
Abstract
Antiphospholipid syndrome (APS) is defined as an autoimmune and prothrombotic disorder in patients with the persistent presence of antiphospholipid antibodies (aPLs). In the classification criteria, aPL expresses lupus anticoagulant (LA) activity, which is detected by prolongation of coagulation assays. The LA detection algorithm is a sequential flow including screening tests, mixing tests, and confirmatory tests to differentiate between LA-positive and other anticoagulant abnormalities. Two types of assays are used, like dilute Russell’s viper venom time (dRVVT) and activated partial thromboplastin time (APTT) because no single test is sensitive to all LAs. The anticoagulant drugs prescribed for the prevention and treatment of thrombosis disorders can interfere with the assays, and it is important to know the effects of these drugs in the assays. Especially, new generation anticoagulant drugs, called direct oral anticoagulants (DOACs), affect the results. In this review, the following points are discussed: i) LA detection flow and data interpretation, ii) the principles of coagulation assays proposed and their characteristics, and iii) the effects of anticoagulant drugs in LA detection.
Keywords
Lupus anticoagulant, thrombosis, antiphospholipid syndrome, antiphospholipid antibodies, activated partial thromboplastin time, dilute Russell’s viper venom timeIntroduction
Antiphospholipid syndrome (APS) is defined as an autoimmune and prothrombotic disorder in patients who exhibit persistent presence of antiphospholipid antibodies (aPLs) and arterial or venous thrombosis and/or miscarriage or pregnancy loss are among the clinical symptoms reported [1]. APS is an important cause of acquired thromboembolic complications in any vasculature: arterial, venous, or small vessels, and it has come to be subcategorized into primary, secondary, and catastrophic APS syndrome. Primary is utilized when there is no associated disorder, secondary is with an associated autoimmune disorder such as systemic lupus erythematosus, and catastrophic when thrombosis occurs at multiple sites in a short space of time [1–3]. Once APS is diagnosed, long-term treatment with anticoagulant drugs is considered because the risk of recurrent thrombosis is high [4]. Since the clinical symptoms of thrombosis and pregnancy morbidity in APS are non-specific, accurate detection of aPL is critical for an effective diagnosis and patient management. Diagnosis of APS relies on the simultaneous presence of characteristic clinical symptoms and persistent presence of one or more aPLs, confirmed at least twice at 12 weeks apart. Three types of aPLs are defined as antibody criteria in the International Society on Thrombosis and Haemostasis (ISTH) classification criteria [5]; those with LA activity and detected with clotting assays; those reactive with anionic phospholipids (PLs)/cardiolipin, and those targeted to human-β2-glycoprotein I (β2GPI), of the immunoglobulin G (IgG) or IgM isotype, and measured with solid phase immunoassays and calibrators for each antibody type. Recently, automated analyzers have been introduced and are currently widely used for the aPL measurements, and the results are reported quantitively [6, 7]. On the other hand, lupus anticoagulants (LAs) are defined by their anticoagulant activity, not by their antigenic targets, and they are detected through the prolongation of clotting times using uncalibrated coagulation assays [8].
The simplest definition of LA relies on its property to prolong the clotting times of PL-dependent coagulation tests. Since LA concerns a group of heterogenous antibodies, no single coagulation assay is enough sensitive to detect all of them and their specificity must be confirmed. Two test systems based on different analytical principles must therefore be employed for evidencing LA, as recommended by ISTH guidelines [8, 9].
The LA detection includes the following tests:
Screening test: conducted using a reagent with a low PL concentration to accentuate the sensitivity to LA. The clotting time is prolonged because LA inhibits coagulation reaction through binding to PLs, which are no longer available for the coagulation cascade, due to the limited amount of PL binding sites.
Mixing test: patient plasma is mixed with normal pooled plasma (NPP) in a 1:1 proportion, and the mixed plasma sample is measured with the screening reagent. In the presence of LA, the clotting time is not corrected because it still inhibits the coagulation reaction in the presence of a limited amount of PL binding sites, while the test is corrected if prolongation results from a coagulation factor deficiency.
Confirmatory test: the clotting test is performed using a reagent with a high PL concentration. In the presence of this high concentration of PLs, LA binding does not mask all PL binding sites, which remain available for coagulation, and shorter clotting times are obtained. Therefore, this test confirms the PL dependence for anticoagulant antibodies.
Test results in patients with LA are expected to show prolonged clotting times in screening tests, not corrected in mixing tests, and shorter clotting times in confirmatory tests. However, this is not always the case because coagulation assays are affected by potential interferences such as those from elevated or reduced coagulation factors, the presence of anticoagulant therapy, and non-PL-dependent inhibitors, like anti-coagulation factor antibodies. These factors introduce a complementary layer of complexity for LA identification and various guidelines are available to guide and optimize laboratory practices [8, 10–12]. Although various guidelines are available for LA detection, there are some differences between them [13, 14]. The comparison of major points for two major guidelines, which are those from ISTH and from the Clinical and laboratory Standard Institute (CLSI), are summarized in Table 1 [11, 12]. This article reviews the LA detection flowchart, principles of laboratory detection systems, interpretation of assays, and their limitations.
Summary of the comparison between ISTH and CLSI guidelines for LA detection
| Category | ISTH 2020 | CLSI 2014 |
|---|---|---|
| Sample preparation | Double centrifugation | Double centrifugation |
| Assay to use | dRVVT and APTT | dRVVT and APTT and/or others |
| Testing order | Screening => mixing & confirmatory | Screening => confirmatory => mixing |
| Calculations for PL-dependence | Normalized ratio or percentage correction | Normalized ratio or percentage correction |
| Mixing test | Perform on: mixture with NPP Interpret with MTC | Perform on: mixture with NPP Interpret with MTC or ICA |
MTC: mixing test-specific cut-off; ICA: index of circulating anticoagulant
Flowchart for testing LAs
Preliminary examination
The routine coagulation tests of prothrombin time (PT), activated partial thromboplastin time (APTT), fibrinogen and thrombin time (TT) should be conducted to detect the presence of coagulation factor deficiencies, coagulation inhibitors, anticoagulant drugs, and acute phase reactant proteins before starting the LA specific investigation [8]. During this preliminary examination, APTT routine tests, using LA insensitive reagent, are recommended to avoid unnecessary evaluations for aPL. Although LA strong-positive can affect the clotting time of LA insensitive APTT reagents, the prolongation indicates the possible presence of other causes, like factor deficiencies or some inhibitors [11].
The three-step procedure
The LA detection algorithm is composed of three steps including screening, mixing, and confirmatory tests. If LA is present in patient’s sample, all test results will be typically LA-positive, while samples with other abnormalities, like low or high coagulation factors’ activity or the presence of other inhibitors, show different profiles. To differentiate LA-positive samples from other abnormalities, three-step procedure that is in line with ISTH guidelines will be helpful (Figure 1). The latest ISTH guidelines recommend performing the mixing test and the confirmatory test at the same time because both results provide useful diagnosis information [8].
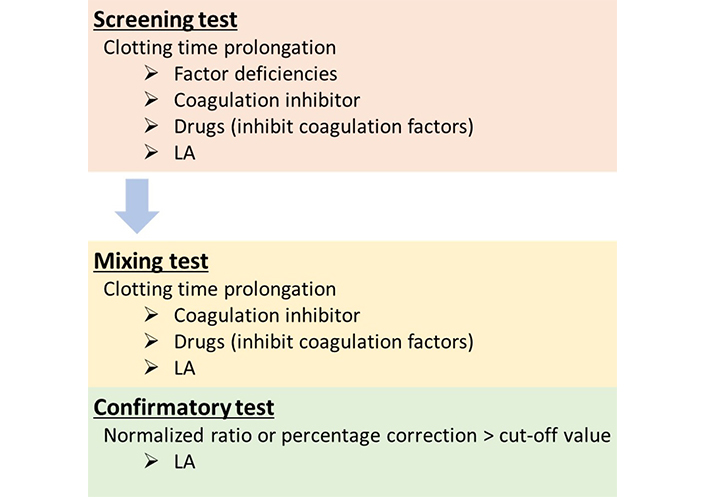
LA detection flowchart. In the screening test, the clotting time is prolonged in the presence of LA, but also in the presence of various conditions, like coagulation factor deficiencies, coagulation inhibitors, or anticoagulant drugs. Mixing tests are performed to investigate whether inhibitors, including LA, are present in samples and they differentiate between the presence of inhibitor and factor deficiencies. The clotting time in the mixing sample is not corrected when inhibitors are present in the tested samples. In the confirmatory test, the PL dependence is confirmed because the coagulation assay is performed in the presence of a high PL concentration, and the clotting time of LA-positive samples is shortened. If the result is above the cut-off value, it indicates that the inhibitor is PL-dependent, and it is interpreted as LA-positive
Screening test
Since LA is a group of heterogenous antibodies that inhibit the binding of coagulation factors to PLs in vitro, the PL concentration used should be low. The reagent with a low PL concentration shows a prolonged clotting time when LA is present in the tested sample [15]. On the other hand, the clotting time prolongation is not always characteristic of a LA-positive sample, since test results can be elevated by numerous other abnormalities [16, 17].
For the expression of results, the latest ISTH guidelines suggest the screening test is positive if the normalized clotting time is prolonged beyond the locally established cut-off and it is also recommended to express the results as screen ratio of patient-to-NPP [8]. This ratio is calculated by dividing the clotting time of the patient by the clotting time of the NPP measured at the same time to reduce the variations resulting from the reagent, the instrument, and the operator. If the screen ratio is higher than the cut-off value, the result is interpreted as positive in the screening test and the mixing test should be performed.
In CLSI guidelines, the screen ratio is proposed with the following formula [11]:
screen ratio = clotting time of patient sample/normal reference interval mean
Although CLSI guidelines recommend the use of the mean of a reference interval as the denominator of the formula [11], the latest ISTH guidelines describe that the mean of the reference interval is less applied because it does not compensate for the day-to-day variations [8].
Mixing test
The mixing test is conducted by mixing the tested patient plasma with NPP at 1:1 proportion and measuring the clotting time. As described above, the clotting time prolongation in screening test does not always indicate an LA-positive sample, because coagulation assays are affected by many other factors like low coagulation factor activity, presence of other inhibitors, or anticoagulant drugs. Clotting times in the mixing tests are shortened in samples with low coagulation factor activities because coagulation factors in NPP compensate for those missing in patient’s plasmas. In this case, the result is interpreted as “corrected”. One of the definitions of “corrected” is that the result is within the normal range. Conversely, the clotting time of the mixing test is not shortened when test plasma samples contain inhibitors, and the result is interpreted as “not corrected”. LA is defined as the group of heterogenous antibodies, which inhibit PL-dependent coagulation assays, and the reaction is not time-dependent [18]. Then, “not corrected” results indicate the presence of an inhibitor in the tested sample, but it does not mean that it is LA, and an additional confirmatory test must be performed to distinguish LA from non-PL-dependent inhibitors.
Historically, there are two types of mixing test interpretation methods. The first one is the use of MTC which is derived from the upper limit of distribution in mix ratio. The mix ratio is calculated as follows:
mix ratio = clotting time of 1:1 mix sample/1:1 mix reference interval mean
Although this formula is recommended in CLSI guidelines [11], it is described to express a normalized ratio and to reduce intra-laboratory variability in the latest ISTH guideline [8].
Another method concerns ICA calculated with the following formula:
ICA = [(clotting time of 1:1 mix sample – clotting timer of NPP)/clotting time of patient sample] × 100
In the mixing test, it is important to report the results with a high sensitivity index since the results may be negative in weak LA-positive samples by dilution. In the previous study, the sensitivity to LA was compared between MTC and ICA and we reported that MTC was superior to ICA for LA detection using multiple reagents and various reagent types [19, 20]. In the latest ISTH guidelines, it is recommended that the mixing test results should be interpreted with MTC expressed as the normalized ratio as same as the screening ratio [8].
NPP for the mixing test is ideally prepared in-house because the residual platelet in plasma must be minimized and all coagulation factors should be at normal levels to show the “corrected” results for factor-deficient plasma samples. In the guidelines, it was described that NPP should be constituted from at least 40 normal donors with coagulation factor activities in the normal range [8, 9]. Alternatively, commercial lyophilized or frozen NPP can also be used if the commercial materials have a similar performance characteristics [8].
Confirmatory test
Although the differentiation between inhibitors and coagulation deficiency is performed in mixing tests, it is difficult to discriminate between a non-specific inhibitor such as LA and some drugs, from coagulation inhibitors. Consequently, confirmatory tests are conducted as the final step in LA diagnosis algorithm. LA insensitive reagents with a high PL concentration are used for confirmatory tests and results are expressed in combination with the screening tests. LA insensitive reagents provide a PL concentration excess to adsorb LA so that the clotting time is shorter than that of the screening test. Non-PL dependent inhibitors like coagulation inhibitors and some drugs are not affected basically, and the result is equivalent to that of screening tests when normalization is obtained for both results. To evidence the PL dependence, the confirmatory test must be performed by increasing the concentration of PLs used in the screening test [11].
The results of LA insensitive reagents are expressed as the confirm ratio by dividing the clotting time of patient by the clotting time of NPP, and the final data is interpreted by normalized ratio. The formulas are shown in Table 2.
Determination of normalized ratios for patients’ samples
| Index names | Formulas |
|---|---|
| Screen ratio | Clotting time patient/normal reference interval mean (calculated with screening reagent) |
| Confirm ratio | Clotting time patient/normal reference interval mean (calculated with confirmatory reagent) |
| Normalized ratio | Screen ratio/confirm ratio |
| Percentage correction | [(Screen ratio – confirm ratio)/screen ratio] × 100 |
If the confirmatory test is prolonged on neat plasma, the test should be performed on a mixing plasma sample at 1:1 proportion of patient plasma: NPP because some anticoagulant drugs prolong the clotting time of the confirmatory test and the prolongation of the confirmatory test indicates the possibility of these anticoagulant drugs [8, 21].
Clinical laboratory tests for LA detection
Sample collection and storage
Blood should be collected into 3.2% trisodium citrate at a ratio of nine parts blood to one part anticoagulant. Double centrifugation should be conducted at 2,000 g for 15 min to yield a platelet count of < 10 × 109/L [8, 9]. The plasma supernatant from the first centrifugation round must be collected and the second centrifugation step must be performed in that process. To reduce the residual platelet is important to obtain reliable testing results because the PL from the residual platelets might bind to LA, and reduce testing sensitivity. False-negative results can then be obtained, especially for weak LA-positive samples [22]. In the operation, it is common to use the frozen plasma samples for the measurements in the batch analysis. Plasmas can be stored at –20°C for 14 days and at –70°C for at least 6 months before testing [11].
Choice of assays
There is evidence that there is no single test sensitive enough for all LA, and two tests based on different principles are required. Dilute Russell’s viper venom time (dRVVT) should be considered as the first screening test, and the second test should be a sensitive APTT with a low PL concentration. It has been reported that the combination of dRVVT and APTT offers high detection rates of LA [9, 23, 24]. dRVVT is recommended due to its specificity and robustness, and APTT is recommended because of its sensitivity by all current guidelines [8, 10, 11]. However, clotting times performed with APTT reagents are affected not only by LA but also by factor deficiencies and the presence of anticoagulants [25]. Therefore, it is important to understand the reagent characteristics before using it and performing the dRVVT tests. Although there are other LA detection assays like kaolin clotting time and dilute PT, use of other tests is not recommended because of too high variability, poor reproducibility, or non-availability of standardized commercial assays [11].
dRVVT
dRVVT is employed as a paired reagent system using an LA screening reagent, with a low PL concentration, and an LA confirmatory reagent, with a high PL concentration. The reagent activator is an enzyme from Russell’s viper venom (RVV) [26]. It directly activates factor X (FX) in a calcium-dependent, but PL-independent manner, and FX is converted to FXa. The FXa then activates prothrombin to thrombin, in a PL-dependent manner, and in the presence of FVa and calcium. Subsequently, thrombin converts fibrinogen to fibrin and leads to the formation of the fibrin clot [27]. PLs are required for the activation of prothrombin (Figure 2). There is a high variability of clotting times generated with the various commercial reagents, but similar results are obtained when expressed as normalized ratios [8, 11, 28]. The procedure is established as a paired test system in dRVVT, and it means that the confirmatory reagent must be designed like the screening reagent, but with a higher PL concentration. The confirmatory performed must always be from the exactly paired reagents [11].
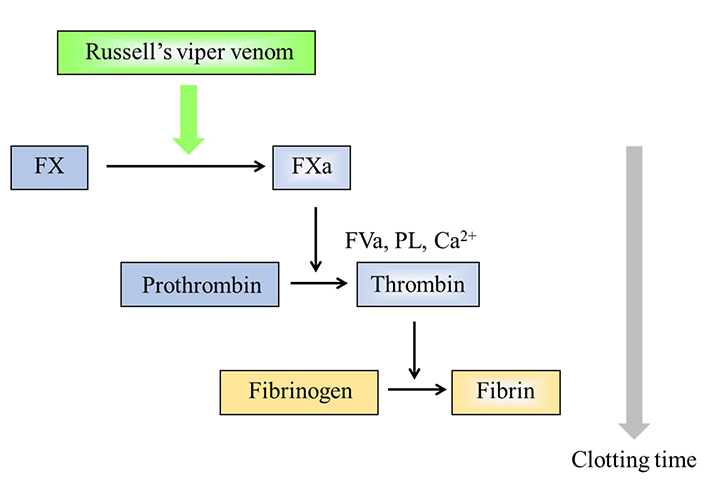
Principle of the dRVVT. RVV directly activates FX in a Ca2+-dependent manner. PL is then required for forming prothrombinase complex. If LA is present in tested plasma samples, it inhibits the PL-dependent reaction and the clotting time is prolonged, only in the screening reagent with a low PL concentration
APTT
APTT is the routine screening test which is composed of two kinds of reagents. The first reagent is composed of a contact phase activator, such as silica and ellagic acid, along with various PLs. The activator activates the contact pathway in the coagulation cascade, and PLs facilitate the coagulation reactions to generate the enzymes from the initial intrinsic pathway of the coagulation cascade, including vitamin K-dependent coagulation factors. The second reagent is calcium chloride, the addition of which initiates the calcium-dependent coagulation reaction. In this procedure, a plasma sample is first mixed with the first reagent at 37°C to allow the contact reactions to proceed. Then, calcium chloride is added to the mixture. As a result, thrombin generated from prothrombin converts fibrinogen to the fibrin clot. The time between the addition of calcium chloride to the reaction mixture and the detection of fibrin formation in the cuvette is monitored, and it is defined and recorded as the clotting time (Figure 3). LA inhibits the PL-dependent reaction in the coagulation cascade, thereby leading to prolongation of clotting time when present in the sample [25].
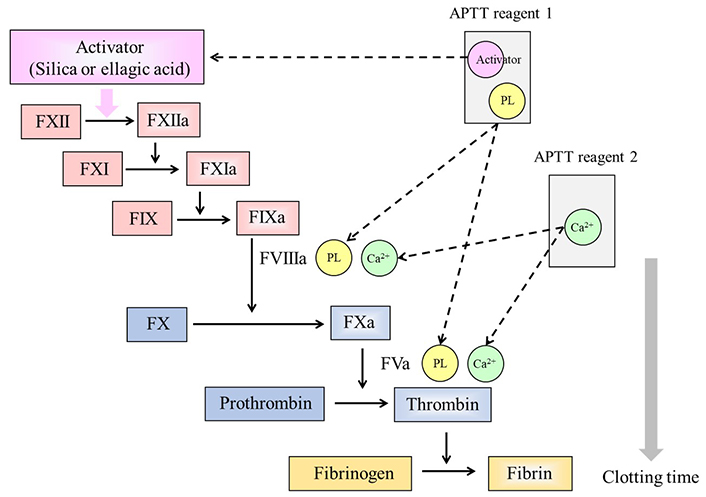
Principle of the APTT. Two reagents are required for APTT measurements. Reagent 1 contains an activator which activates the contact phase factors, and PLs. Reagent 2 is calcium chloride (Ca2+) required for the PL-dependent coagulation reactions in the cascade. LA inhibits the PL-dependent reactions, and the clotting time is prolonged in reagents with a low PL concentration
APTT is used not only for LA screening, but also for the screening of coagulation factor deficiencies, coagulation inhibitors, and heparin monitoring [25]. There are many commercially available APTT reagents, and the characteristics of coagulation factor and heparin sensitivity are variable among the commercial reagents [25, 29–35]. It has also been reported that LA sensitivity is largely dependent on the PL concentrations and compositions of the relative amounts of individual PLs, such as phosphatidylcholine, phosphatidylserine, and phosphatidylethanolamine [15, 36–38]. For the effect of activator, the LA sensitivity was compared among the commercially available APTT reagents, and silica activator which is currently used as the reagent activator has the highest sensitivity to LA, and ellagic acid has a lower sensitivity [38]. Although some studies reported an acceptable sensitivity and usefulness for LA detection using ellagic acid-based reagents [39], the former ISTH guidelines do not recommend the use of ellagic acid activators in APTT reagents used for LA detection [9]. In a subsequent study, ellagic acid-based APTT reagent with optimized PL composition and concentrations was compared with silica-based APTT reagent with the same PL level and LA sensitivity of ellagic-based APTT reagent was equivalent to that of silica-based APTT reagent [40]. Then, it was considered that the sensitivity to LA was largely dependent on the PL composition and concentrations and most commercially available ellagic acid activator reagents have relatively LA insensitive PL components. Consequently, the CLSI guidelines do not restrict APTT reagent choice based on the activator alone [11]. In the latest ISTH guidelines, APTT reagent with a suitable PL composition, a low concentration, and preferably silica as activator is recommended, but it is also reported that ellagic acid as APTT activator may show acceptable sensitivity, at least in some APTT reagents, and laboratories should evaluate the sensitivity of their APTT reagents using well-characterized LA-positive samples [8].
There are various types of APTT confirmatory tests, and two types of confirmatory tests like the platelet neutralization procedure (PNP), and LA insensitive APTT reagents, which are described in this review. In PNP testing, PLs derived from platelets are used because anionic PLs are efficient in adsorbing LA antibodies with high specificity [41]. These PLs are prepared by washing platelets to remove plasma, and they are then exposed to repeated freezing-thawing cycles to disrupt these platelets and to release membrane-derived PLs into suspension. This procedure is conducted to test plasma samples with APTT prolonged clotting times in the screening test. In detail, the patient’s plasma sample is added to two tubes. In the first tube, buffer and APTT screening reagent are mixed with the plasma sample and clotting time is measured. In the second tube, the platelet-PL reagent is used instead of buffer, and the clotting time is also measured (Figure 4). When LA is present in the tested plasma sample, the clotting time of the second tube is significantly shortened because the anionic PLs adsorb LA antibodies effectively. In LA insensitive APTT reagent method, the insensitive confirmatory reagent used contains a high PL concentration. In detail, the clotting time of LA insensitive reagent is detected, and the time is compared with that of LA sensitive APTT reagent when a sample shows a prolonged clotting time (Figure 5). Pairing of LA sensitive APTT and LA insensitive APTT has been shown to be effective for LA detection because LA is highly adsorbed by the PLs of the LA insensitive reagent. If the clotting time of LA insensitive reagent is significantly shortened as compared to that of LA sensitive reagent, the sample is considered to be LA-positive [42]. APTT insensitive reagent with the same activator and a high PL concentration should be used as the pair of LA sensitive APTT reagents because it is important to show the difference between clotting times obtained with the two reagents is due to the differences from the PL concentration. Although a different activator is used in the APTT reagent pair for the screening and the confirmatory tests, laboratories should evaluate their characteristics and suitability for LA detection [37]. One of the recommended paired reagent systems uses silica as activator, for both LA sensitive and insensitive reagents. This system is well-characterized and considered one of the most effective tests for LA detection [43–45].
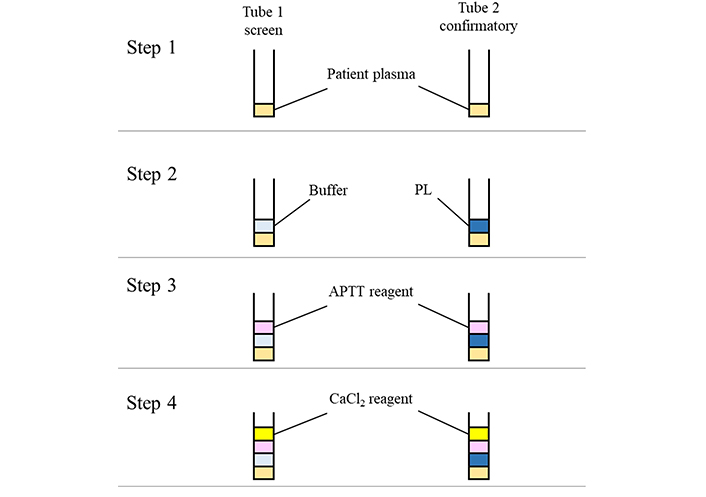
Method principle of the PNP. The PNP is conducted in 4 steps. In step 1, patient’s plasma sample is dispensed into each tube. Buffer or PL is mixed with the sample in each tube in step 2. APTT reagent is added to each tube in step 3. Finally, CaCl2 reagent is added to each tube, and the clotting times are detected. The PL dependence of LA is confirmed by the clotting time difference between buffer and PL solutions
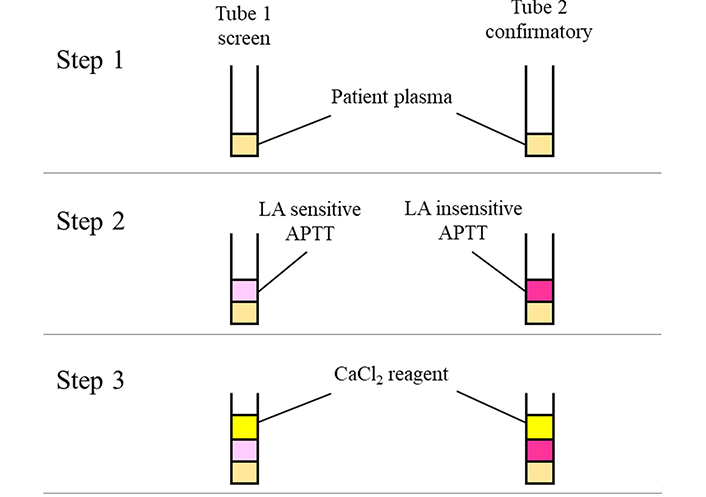
LA detection method by LA sensitive and insensitive APTT reagents. Two types of APTT reagents are used for this assay. In step 1, patient’s plasma sample is dispensed into each tube. LA sensitive and LA insensitive APTT reagents are added to tube 1 and tube 2, respectively, in step 2. Finally, CaCl2 reagent is added to each tube, and the clotting times are detected. The PL dependence of LA is confirmed by the clotting time difference between APTT reagents
Standardization with reference materials
Although difficult to establish, the standardization activity has been conducted for a long time, and LA international reference preparation or sets have been proposed by pooling APS patient plasmas [46]. One of the standardization materials concerns the Harris standards, which were prepared by pooling plasmas from APS patients, and the values were assigned for anti-cardiolipin (aCL) IgG or IgM and expressed as GPL or MPL [46–49]. In parallel, monoclonal antibodies, targeted to β2GPI and presenting the characteristics of aPL, were developed by Ichikawa et al. [50] and these monoclonal antibodies are called as Koike standards [51]. One of the famous IgG-type monoclonal antibodies is HCAL, which can be used for solid phase immunoassay of aCL and anti-β2GPI. For IgM-ype monoclonal antibody, EY2C9 was used as the reference monoclonal antibody. Although these monoclonal antibodies have been used for the standardization of solid-phase immunoassays [6, 7], there is no golden standard materials for LA detection. National Institute for Biological Standards and Control (NIBSC) is proposing an LA reference preparation set, with two kinds of LA panels. The first one is a World Health Organization (WHO) reference panel, which is the 1st international reference panel, and it includes an LA-negative, a moderate LA-positive, and a strong LA-positive freeze-dried sample [52]. These plasma samples were freshly prepared at NIBSC using mixtures of frozen platelet-poor normal plasmas and platelet-poor LA-positive plasmas. LA-negative and LA-positive pools were made using samples from 10 donors and 17 donors, respectively. The LA-positive pool was diluted 1:1 in LA-negative pool to generate a strong LA-positive sample and 1:3:3 to generate a moderate LA-positive sample. The second one is 1st British reference plasma panel for LA, including an LA-negative, a weak LA-positive, and a moderate LA-positive freeze-dried sample. The LA-negative sample was prepared from 19 donors. The weak LA-positive pool was from two patients’ plasmas diluted 1:3 with an LA-negative sample. The moderate LA-positive sample was prepared with the LA-positive pool diluted 1:2 with the LA-negative sample [53]. This LA panel is useful for at least reagent manufacturers, and its use can highly contribute to better reagent standardization.
Interference of anticoagulant drugs in LA testing
Interference of anticoagulant drugs
LA assays are coagulation assays based on clotting times. Therefore, anticoagulants can interfere with the assays and prolong the test clotting times, and this may increase the rate of false-positive. Then, LA testing in anticoagulated patients should be undertaken with great care. To avoid this interference, clinicians often perform clinical laboratory tests to understand the patient’s background before any anticoagulant therapy. In particular, the coagulation and fibrinolysis states during the acute disease phase in disorders associated with thrombosis, bleeding, and inflammation, change with time. It is important to perform these tests as soon as possible and before any administration of anticoagulant therapy. In the guidelines, it is suggested that blood for LA detection should be collected before starting the anticoagulant therapy, whenever possible [12]. It is also recommended to perform PT, APTT, and TT to understand the patient’s background information about unexpected coagulopathies, or undocumented anticoagulation for patients with unknown clinical history. Anticoagulant therapy with heparin or direct thrombin inhibitors (DTIs) prolongs TT: when prolonged it is necessary to remove or neutralize the drugs before investigating for LA [12]. For direct FXa inhibitors, anti-FXa assays can be used for their detection and measurement in plasma.
Heparins
Heparin, which is one of the oldest biological anticoagulant drugs, is a mixture of branched glycosaminoglycans, and it has a large number of negatively charged sulfate groups. Heparin binds to antithrombin (AT) through the irregular pentasaccharide sites, and it accelerates the interaction between AT and thrombin or FXa [54]. Mainly, two types of heparins are used clinically. The first one is unfractionated heparin (UFH) which has an average molecular weight of about 30,000–35,000 Da. The other one is low molecular weight heparin (LMWH) with an average molecular weight of about 4,000–5,000 Da. The ability to inactivate thrombin and other activated coagulation factors is correlated with the chain length of glycosaminoglycans [55–58].
UFH has the potential to interfere with LA assays like APTT. On the other hand, most commercial dRVVT reagents contain heparin neutralizers capable of quenching heparin levels up to 0.8–1.0 U/mL [9, 12, 59]. The same amounts of neutralizers are included in both screening and confirmatory reagents in most of the dRVVT reagents. It means that the prolonged clotting time in the screening reagent is due to LA when the clotting time of the confirmatory reagent is corrected. Because the effect of UFH is neutralized up to the quenching heparin concentration and the prolongation would be at the same levels for both screening and confirmatory reagents, in case the UFH levels are above the quenching levels [18]. LA tests are less affected by LMWH than by UFH. Nonetheless, the interference possibility should be considered when LA assay results are interpreted in patients treated with LMWH [60]. Characteristics are different from reagent to reagent. Then, it is recommended that laboratories confirm the characteristics of reagents used for testing and the interferences of anticoagulants [12]. In addition, it is also important to confirm whether the heparin concentration in samples from patients treated with UFH or LMWH is within the range of the reagent neutralization by anti-FXa assay [58].
Vitamin K antagonists
Warfarin is used as an anticoagulant drug due to its structural resemblance with vitamin K. Therefore, this group of drugs lowering the coagulation potential by reducing vitamin K concentration is called vitamin K antagonists. Vitamin K is an essential cofactor for g-carboxylation of glutamate residues on coagulation factors II, VII, IX, and X. These coagulation factors are synthesized in an inactive form and are carboxylated in the liver. The g-carboxylation formation induces a calcium-dependent conformational change to promote the binding between the coagulation factors and PL surface through calcium ions. Warfarin can bind to vitamin epoxide reductase which is the enzyme for g-carboxylation formation, in which warfarin reduces the presence of g-carboxylation form in coagulation factors II, VII, IX, and X [61–63].
Warfarin induces a multiple, acquired factor deficiency, in which the clotting times of dRVVT and APTT are prolonged and dRVVT is more likely to be affected than APTT because several non-vitamin K-dependent factors are associated with the coagulation reaction in APTT. It is reported that false-positive results in dRVVT are obtained in samples from patients taking warfarin [64, 65]. Then, the utility of dRVVT testing has been discussed on neat plasma from patients taking warfarin, LA can be confidently detected in the mixing tests for dRVVT and APTT if the antibody is sufficient to overcome the dilution effect by the mixing test [66–71]. However, the latest ISTH guidelines described that the mixing test was not a reliable solution in samples from patients under warfarin therapy [12]. One solution is to use Taipan snake venom time (TSVT) and the ecarin clotting time (ECT) tests because these reagents are insensitive to the effect of warfarin, the elevated screening test in TSVT being corrected by ECT in LA-positive samples [72, 73]. It has also been reported that some dRVVT reagents are insensitive to the effects of warfarin, and it is also useful to employ these reagents [74].
DOACs
Direct oral anticoagulants (DOACs) are a new class of oral anticoagulant drugs, especially, dabigatran (one of DTIs), rivaroxaban, apixaban, and edoxaban (all 3, direct FXa inhibitors). These anticoagulant drugs have been widely prescribed for the prevention and treatment to several kinds of thromboembolic diseases. Then, a sample addressed for LA testing might contain these drugs when the patients treated for thrombosis disorders, and laboratories should understand the interferences of DOACs in LA testing. Whilst warfarin inhibits the synthesis of coagulation factors and reduces the presence of g-carboxylation form, DOACs directly inhibit coagulation factors, thrombin, or FXa. Dabigatran, which is the active form of the prodrug dabigatran etexilate, binds to the thrombin active site and inhibits free and fibrin-bond thrombin [75]. Rivaroxaban is a competitive inhibitor of FXa and it inhibits both free FXa and prothrombinase complex [76]. Apixaban is a direct and reversible inhibitor of FXa, and it inhibits free FXa, prothrombinase complex, and FXa bound to platelets [77]. Edoxaban is also a highly selective, direct, and reversible inhibitor of FXa and it inhibits free FXa as well as that within the prothrombinase complex [78].
Dabigatran has the potential to interfere with all LA assays because it inhibits thrombin in the common pathway [60, 79]. The presence of direct FXa inhibitors prolongs the clotting times for both screening and confirmatory LA tests. For dRVVT reagents, the clotting time difference between screening and confirmatory tests is important and false-positive results may be obtained, especially for rivaroxaban. Conversely, with apixaban and edoxaban, this difference is low and the normalized ratios are not that changed or even reduced. Then, the frequency of false-positive results may be lower than with rivaroxaban [21, 80, 81]. It was also reported that some dRVVT reagents were not influenced by DOACs, and using them could be a solution to avoid DOACs interference [74]. Assays based on APTT are known to be less affected by direct FXa inhibitors than dRVVT or PT-based assays, but caution is required when interpreting results as there is some reagent-to-reagent variability [79–81]. The interference in assays including mixing tests is also different among direct FXa inhibitors [21]. The latest ISTH guidelines keep the attention on DOAC effect in LA testing and they do not recommend performing LA tests, including mixing tests, in patients under DOAC therapy [12]. Universal adsorbents against DOACs are now available, which eliminate the effects of all DOACs by removing these drugs, and they provide a convenient solution [82, 83]. These substances are hydrophobic-binding agents, which are apparently able to neutralize any type of DOACs including dabigatran, rivaroxaban, apixaban, and edoxaban [84]. Guidelines report that these adsorbents could be promising tools to help detecting LA in patients anticoagulated with DOACs [12]. The pairing of TSVT and ECT testing can be used to detect LA in patients taking direct FXa inhibitors because these venom assays directly activate prothrombin and bypass the anticoagulant effect of direct FXa inhibitors [85, 86].
Conclusions
APS is an autoimmune disorder associated with the occurrence of thrombosis, and LA detection is not simple due to its heterogeneity. In this review, we describe the LA testing chart flow for screening, mixing and confirmatory tests, LA assays, and the interference of anticoagulants. LA testing should be conducted based on the correct procedures of i) screening; ii) mixing; and iii) confirmatory testing, and the formulas expressing results with the normalized ratios are important for the data interpretation. In LA assays, no single test is sensitive for all LA, and two tests, like dRVVT and APTT, should be employed for its detection. The characteristics are different between dRVVT and APTT, and there are some variabilities among reagents. Laboratory technicians should understand the characteristics of reagents used in their laboratories. To understand the interference of anticoagulants, which is of the essence for data interpretation, results should be reported and interpreted along with the possibility of false-negative or false-positive results, whenever the results are doubtful. The understanding of assays and the possible interferences are helpful and useful for data interpretation. Although there are limitations to understanding all mechanisms of LA effects, LA detection practices will continue to refine and evolve. Further studies and developments are expected for better practice and data interpretation.
Abbreviations
| aPLs: |
antiphospholipid antibodies |
| APS: |
antiphospholipid syndrome |
| APTT: |
activated partial thromboplastin time |
| CLSI: |
Clinical and laboratory Standard Institute |
| DOACs: |
direct oral anticoagulants |
| dRVVT: |
dilute Russell’s viper venom time |
| ECT: |
ecarin clotting time |
| FX: |
factor X |
| ICA: |
index of circulating anticoagulant |
| IgG: |
immunoglobulin G |
| ISTH: |
International Society on Thrombosis and Haemostasis |
| LAs: |
lupus anticoagulants |
| LMWH: |
low molecular weight heparin |
| MTC: |
mixing test-specific cut-off |
| NPP: |
normal pooled plasma |
| PLs: |
phospholipids |
| PNP: |
platelet neutralization procedure |
| PT: |
prothrombin time |
| TSVT: |
Taipan snake venom time |
| TT: |
thrombin time |
| UFH: |
unfractionated heparin |
| β2GPI: |
β2-glycoprotein I |
Declarations
Author contributions
OK: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. MP: Investigation, Writing—review & editing. JA: Supervision, Validation, Writing—review & editing. All authors read and approved the submitted version.
Conflicts of interest
OK was an employee of HYPHEN BioMed at the time the work carried out. MP is an employee of HYPHEN BioMed. JA is a consultant for HYPHEN BioMed and Sysmex Corporation.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2023.