluorescence quenching mechanism. Based on the binding constants, it was observed that 4-F-3-PBA (1.53 × 105 L/mol) exhibited a stronger binding affinity to HSA than 3-PBA (1.42 × 105 L/mol) at subdomain IIA (site I) (Figure 3). UV absorption and CD spectra indicated that the metabolites induced subtle changes in the microenvironment and conformation of HSA. Moreover, ITC revealed that the metabolites and HSA exhibited spontaneous combination primarily through hydrogen bonding and van der Waals interactions. Also, molecular docking analyses validated the aforementioned findings. To sum up, the toxicity characteristics of the metabolites were subjected to additional analysis using software, revealing that 4-F-3-PBA exhibited higher toxicity compared to 3-PBA. Given the widespread exposure of these metabolites in food, the environment, and the human body, there is a compelling need for further investigation into the toxicity of pyrethroid insecticide metabolites [45].
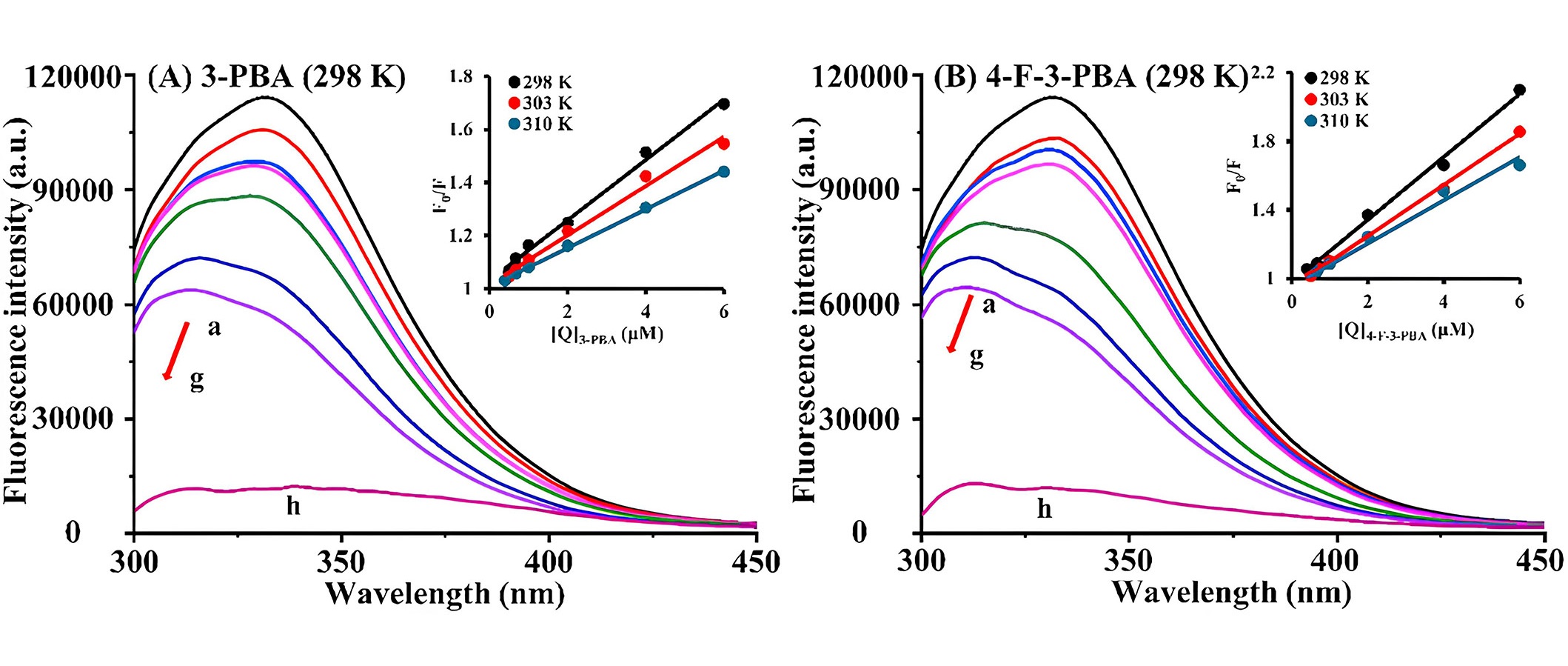
Fluorescence emission spectra of HSA with varying concentrations of 3-PBA and 4-F-3-PBA. (A) 3-PBA, temperature = 298 K, HSA = 2 μml/L (a–g), 3-PBA = 0–6 μmol/L (a–g), 3-PBA = 2 μmol/L (h); (B) 4-F-3-PBA, temperature = 298 K, HSA = 2 μml/L (a–g), 4-F-3-PBA = 0–6 μmol/L (a–g), 4-F-3-PBA = 2 μmol/L (h). a.u.: arbitrary units
Note. Reprinted from “Exploring the binding mechanism and adverse toxic effects of degradation metabolites of pyrethroid insecticides to human serum albumin: multi-spectroscopy, calorimetric and molecular docking approaches,” by Cui Y, Sun Y, Yu H, Guo Y, Yao W, Xie Y, et al. Food Chem Toxicol. 2023;179:113951 (https://doi.org/10.1016/j.fct.2023.113951). © 2023 Elsevier Ltd.
In another study conducted in 2023, Yang et al. [46] aimed to investigate the pH-dependent binding mechanisms between cyandin-3-O-glucoside (C3G) and BSA, exploring the interactions of natural pigments, anthocyanins, with proteins under varying pH conditions. Fluorescence quenching experiments and microscale thermophoresis (MST) analyses revealed a pH-dependent binding affinity, with the order pH 7 > pH 5 > pH 3, and dissociation constants (Kd) of 43.1 μmol/L (fluorescence quenching) and 33.0 μmol/L (MST) at pH 7. The prevailing forms of C3G were determined through UV-Vis absorption spectra with pH-jump experiments. Moreover, CD, Trp fluorescence, zeta potential, and particle size measurements indicated the “molten globule” state of BSA at pH 3, with C3G having a negligible effect on BSA conformation. MD simulations were employed to investigate the binding mechanisms, emphasizing the significance of electrostatic interaction. The flavylium cation exhibited limited binding to BSA at pH 3 due to electrostatic repulsion. In contrast, uncharged forms of C3G at all pH values bound to the BSA surface through hydrophobic interactions and hydrogen bonds, possibly in a weak and nonspecific manner. Anionic quinoidal bases, prevalent C3G forms at pH 7, predominantly bound to positively charged pockets on BSA, establishing several hydrogen bonds with surrounding amino acids, resulting in enhanced binding affinity [46].
Luteolin and naringenin are flavonoids found extensively in various foods and beverages, and they are also constituents of specific dietary supplements. With a significant consumption of these flavonoids, their sulfate and glucuronide conjugates can accumulate to micromolar concentrations in the bloodstream. Some investigations have delved into specific pharmacokinetic interactions associated with luteolin and naringenin; however, detailed information on their metabolites is limited in existing studies. In a study conducted by Kaci et al. [47], the interactions involving sulfate and glucuronic acid conjugates of luteolin and naringenin were explored concerning their binding to HSA, cytochrome P450 enzymes (CYP2C9, 2C19, and 3A4), and organic anion-transporting polypeptide transporters (OATP1B1 and OATP2B1). In fluorescence experiments (Figure 4), each examined flavonoid exhibited a concentration-dependent reduction in the emission signal of HSA at 340 nm. Both Stern-Volmer plots and Hyperquad assessments indicated a 1:1 stoichiometry of complex formation. Luteolin exhibited a higher affinity for protein binding compared to naringenin. Among the tested flavonoids, luteolin-7-glucuronide (L7G) displayed a significantly decreased binding affinity, which further diminished in the presence of a second glucuronic acid substitution at position 3’ (LdG). However, luteolin-3’-glucuronide (L3’G) exhibited a binding constant similar to that of the parent flavonoid. The presence of a sulfate substituent at position 3’ (LS) increased the binding affinity toward HSA. For naringenin, glucuronide conjugation markedly reduced the stability of albumin complexes, while sulfate substitution led to a slight enhancement in the binding affinity. Finally, researchers revealed that conjugated metabolites of luteolin and naringenin may exert a significant influence on the pharmacokinetic interactions of these flavonoids [47].
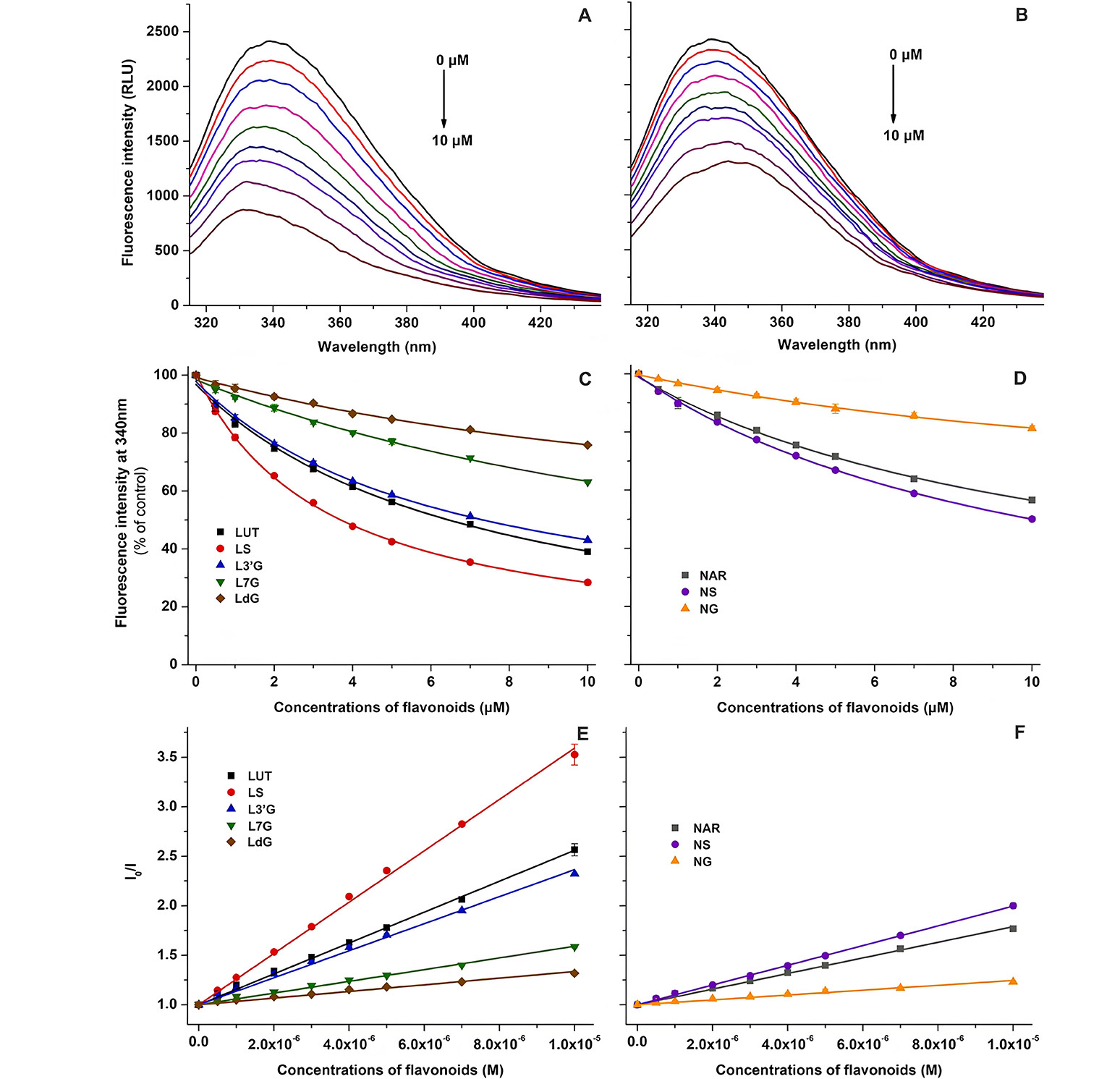
Fluorescence experimental results. (A) Representative fluorescence emission spectra of HSA (2 µmol/L) in the presence of increasing concentrations (0–10 µmol/L) of luteolin (LUT) in phosphate buffer saline [PBS, pH 7.4; excitation wavelength (λex) = 295 nm]; (B) representative fluorescence emission spectra of HSA (2 µmol/L) in the presence of increasing concentrations (0–10 µmol/L) of naringenin (NAR) in PBS (pH 7.4; λex = 295 nm); (C) flavonoid-induced decrease in the fluorescence emission signal of HSA at 340 nm for LUT, LS, L3’G, L7G, and LdG; (D) flavonoid-induced decrease in the fluorescence emission signal of HSA at 340 nm for NAR, NS, and NG; (E) Stern-Volmer plots of flavonoid-HSA complexes for LUT, LS, L3’G, L7G, and LdG; (F) Stern-Volmer plots of flavonoid-HSA complexes for NAR, NS, and NG. NS: naringenin-4’-O-sulfate; NG: naringenin-7-O-glucuronide; RLU: relative light unit; I0/I: fluorescence intensity 0/fluorescence intensity
Note. Reprinted from “Interaction of luteolin, naringenin, and their sulfate and glucuronide conjugates with human serum albumin, cytochrome P450 (CYP2C9, CYP2C19, and CYP3A4) enzymes and organic anion transporting polypeptide (OATP1B1 and OATP2B1) transporters’ by Kaci H, Bodnárová S, Fliszár-Nyúl E, Lemli B, Pelantová H, Valentová K, et al. Biomed Pharmacother. 2023;157:114078 (https://doi.org/10.1016/j.biopha.2022.114078). CC BY.
Despite the recognized health benefits of phenolic acids, their interactions with proteins remain unclear. In 2023, Zhang et al. [48] investigated the interactions between BSA and chlorogenic acid (CHA), caffeic acid (CA), as well as their Al3+ and Cu2+ complexes, utilizing UV-Vis, fluorescence, and CD spectroscopy. Significantly, the binding affinities for BSA were increased with the esterification of the carboxyl group of CA with quinic acid. Furthermore, upon chelation with Cu2+ and Al3+, both CHA and CA demonstrated enhanced binding affinities for BSA. Additionally, CHA demonstrated the ability to form CHA-Cu2 and CHA-Al2 complexes with Cu2+ and Al3+. The results from CD spectroscopy suggested that the interaction between CHA and Al3+ with BSA contributed to the folding of BSA’s secondary structure. Furthermore, the presence of CHA induced conformational changes in BSA when binding with Al3+ [48].
In 2022, Hoseyni et al. [49] explored the interaction between ten synthetic food dyes (Quinoline Yellow, Sunset Yellow, Carmoisine, Amaranth, Red 2G, Allura Red AC, Patent Blue V, Brilliant Blue FCF, Food Green S and Fast Green) and HSA through fluorescence spectroscopy, bio-partitioning micellar chromatography (BMC), and molecular docking analyses. The findings from fluorescence spectroscopy revealed a pronounced quenching effect on the intrinsic fluorescence of HSA, indicating a strong interaction near the Trp-214 residue of HSA. The modified Stern-Volmer equation was employed to calculate the Kb values and the number of binding sites in HSA. Utilizing BMC with polyoxyethylene 23 lauryl ether (Brij-35) as a micellar mobile phase, an in vitro system was established to predict the Kb values of food dyes to HSA. A model was developed to assess the relationship between BMC retention data and the Kb of food dyes, and the predictive capability of the model was evaluated. Moreover, molecular docking studies indicated that synthetic food dyes have the potential to bind within the extensive hydrophobic cavity of site I (subdomain IIA) in HSA [49].
Monascus pigments, secondary metabolites generated by Monascus species, have found widespread use as food colorants in China, Japan, Korea, and Southeast Asia for many years. Ankaflavin (AK) stands out as a representative yellow pigment derived from Monascus-fermented rice, known for its various biological effects. However, due to its limited solubility, investigations into AK delivery systems, particularly those built from protein-polysaccharide complexes, have garnered significant interest. The study conducted by Wu et al. [50] centered on examining the interactions between AK and BSA, with a specific emphasis on investigating how carrageenan (Car) affects the binding of AK to BSA. Findings indicated that the quenching of BSA by AK occurred through a static quenching mechanism. The resulting BSA-AK complexes were predominantly stabilized by hydrophobic forces, with AK positioned within the hydrophobic cavity of BSA. In contrast to free AK or AK solely complexed with BSA, the BSA-AK-Car complexes exhibited a greater absorption intensity of AK, suggesting alterations in the microenvironment of BSA. This was validated by the rise in the α-helix content of BSA following the creation of BSA-AK-Car complexes. Hydrogen bonding, van der Waals, and electrostatic interactions were identified as the main forces maintaining the BSA-AK-Car complexes. Additionally, the antioxidant activity of Monascus-fermented products was adversely affected by BSA, but the introduction of Car could enhance the antioxidant capacity of BSA-Monascus-fermented products-Car complexes [50].
Bioallethrin, a commonly used household insecticide, poses a risk of human exposure. Therefore, its cytotoxic effects on human erythrocytes prompted an investigation into its interaction with Hb through both in silico and biophysical methods. In the study conducted by Arif et al. [51] when Hb was incubated with increasing concentration of bio-allethrin, an increase in absorbance value was observed, which was accompanied by a slight reduction in the Soret band. Additionally, the intrinsic fluorescence of Hb increased, confirming that a new peak was observed. In this study, synchronous fluorescence analysis suggested that the interaction of bio-allethrin with Hb primarily affected the microenvironment around tyrosine residue. The alterations in Hb structure were validated by a notable shift in CD spectra, accompanied by approximately 25% reduction in α-helical content. Molecular docking, complemented by visualization in Discovery Studio, confirmed the establishment of a Hb-bio-allethrin complex with a binding energy of –7.3 kcal/mol. The structural modifications caused by bio-allethrin resulted in the suppression of the esterase activity of Hb. As a result, this investigation indicates that bio-allethrin establishes a stable complex with human Hb, potentially causing a decline in Hb functionality within the body [51].
Phenolic compounds constitute a vital component of the human diet, garnering interest for their antioxidant properties and potential health benefits. The impact of these compounds on human health is contingent upon consumption levels and bioavailability. Numerous studies have underscored the positive effects of polyphenols on the vascular system, including blood pressure reduction, enhanced endothelial function, bolstered antioxidant defenses, inhibition of platelet aggregation and low-density lipoprotein oxidation, as well as diminished inflammatory responses. Despite several reports highlighting the health benefits of moderate wine consumption, the specific contribution of the primary phenolics in red wine to the quenching properties of major human serum proteins remains unexplored. Shafreen et al. [52] aim to investigate red wine samples for their antioxidant activities, bioactive compounds, and interactions between wine polyphenols and key serum proteins. In this research, the interactions of fibrinogen and HSA with compounds including epicatechin, epigallocatechin, resveratrol, rutin, quercetin, gallic acid, tannic acid, myricetin, CA were examined in detail. As per the findings from fluorescence and molecular docking analyses (Figure 5), tannic acid demonstrated the highest binding affinity with HSA at –10.4 kcal/mol, succeeded by routine with a binding affinity of –9.9 kcal/mol. Additionally, nearly all compounds present in the investigated wine showcased interactions with HSA, as evidenced [52].
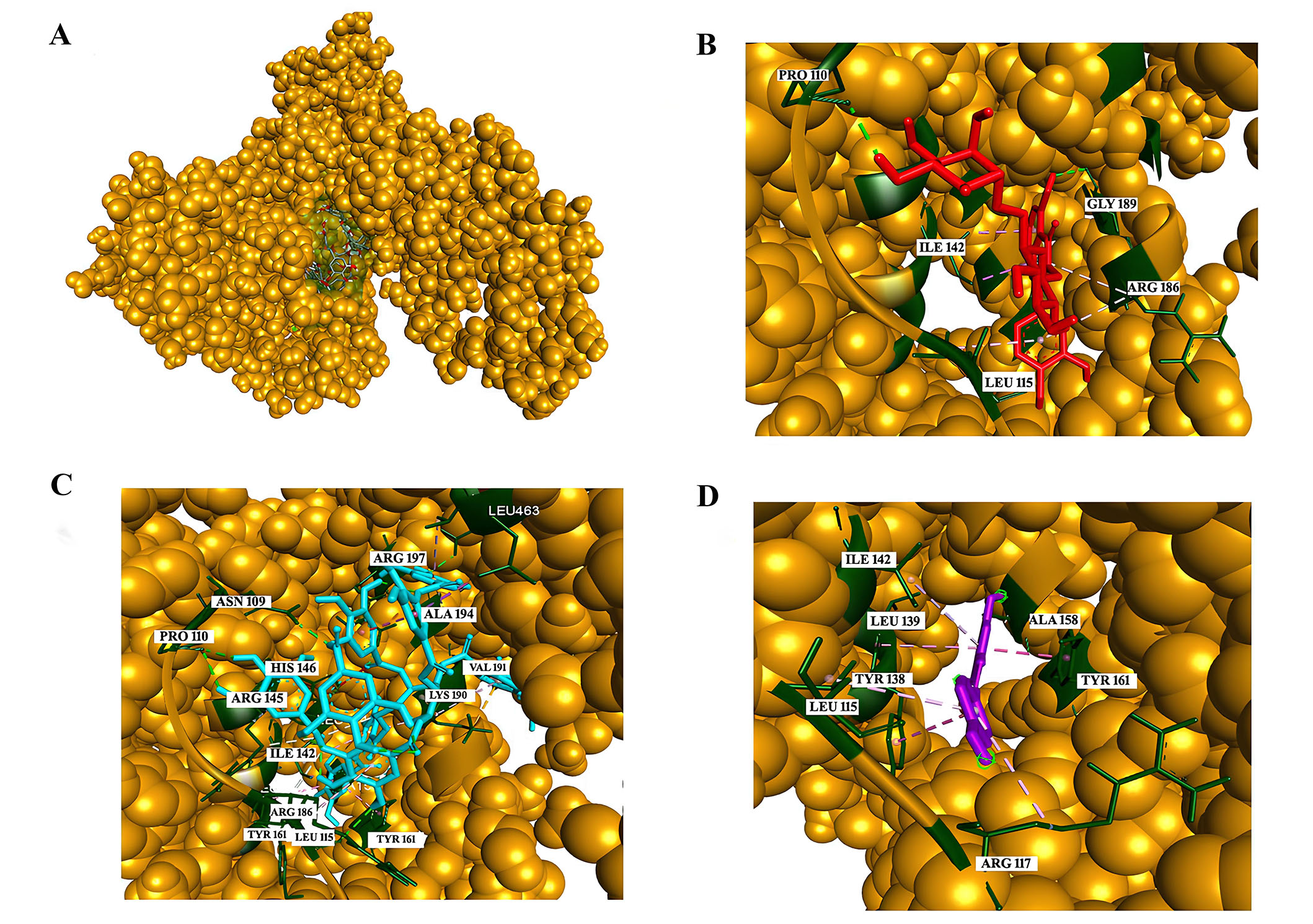
Analysis of interactions with HSA. (A) The green-colored surface represents the binding pocket of the HSA protein [Corey-Pauling-Koltun (CPK) model]; (B) interaction of rutin with the amino acids in the binding pocket; (C) interaction of tannic acid with the amino acids in the binding pocket; (D) interaction of resveratrol with the amino acids in the binding pocket. PRO: proline; ILE: isoleucine; GLY: glycine; ARG: arginine; LEU: leucine; ALA: alanine; VAL: valine; LYS: lysine; TYR: tyrosinase; HIS: histidine; ASN: asparagine
Note. Reprinted from “In vitro and in silico interaction studies with red wine polyphenols against different proteins from human serum†,” by Shafreen RMB, Lakshmi SA, Pandian SK, Kim YM, Deutsch J, Katrich E, et al. Molecules. 2021;26:6686 (https://doi.org/10.3390/molecules26216686). CC BY.
Various spectroscopic and computational methods play a pivotal role in elucidating the interactions between serum proteins and environmental compounds like food additives, pesticides, and pollutants. UV-Vis spectroscopy offers advantages in its simplicity and rapid data acquisition, allowing for the assessment of protein structural changes. Fluorescence spectroscopy, with its high sensitivity, provides detailed information on protein conformation alterations and binding affinities. CD spectroscopy is valuable for probing changes in protein secondary structure, although it has limitations in providing precise quantitative data. FTIR spectroscopy allows for the investigation of molecular vibrations and structural modifications in proteins, although it might lack the sensitivity observed in other methods. Molecular docking, a computational tool, is advantageous for predicting binding modes, binding energies, and affinities but is reliant on accurate structural information and may oversimplify the dynamic nature of protein interactions. Combining these techniques offers a multi-faceted understanding of the intricate interactions between serum proteins and various environmental substances, contributing to a comprehensive assessment of their potential physiological effects. Moreover, chromatographic methods are particularly valuable for their ability to separate and quantify individual components in complex mixtures, allowing for the identification of protein-bound contaminants and the assessment of binding affinities. Size-exclusion chromatography (SEC) within high-pressure liquid chromatography (HPLC) is specifically effective in studying changes in protein conformation and interactions. Moreover, gas chromatography is well-suited for volatile compounds, offering high sensitivity in the analysis of pesticide-protein interactions. While UV-Vis, fluorescence spectroscopy, CD, FTIR, and molecular docking offer valuable information, chromatographic methods significantly contribute to the analytical toolbox for a comprehensive understanding of serum protein interactions with environmental contaminants. To sum up, while these methods collectively offer a comprehensive understanding, it is crucial to recognize the specific strengths and limitations of each technique to draw meaningful conclusions about the intricate interactions between serum proteins and environmental substances.
Within the scope of this review, the interactions of various food additives, pesticides, and contaminants with different serum proteins were examined. Examples of selected studies carried out on this subject since 2020 were presented to researchers. The methods, quenching mechanism, quenching/binding constant, thermodynamic results, and binding region used in these studies were tabulated in detailed (Table 1)
Interaction studies with various food additives, pesticides, and contaminants against different serum proteins.
| Compound | Protein | Methods | Quenching mechanism | Quenching/Binding constant | Thermodynamic results | Binding region | Reference |
|---|---|---|---|---|---|---|---|
| Amaranth | HSA | UV-Vis, fluorescence spectroscopy, CD, molecular docking, and molecular dynamics simulations (MDS) | Static | KSV: 2.78 × 104−3.76 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = −11.8 kJ/molΔS° = 0.047 kJ‧mol–1‧K–1) | Subdomain IIA (site I) | [53] |
| New coccin | HSA | KSV: 4.1 × 104−5.53 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = −11.7 kJ/molΔS° = 0.050 kJ‧mol–1‧K–1(Hydrogen bonding and electrostatic forces) | ||||
| Myricitrin | HSA | UV-Vis, fluorescence spectroscopy, CD, micro-FTIR, molecular docking, and MDS | Static | KSV: 4.73 × 104−5.76 × 104 M–1Ka: 0.5 × 105−3.48 × 105 M–1 | ΔG° < 0 (spontaneous)ΔH° = −16.23 kJ/molΔS° = 0.049 kJ‧mol–1‧K–1(Hydrogen bonding, hydrophobic interactions, and electrostatic forces) | Subdomain IIA (site I) | [54] |
| Sodium tripolyphosphate | BSA | UV-Vis, fluorescence spectroscopy, FTIR, and molecular docking | Static | KSV: 4.5 × 103−9.38 × 103 M–1Ka: 4.25 × 101 −2.23 × 106 M–1 | ΔG° < 0 (spontaneous)ΔH° = −341.3 kJ/molΔS° = −1.064 kJ‧mol–1‧K–1(Hydrogen bonding and van der Waals forces) | Subdomain IIA (Sudlow’s site I) | [55] |
| 2,4,6-Trichlorophenol | BSA | UV-Vis, fluorescence spectroscopy, ITC, and molecular docking | Static | KSV: 1.018 × 106−1.692 × 106 M–1Ka: 1.962 × 105−5.701 × 105 M–1 | ΔG° < 0 (spontaneous)ΔH° = −58.06 kJ/molΔS° = −85.67 J‧mol–1‧K–1(Hydrogen bonding and van der Waals forces) | Subdomain IIIA(site II) | [56] |
| 2,4,6-Tribromophenol | KSV: 1.597 × 106−2.941 × 106 M–1Ka: 2.501 × 105−15.385 × 105 M–1 | ΔG° < 0 (spontaneous)ΔH° = −98.97 kJ/molΔS° = −215.76 J‧mol–1‧K–1(Hydrogen bonding and van der Waals forces) | |||||
| Sodium hydrosulfite | BSA | UV-Vis, fluorescence spectroscopy, FTIR, molecular docking, and surface plasmon resonance (SPR) | Static and dynamic | KSV: 5.13 × 103−6.12 × 103 M–1Ka: 0.313 × 102−44.545 × 102 M–1 | ΔG° < 0 (spontaneous)ΔH° = −139,783 kJ/molΔS° = −404.592 J‧mol–1‧K–1(Hydrogen bonding and van der Waals forces) | Subdomains IIA and IIIA (sites I and II) | [57] |
| Natamycin | BSA | UV-Vis, fluorescence spectroscopy, molecular docking, and SPR | Static and dynamic | KSV: 5.32 × 103−10.01 × 103 M–1Ka: 2.13 × 102−18.73 × 102 M–1 | ΔG° < 0 (spontaneous)ΔH° = −87.16 kJ/molΔS° = −237.6 J‧mol–1‧K–1(Hydrogen bonding and van der Waals forces) | Subdomain IIIA (Sudlow’s site I) | [58] |
| Sunset yellow | HSA | UV-Vis, fluorescence spectroscopy, and molecular docking | Static | KSV: 5.15 × 104−6.8 × 104 M–1Ka: 0.2 × 106−3.11 × 106 M–1 | ΔG° < 0 (spontaneous)ΔH° = −52.24 kJ/molΔS° = −50.07 J‧mol–1‧K–1(Hydrogen bonding and van der Waals forces) | Sudlow’s site I | [59] |
| Allura red | KSV: 3.75 × 104−4.21 × 104 M–1Ka: 0.04 × 106−0.3 × 106 M–1 | ΔG° < 0 (spontaneous)ΔH° = −58.79 kJ/molΔS° = −115.1 J‧mol–1‧K–1(Hydrogen bonding and van der Waals forces) | |||||
| Propazine | BSA | UV-Vis, fluorescence spectroscopy, and molecular docking | Static | KSV: 1.46 × 103−1.60 × 103 M–1Ka: 0.6 × 10–3−9.55 × 10–3 M–1 | ΔG° < 0 (spontaneous)ΔH° = −103.45 kJ/molΔS° = −0.05 kJ‧mol–1‧K–1(Hydrogen bonding, hydrophobic interactions, and van der Waals forces) | Subdomains IIA and IIIA(sites I and II) | [60] |
| Quinoxyfen | KSV: 4.17 × 103−6.39 × 103 M–1Ka: 5.01 × 102−7.08 × 102 M–1 | ΔG° < 0 (spontaneous)ΔH° = −12.84 kJ/molΔS° = 0.01 kJ‧mol–1‧K–1(Hydrogen bonding, hydrophobic interactions, and van der Waals forces) | |||||
| Aclonifen | HSA | UV-Vis, fluorescence spectroscopy, and molecular docking | Dynamic | KSV: 1.62 × 105−3.05 × 105 M–1Ka: 0.0174 × 106−1.95 × 106 M–1 | ΔG° < 0 (spontaneous)ΔH° = 225.43 kJ/molΔS° = 0.864 kJ‧mol–1‧K–1(Hydrophobic interactions) | Subdomains IIA and IIIA (sites I and II) | [61] |
| Bifenox | KSV: 1.6 × 105−2.10 × 105 M–1Ka: 0.002 × 106−1.02 × 106 M–1 | ΔG° < 0 (spontaneous)ΔH° = 304.63 kJ/molΔS° = 1.11 kJ‧mol–1‧K–1(Hydrophobic interactions) | |||||
| Phosmet | Bovine hemoglobin (BHb) | UV-Vis, fluorescence spectroscopy, CD, FRET, and molecular docking | Dynamic | KSV: 3.5 × 10–6−4.8 × 10–6 M–1Ka: 0.004 × 103−6.4 × 103 M–1 | ΔG° < 0 (spontaneous)ΔH° = −284.97 kJ/molΔS° = −88.29 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | NS | [62] |
| Isoflucypram | HSA | UV-Vis, fluorescence spectroscopy, CD, FTIR, molecular docking, and MDS | Static and dynamic | KSV: 1.593 × 104–1.832 × 104 M–1Ka: 0.158 × 103−4.923 × 103 M–1 | ΔG° < 0 (spontaneous)ΔH° = −187.549 kJ/molΔS° = −563.59 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | Sudlow’s site I | [63] |
| Cuminaldehyde (4-isopropyl benzaldehyde) | HSA | UV-Vis, fluorescence spectroscopy, CD, and molecular docking | Static | KSV: 5.5 × 103–8.3 × 103 M–1 | ΔG° < 0 (spontaneous)ΔH° = −16.0 kJ/molΔS° = 21.6 J‧mol–1‧K–1(Hydrophobic forces, and hydrogen bonding) | Subdomain IIA (site I) | [64] |
| Cuminol (4-isopropyl benzyl alcohol) | KSV: 6.3 × 102–9.4 × 102 M–1 | ΔG° < 0 (spontaneous)ΔH° = −15.9 kJ/molΔS° = 3.3 J‧mol–1‧K–1(Hydrophobic forces, and hydrogen bonding) | |||||
| Potassium bromate | BSA | UV-Vis, fluorescence spectroscopy, and molecular docking | Static and dynamic | KSV: 1.14 × 104−1.36 × 104 M–1Ka: 9.34 × 103−2.93 × 106 M–1 | ΔG° < 0 (spontaneous)ΔH° = −122.8 kJ/molΔS° = −320.51 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | Subdomain IB (site III) | [65] |
| Quinoline yellow | Lysozyme | UV-Vis, fluorescence spectroscopy, CD, molecular docking, and MDS | Static | KSV: 94.55 × 103−125.83 × 103 M–1Ka: 5.69 × 106−23.76 × 106 M–1 | ΔG° < 0 (spontaneous)ΔH° = −49.02 kJ/molΔS° = −32.69 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | NS | [66] |
| Carbofuran | BSA | UV-Vis, fluorescence spectroscopy, CD, and molecular docking | Static | KSV: 2.02 × 104 M–1Ka: 1.17 × 108 M–1 | NS | Site I | [67] |
| Naringenin | Lysozyme | UV-Vis, fluorescence spectroscopy, CD, molecular docking, and MDS | Static | KSV: 24.28 × 103−66.94 × 103 M–1Ka: 53.74 × 103−2,803.14 × 103 M–1 | ΔG° < 0 (spontaneous)ΔH° = 259.13 kJ/molΔS° = 953.11 J‧mol–1‧K–1(Hydrophobic interactions) | Trp 62, Trp 63, and Trp 108 | [68] |
| Azinphos-methyl | BSA | UV-Vis, fluorescence spectroscopy, CD, FTIR, and molecular docking | Dynamic | KSV: 0.6 × 104−1.46 × 104 M–1Ka: 0.099 × 105−0.209 × 105 M–1 | ΔG° < 0 (spontaneous)ΔH° = −133.25 kJ/molΔS° = −0.378 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | Subdomain IB (site III) | [69] |
| Flupyrimin | BSA | UV-Vis, fluorescence spectroscopy, CD, FTIR, and molecular docking | Static | KSV: 1.664 × 104−1.921 × 104 M–1Ka: 1.579 × 105−1.907 × 105 M–1 | ΔG° < 0 (spontaneous)ΔH° = −14.36 kJ/molΔS° = 52.95 J‧mol–1‧K–1(Hydrophobic forces, and hydrogen bonding) | Subdomain IIIA(site II) | [70] |
| HSA | KSV: 1.909 × 104−2.361 × 104 M–1Ka: 1.706 × 105−2.11 × 105 M–1 | ΔG° < 0 (spontaneous)ΔH° = −16.26 kJ/molΔS° = 47.31 J‧mol–1‧K–1(Hydrophobic forces, and hydrogen bonding) | |||||
| Nitenpyram | BSA | KSV: 2.16 × 104−2.349 × 104 M–1Ka: 1.911 × 105−2.108 × 105 M–1 | ΔG° < 0 (spontaneous)ΔH° = −7.51 kJ/molΔS° = 76.76 J‧mol–1‧K–1(Hydrophobic forces, and hydrogen bonding) | ||||
| HSA | KSV: 2.225 × 104−2.519 × 104 M–1Ka: 1.994 × 105−2.346 × 105 M–1 | ΔG° < 0 (spontaneous)ΔH° = −12.40 kJ/molΔS° = 61.16 J‧mol–1‧K–1(Hydrophobic forces, and hydrogen bonding) | |||||
| Formetanate hydrochloride | HSA | Fluorescence and CD spectroscopy, molecular docking, and MDS | Static | KSV: 0.01−0.02 M–1Ka: 3.75 × 10–5−5.14 × 10–5 M–1 | ΔG° < 0 (spontaneous)ΔH° = 13.46 kJ/molΔS° = 0.15 J‧mol–1‧K–1(Hydrophobic forces) | Sudlow’s site I and site II | [71] |
| Mancozeb | Hb | UV-Vis, fluorescence spectroscopy, CD, molecular docking, and MDS | Static | KSV: 2.09 × 104−3.27 × 104 M–1Ka: 1.76 × 104−5.33 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = −10.35 kcal/molΔS° = −0.013 kcal‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | NS | [72] |
| Dicofol | HSA | UV-Vis, fluorescence spectroscopy, CD, molecular docking, and MDS | Static | KSV: 0.73 × 105−1.3 × 105 M–1Ka: 0.82 × 105−2.77 × 105 M–1 | ΔG° < 0 (spontaneous)ΔH° = −55.35 kJ/molΔS° = −84.7 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | Suldow’s site I | [73] |
| Salicylic acid | BSA | UV-Vis, fluorescence spectroscopy, CD, and molecular docking | Static and dynamic | KSV: 10.26 M–1 | NS | Near the Trp-213 | [74] |
| Beauvericin | HSA | UV-Vis, fluorescence spectroscopy, and molecular docking | NS | NS | NS | NS | [75] |
| Cyclopiazonic acid | logKSV: 4.37logKa: 4.38 | Sudlow’s site I | |||||
| Sterigmatocystin | logKSV: 4.32logKa: 3.98 | NS | |||||
| Butylated hydroxyanisole | BSA | UV-Vis, fluorescence spectroscopy, and molecular docking | Static | KSV: 5.96 × 103−9.3 × 103 M–1Ka: 0.57 × 104−3.18 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = 110.8 kJ/molΔS° = 443.3 J‧mol–1‧K–1(Hydrophobic forces) | Subdomain IIA (site I) | [76] |
| Sorbic acid | HSA | UV-Vis, fluorescence spectroscopy, CD, FTIR, molecular docking, and MDS | Static | KSV: 5.555 × 104−6.218 × 104 M–1Ka: 4.033 × 105−5.046 × 105 M–1 | ΔG° < 0 (spontaneous)ΔH° = −29.366 kJ/molΔS° = 108.149 J‧mol–1‧K–1(Hydrogen bonding, and hydrophobic forces) | Subdomain IIA (site I) | [77] |
| Dicyclohexyl phthalate | HSA | UV-Vis, fluorescence spectroscopy, CD, FTIR, and molecular docking | Static | KSV: 2.05 × 105 M–1Ka: 13.47 × 104−39.54 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = −39.26 kJ/molΔS° = −29.14 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | Subdomain IIA (site I) | [78] |
| Monocyclohexyl phthalate | KSV: 1.24 × 105 M–1Ka: 0.7 × 104−1.72 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = −43.18 kJ/molΔS° = −68.34 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | |||||
| Malachite green oxalate | HSA | UV-Vis, fluorescence spectroscopy, CD, molecular docking, and MDS | Static | KSV: 374.9 M–1Ka: 4.35 × 106 M–1 | ΔG° < 0 (spontaneous)(Hydrogen bonding, and van der Waals forces) | NS | [79] |
| Lutein dipalmitate | BSA | UV-Vis, fluorescence spectroscopy, CD, and molecular docking | Dynamic | KSV: 2.17 × 104−3.22 × 104 M–1Ka: 0.63 × 104−2.75 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = −56.82 kJ/molΔS° = −106.02 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | Subdomain IIA (site I) | [80] |
| Chlorpyrifos | α2M | UV-Vis, fluorescence spectroscopy, CD, and molecular docking | Static | KSV: 1.017 × 104−1.656 × 104 M–1Ka: 5.432 × 10–4−6.181 × 10–4 M–1 | ΔG° < 0 (spontaneous)ΔH° = 15.62 kJ/molΔS° = 60.25 J‧mol–1‧K–1(Hydrophobic forces) | Receptor-binding domain of the α2M | [81] |
| Epoxiconazole | BSA | UV-Vis, fluorescence spectroscopy, CD, and molecular docking | Static | Ka: 0.79 × 104−3.8 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = −81.54 kJ/molΔS° = −199.35 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | Subdomain IIA (site I) | [82] |
| HSA | Ka: 0.814 × 104−6.22 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = −105.74 kJ/mol ΔS° = −265.88 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | |||||
| Prothioconazole | BSA | Ka: 1.66 × 105−6.45 × 105 M–1 | ΔG° < 0 (spontaneous)ΔH° = −84.85 kJ/mol ΔS° = −174.18 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | ||||
| HSA | Ka: 2.08 × 105−5.75 × 105 M–1 | ΔG° < 0 (spontaneous)ΔH° = −64.39 kJ/mol ΔS° = −105.50 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | |||||
| Dicofol | HSA | UV-Vis, fluorescence spectroscopy, CD, ITC, and molecular docking | Static | KSV: 1.18 × 104−1.37 × 104 M–1Ka: 4.38 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = −5.42 kJ/molΔS° = 21.08 J‧mol–1‧K–1(Hydrogen bonding, and hydrophobic forces) | Subdomain IIA (site I) | [83] |
| Thiamethoxam | Lysozyme | UV-Vis, fluorescence spectroscopy, molecular docking, and MDS | Static | KSV: 0.2 × 104−0.25 × 104 M–1Ka: 0.12 × 104−0.45 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = 58.5 kJ/molΔS° = 372.55 J‧mol–1‧K–1(Hydrophobic interactions) | NS | [84] |
| BSA | KSV: 1.1 × 104−1.15 × 104 M–1Ka: 1.93 × 104−4.99 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = 11.94 kJ/mol ΔS° = 231.88 J‧mol–1‧K–1(Hydrophobic interactions) | Site I | ||||
| HSA | KSV: 1.24 × 104−1.35 × 104 M–1Ka: 1.91 × 104−2.41 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = 12.84 kJ/mol ΔS° = 235.20 J‧mol–1‧K–1(Hydrophobic interactions) | |||||
| Phosmet | BSA | UV-Vis, fluorescence spectroscopy, CD, and molecular docking | Static | Ka: 0.15 × 104−3.68 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = −16.33 kJ/molΔS° = −469 kJ‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | Sudlow’s site II | [85] |
| Phosmet | Lysozyme | UV-Vis, fluorescence spectroscopy, CD, FTIR, and molecular docking | Static | KSV: 0.42 × 104−1.51 × 104 M–1Ka: 0.0168 × 104−9.14 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = −60.2 kJ/mol ΔS° = −187.78 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | NS | [86] |
| β-Resorcylic acid | Lysozyme | UV-Vis, fluorescence spectroscopy, CD, FRET, and molecular docking | Static | KSV: 1.69 × 103−5.15 × 103 M–1Ka: 1.13 × 103−4.68×103 M–1 | ΔG° < 0 (spontaneous)ΔH° = −13.97 kcal/molΔS° = −29.42 cal‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | Amino acid residues: Arg115, Arg119, Try124, and Gln123 | [87] |
| Acenaphthene | BSA | UV-Vis, fluorescence spectroscopy, CD, FTIR, and molecular docking | Static | KSV: 1.98 × 105 M–1Ka: 3.82 × 105 M–1 | NS | Subdomain IB(site III) | [88] |
| Quinoline yellow | αLA | UV-Vis, fluorescence spectroscopy, CD, molecular docking, and MDS | Dynamic | KSV: 4.0 × 10–4−4.4 × 10–4 M–1Ka: 0.091 × 10–5−0.955 × 10–5 M–1 | ΔG° < 0 (spontaneous)ΔH° = 18.79 kcal/molΔS° = 83.71 cal‧mol–1‧K–1(Hydrophobic interactions) | Central binding site of αLA | [89] |
| Monosodium glutamate | BSA | UV-Vis, fluorescence spectroscopy, CD, molecular docking, and MDS | Static and dynamic | KSV: 1.873 × 103−2.836 × 103 M–1Ka: 1.151 × 101 −4.05 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = 243.903 kJ/molΔS° = 888.291 J‧mol–1‧K–1(Hydrophobic interactions) | Sudlow’s site II | [90] |
| Calcium lactate | BSA | UV-Vis, fluorescence spectroscopy, CD, and molecular docking | Static and dynamic | KSV: 2.06 × 103−3.21 × 103 M–1Ka: 1.44 × 102−2.9 × 102 M–1 | ΔG° < 0 (spontaneous)ΔH° = −7.493 kJ/molΔS° = 24.61 J‧mol–1‧K–1(Electrostatic forces) | Sudlow’s site II (subdomain IIIA) | [91] |
| Sudan III | BSA | UV-Vis, fluorescence spectroscopy, CD, and molecular docking | Static | Ka: 5.83 × 102−6.41 × 102 M–1 | ΔG° < 0 (spontaneous)ΔH° = −5.65 kJ/molΔS° = 53.8 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | Subdomain IIA(site I) | [92] |
| Rhodamine B | HSA | UV-Vis, fluorescence spectroscopy, CD, FTIR, nuclear magnetic resonance (NMR), and molecular docking | Static | KSV: 5.86 × 104−6.23 × 104 M–1Ka: 6.06 × 104−6.35 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = −2.99 kJ/mol ΔS° = 81.91 J‧mol–1‧K–1(Electrostatic forces) | Subdomain IIA (site I) | [93] |
| Rosmarinic acid | HSA | UV-Vis, fluorescence spectroscopy, CD, ITC, molecular docking, and MDS | Static | KSV: 1.5 × 104−2.7 × 104 M–1Ka: 0.36 × 107−1.1 × 107 M–1 | ΔG° < 0 (spontaneous)ΔH° = 11.7016 kcal/molΔS° = 71.1303 cal‧mol–1‧K–1(Hydrophobic forces) | NS | [94] |
| 5-Hydroxymethyl-2-furaldehyde | HSA | UV-Vis, fluorescence spectroscopy, CD, and molecular docking | Static | KSV: 3.25 × 104−4.91 × 104 M–1Ka: 3.72 × 104−5.25 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = −30.02 kJ/molΔS° = −10.14 J/‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | Subdomain IIA(site I) | [95] |
| 3,5,6-Trichloro-2-pyridinol | BSA | Fluorescence spectroscopy, NMR, and molecular docking | Static | KSV: 2.1 × 105 M–1 | ΔG° < 0 (spontaneous)ΔH° = 23.77 kJ/molΔS° = 146.98 J‧mol–1‧K–1(Hydrophobic forces) | Subdomain IIA(site I) | [96] |
| Paraoxon methyl | KSV: 4.09 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = 94.74 kJ/molΔS° = 372.93 J‧mol–1‧K–1(Hydrophobic forces) | Subdomain IIA and IIIA(sites I and II) | ||||
| Chlorpyrifos | HSA | Solid-phase microextraction (SPME), and molecular docking | NS | Ka: 1.42 × 105 M–1 | NS | NS | [97] |
| Parathion-methyl | Ka: 1.45 × 104−8.19 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = −193.3 kJ/molΔS° = −543.7 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | Subdomain IIA(site I) | ||||
| Malathion | Ka: 1.07 × 104−4.02 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = −147.7 kJ/molΔS° = −399.2 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | Subdomain IIIA(site II) | ||||
| Benthiavalicarb-isopropyl | HSA | UV-Vis, fluorescence spectroscopy, CD, molecular docking, and MDS | Static | KSV: 4.41 × 103−8.66 × 103 M–1Ka: 0.032 × 102−7.965 × 102 M–1 | ΔG° < 0 (spontaneous)ΔH° = −206.39 kJ/molΔS° = −654.93 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | Hydrophobic cavity of HSA | [98] |
| Pendimethalin | HSA | UV-Vis, fluorescence spectroscopy, CD, molecular docking, and MDS | Static | KSV: 7.17 × 104−9.92 × 104 M–1Ka: 8.47 × 104−10.63 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = −16.17 kJ/molΔS° = 45.78 J‧mol–1‧K–1(Hydrophobic forces) | Subdomain IIA (Sudlow’s site I) | [99] |
| Tebuconazole | BSA | UV-Vis, fluorescence spectroscopy, and CD | Static | Ka: 2.25 × 102−4.67 × 102 M–1 | ΔG° < 0 (spontaneous)ΔH° = −46.72 kJ/molΔS° = −105.67 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | NS | [100] |
| Perfluorooctanoic acid | HSA | UV-Vis, fluorescence spectroscopy, FTIR, and molecular docking | Static | KSV: 1.076 × 104−1.328 × 104 M–1Ka: 0.4463 × 104−0.6153 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = −17.48 kJ/molΔS° = 13.53 J‧mol–1‧K–1(Electrostatic forces) | Site I | [101] |
| Perfluorodecanoic acid | KSV: 1.431 × 104−1.731 × 104 M–1Ka: 1.4514 × 104−2.6788 × 104 M–1 | ΔG° < 0 (spontaneous)ΔH° = −33.37 kJ/molΔS° = −27.91 J‧mol–1‧K–1(Hydrogen bonding, and van der Waals forces) | |||||
| Acesulfame | HSA | UV-Vis, fluorescence spectroscopy, CD, and molecular docking | Static | KSV: 0.81 × 103−1.77 × 103 M–1Ka: 1.74 × 102−1.82 × 102 M–1 | ΔG° < 0 (spontaneous)ΔH° = −2.88 kJ/molΔS° = 33.66 J‧mol–1‧K–1(Electrostatic forces) | Subdomain IIA(site I) | [102] |
Ksv is Stern-Volmer or quenching constant. Ka is binding constant. ΔG°: Gibbs free energy change; ΔH°: enthalpy change; ΔS°: entropy change. α2M: alpha-2-macroglobulin; αLA: alpha-lactalbumin; NS: not stated
Conclusions
In recent years, the surge in published articles dedicated to protein-ligand interactions has been significant, thanks to the integration of diverse analytical, and computational techniques in research. These studies utilize various methods to validate the interaction and binding modes of both novel and established ligands with proteins, offering valuable insights into their mechanisms of action. This review presents a comprehensive overview of commonly employed techniques in this field and their interaction applications of various food additives, pesticides, and contaminants with serum proteins.
Abbreviations
| 3-PBA: | 3-phenoxybenzoic acid |
| 4-F-3-PBA: | 4-fluoro-3-phenoxybenzoic acid |
| AK: | ankaflavin |
| BMC: | biopartitioning micellar chromatography |
| BSA: | bovine serum albumin |
| C3G: | cyandin-3-O-glucoside |
| CA: | caffeic acid |
| Car: | carrageenan |
| CD: | circular dichroism |
| CHA: | chlorogenic acid |
| FRET: | Förster (fluorescence) resonance energy transfer |
| FTIR: | Fourier transform infrared |
| Hb: | human hemoglobin |
| HSA: | human serum albumin |
| ITC: | isothermal titration calorimetry |
| MD: | molecular dynamics |
| MDS: | molecular dynamics simulations |
| Trp: | tryptophan |
| UV-Vis: | Ultraviolet-visible |
Declarations
Author contributions
CE and MZK: Conceptualization, Investigation, Writing—original draft, Writing—review & editing.
Conflicts of interest
Cem Erkmen is the GE of Exploration of Foods and Foodomics, but he had no involvement in the journal review process of this manuscript. Both authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2024.