
Abstract
The skin, as the first physical barrier for invading pathogens, also serves as an immunologically active organ. Breaching the skin barrier is thus essential for pathogens to enter the body. The skin contains various immune responsive cells that initiate both the innate and adaptive immune response upon invasion. Activated immune cells help to regulate cytokine response and their differentiation to promote or suppress the crucial immune response against invading pathogens. Human herpes simplex viruses (HHSVs) are the oldest pathogen that can escape immune surveillance of the human host by a well-developed escape mechanism within ganglia, as their evolutionary strategy. In primary infection, a non-specific defense of the host initiates the response against the invading virion. The initial direct antiviral action of the host is regulated by activated macrophages, via the release of cytokines like tumor necrosis factor (TNF), and type-1 interferon (IFN-1). The host-derived cytokines including IFN-12, TNF, and IFN-1 in turn induce natural killer (NK) cells to release IFN-γ. Their positive feedback with synergistic interactions collectively releases nitric oxide (NO) and reactive oxygen species (ROS) against the invading virus. Simultaneously, the combination of cytokines, macrophages, and other cells activates the immune system to eliminate the pathogen. However, the virus has also evolved various mechanisms to counter the host defense strategies. This review will highlight virus-mediated skin infections, especially by HSV, and portray a detailed role of virus-induced cytokines in host-immunity to challenge the invading virion during mucocutaneous HSV infection. Further, this review will discuss the viral-interference on host defense to provide a simplistic overview of the complications of cutaneous HSV infection.
Keywords
Skin diseases, herpes simplex virus, cytokines, immune activation and evasionIntroduction
Skin, being the largest living organ of the human, covers and protects the body with diverse functions, including (i) sensations of pain, temperature, and touch; (ii) preventing dehydration by holding fluid; (iii) preventing entry of infectious pathogens including viruses, bacteria, fungi, and parasites; (iv) maintenance of body temperature; and (v) synthesis of vitamin-D using solar energy. Skin as the outermost living cover is exposed to various environmental stressors including diseases of a myriad type that result in clogging, irritation, inflammation, rashes, infection, and other changes. Human body is subjected to hundreds of skin conditions that represent 1.79% global burden [1], mostly minor; with only about one-in-four people developing a fatal problem. The common types of skin disorders, including infections, their causes and symptoms are summarized in Table S1 [2].
Skin infections of viral origin
The skin being the outermost living cover, may harbor and get infected by diverse pathogens like viruses, bacteria, fungi, and protozoa. Viruses being ultramicroscopic, metabolically inert, acellular nucleoprotein particles can frequently infect skin cells, as presented in Table 1 and Table S1. There are several viruses that may affect the skin and mucocutaneous layers of human body: while the most common skin viruses include herpes, papilloma, and pox, as presented in Table 1. The primary viral disease of the skin is caused by the members of the Herpesviridae family including herpes caused by herpes simplex virus (HSV), virus that causes molluscum contagiosum (MC), herpes zoster caused by varicella zoster virus (VZV), and the vaccinia viruses (VACVs) that infect skin epidermal keratinocytes. However, the most common viral skin diseases are chickenpox, reactivated chicken pox (shingles) caused by VZV; MC caused by the pox virus or MC virus; and roseola caused by herpes virus 6 and 7 [2]. It is also interesting to note that HSV may alter the prognosis and susceptibility of other apparently unrelated skin diseases [3].
Major viral infection of the skin and mucocutaneous layer of the skin
| S No | Infection | Causative virus |
|---|---|---|
| 01 | Chickenpox | Varicella virus |
| 02 | Measles | Morbilli virus |
| 03 | German measles | Rubella virus |
| 04 | Erythema infectiosum | Parvo virus |
| 05 | Purpuric gloves-sock syndrome | Parvo virus |
| 06 | Infectious mononucleosis | Epstein-Barr virus (EBV) |
| 07 | EBV-lymphoproliferative disorder | EBV |
| 08 | Oral hairy leukoplakia | EBV |
| 09 | Acrodermatitis Gianotti-Crosti syndrome | EBV, hepatitis B virus |
| 10 | Erythema subitum or roseola | Human herpes virus 6 and/or human herpes virus 7 |
| 11 | Mucocutaneous pityriasis rosea | Human herpes virus 6, 7 |
| 12 | Kaposi sarcoma | Human herpes virus 8 |
| 13 | Vesicular stomatitis, vesicular eruptions | Enterovirus |
| 14 | Hand foot and mouth disease | Coxsackie virus |
| 15 | Smallpox, cowpox, monkeypox | Pox viruses |
| 16 | Para pox infection include Orf, Milker’s nodules | Cowpox virus |
| 17 | Hemorrhagic fevers | Toga, bunya, and arena viruses |
| 18 | Chikungunya | Chikungunya virus |
| 19 | Dengue-hemorrhagic fever | Dengue viruses |
| 20 | Zika fever | Zika virus |
| 21 | Epidermodysplasia verruciformis | Human papilloma virus |
| 22 | Penile, vulval, oral, oral intraepithelial, neoplasia, epidermodysplasia verruciformis | Human papilloma virus |
| 23 | Wart (verrucas, anogenital), squamous cell papilloma | Papilloma viruses |
| 24 | Skin complaints with HIV/AIDS | Human immunodeficiency virus |
| 25 | Rickettsial diseases, yellow fever | Flavi virus |
| 26 | Localized skin and mucosal infections: cold-sore, orofacial herpes, genital herpes, neonatal herpes, encephalitis, corneal keratitis leading to blindness, eczema, herpeticum, herpes zoster | Herpes simplex virus type-1 (HSV-1), HSV-2, vesicular stomatitis virus, and molluscum contagiosum (MC) |
| 27 | Erythema multiforme, nodosum, Kawasaki | Viral hepatitis |
| 28 | Laterothoracic exanthem | Many viruses |
Herpes simplex viruses
HSV type-1 (HSV-1) and HSV-2 are neurotropic double stranded (ds) DNA viruses, responsible for the most common skin infections. However, only a low proportion of infected individuals are clinically active. Activation of HSV-2 may lead to painful oral or genital lesions, encephalitis, invasive neonatal infections, primary hepatitis, and cervical carcinoma [4]. WHO estimated that nearly 491 million people, aged 15–49 years, have HSV-2 [4]. In 2016, a global estimate in 2016 revealed that around 67% people, under the age of 50, are infected with HSV-1. The virus is mainly spread via oral contact, and around 13% people between ages 13–49 have HSV-2 infection, manifested as genital herpes [5, 6]. A US based study by National Health and Nutrition Examination Survey showed that the prevalence of HSV-1 is the highest in the American-Mexican population and the lowest in non-Hispanic white people [6]. However, HSV infection has declined over the years; and prevalence has decreased with age but in general, it is higher in females [7]. Epidemiological data from Southeast Asia also showed decreased HSV seroprevalence with age, the highest (80%) in age group > 40 and the lowest (55.5%) in age group < 20 [8]. A recent epidemiological report from Europe suggests approximately 12% of the adult population is chronically infected with HSV-2 [9]. However, more epidemiological data is needed to get a complete global picture of HSV infection. Primary infection of HSV is initiated by the direct entry of the virion in the hosts’ mucosal tissues. After establishing primary infection, the progeny virion disseminates from the infection site to the neuronal tissue, to establish a latent state in the sensory ganglia. The sporadic reactivation of latency helps the virion to replicate and is transported back from the infected ganglia to the peripheral mucosal tissue [4, 5] and multiply to develop blisters or painful lesions.
Diverse viral proteins are involved in HSV replication. The 152 kb long HSV genome encodes over 90 unique transcriptional units; which are composed of unique long (UL) and unique short (US) regions, flanked by repeated regions. Gene expression is an ordered event, initiated with the synthesis of three gene products: immediate early (IE), early (E), and late (L). The HSV virion surface is studded with about 14 surface proteins, mostly glycoproteins. The first contact is usually between the glycoprotein C (gC) and glycosaminoglycans. The rest of the glycoproteins play important roles in viral entry which is still not completely understood. The entry receptors are known to be nectin-1 and herpesvirus entry mediator (HVEM) [10]. Skin is facilitated by wounds that expose the lower epidermal layers [11]. The purine analogue acyclovir is the drug of choice against HSV [12], but there are many medicinal plant extracts or phytocompounds reported to be effective against HSV as well [13].
Cytokines: the immune mediators
Cytokines are a group of low-molecular-weight telemorphic polypeptides (proteins) act as mediators that provide inflammatory responses with critical immunoregulatory proteins. During diseases, chronic or infectious, innate and adaptive immune responses play a central role but their balance is regulated by cytokines [11]. Cytokines regulate the repair and development of tissues, inflammation, hematopoiesis, etc., through diverse signals via cellular receptor binding. To induce an immune response, localized and/or systemic, the cytokines act on target cells in three different ways: autocrine, endocrine, and/or paracrine. Cytokines can also act on its target cells and influence the functionality of other cytokines by additive, antagonistic, or synergistic fashion. During cellular damage or infection, cytokines are produced by diverse cells, including immune cells. These proteins act through a number of conserved signaling pathways which help to program the required transcriptional factors to regulate cellular apoptosis, growth, development, differentiation, reprograming, and survival in the local tissue for improved immune responses. Thus, cytokines are mediators of immune response and communication, and are essential to defend the host against pathogens [14]. Although cells produce cytokine to protect themselves, sometimes it acts as a sword against itself. It works through positive feedback loops and this often leads to cytokine storm. This storm leads to severe damage to host tissues and can even lead to death [15]. Viral infection, like all infections, involves the conflict between the host and the virus. Thus, the outcome of viral infection depends on host-virus interactions, for which viruses utilize diverse evasive strategies to overcome the host defense while the host mounts an elaborate cytokine response. Viral evasion is usually balanced with disease pathology and possibilities of new transmission. Generally, mammals use a redundant and ubiquitous antiviral defense, and various aspects of host defense are crucial in different viral infections. On the other hand, the redundancy indicates that if one fails the other systems can take over the fight; and thus, the outcome depends on the timing and regulation of antiviral effectors with the infecting virus.
Cytokine families
The cellular release of cytokines and its pattern not only depend on the antigen type and its nature but also on the cell types being stimulated. Presently cytokines are classified into one family—interleukin-17 (IL-17), and five superfamilies: (i) tumor necrosis factor (TNF) superfamily, (ii) IL-6 superfamily, (iii) IL-1 superfamily, with (iv) type-I and (v) type-II superfamily [16, 17]. Broadly they are classified into pro-inflammatory and anti-inflammatory mediators. Pro-inflammatory cytokines include TNF-α, TNF-β, interferon (IFN)-α, IFN-β, IFN-γ, IL-1, IL-6, IL-8, IL-11, IL-17, IL-18, and granulocyte colony stimulating factor (G-CSF). While anti-inflammatory cytokines include transforming growth factor (TGF)-β, IL-10, IL-12, IL-22, IL-37 (1L-1F7), and IL-38 (IL-1F10). Interestingly a few cytokines including IL-2, IL-3, IL-4, IL-5, IL-7, IL-9, granulocyte macrophage CSF (GM-CSF), and M-CSF are involved in the adaptive response. Details of the selected cytokines and their functions are available at: https://www.thermofisher.com/in/en/home/life-science/cell-analysis/cell-analysis-learning-center/immunology-at-work/proinflammatory-cytokines-overview.html.
Cytokine profile in viral infections
In viral infections, cytokines are the first line of non-specific defense, with virus-specific response. Recognition of viral particles by pattern recognition receptors (PRRs) activates the signaling cascade for their synthesis and release. The structural changes of PRRs initiate the signal transduction cascade that ultimately activates transcription factors in host cell cytoplasm to translocate into the nuclei which promote the synthesis and release of diverse cytokines. However, the cytokine type not only depends on cell types but also on the infecting virus and the signaling pathway involved [18, 19].
Pattern recognition receptors versus virus
Almost all cells can be infected by pathogens (including viruses) including but not exclusive of epithelial, sub-epithelial, endothelial, neurons, fibroblasts, and even innate and adaptive immune cells. Both non-hematopoietic and immune cells possess PRRs; and some can recognize viral proteins, while others scan and identify structural peculiarities in the viral genome. Out of about 10 toll-like receptors (TLRs) present in human plasma- and endosome-membranes, only two (TLR-4 and TLR-2) can detect surface glycoproteins of viruses before viral penetration to the host cell. On the other hand, three TLRs (3, 8, and 9) can sense viral nucleic acids in endosomes, after viral entry. TLR-3 is known to detect dsRNA, TLR-8 can sense single stranded (ss) RNA of viral genome; while TLR-9 senses nonmethylated CpG-viral DNA (vDNA). The receptors like retinoic acid-inducible gene (RIG)-1 and melanoma differentiation-associated gene 5 (MDA5) are the RNA-helicases and other types of receptors that sense viral-RNA and detect viral-genomic or intermediate dsRNA [20, 21] formed during replication of ss or dsRNA viruses [22]. Evidence reveals that some dsRNA intermediates travel to endosomes, where TLRs detect and trigger the signal [23]. Recognition by the PRRs triggers a signal cascade that culminates in the secretion of IFNs from the infected cell. Secreted IFNs in turn act to stimulate other immune responsive cells to secrete more chemokines and cytokines as well, to trigger the adaptive immune response.
Figure 1 depicts how molecular patterns (DNAs and RNAs) produced by viral infection trigger PRRs (green) and transfer innate immune signaling via different adaptor proteins (blue). This results in the development of antiviral responses, which include necroptosis, cytokine production, inflammasome activation, and translation inhibition.
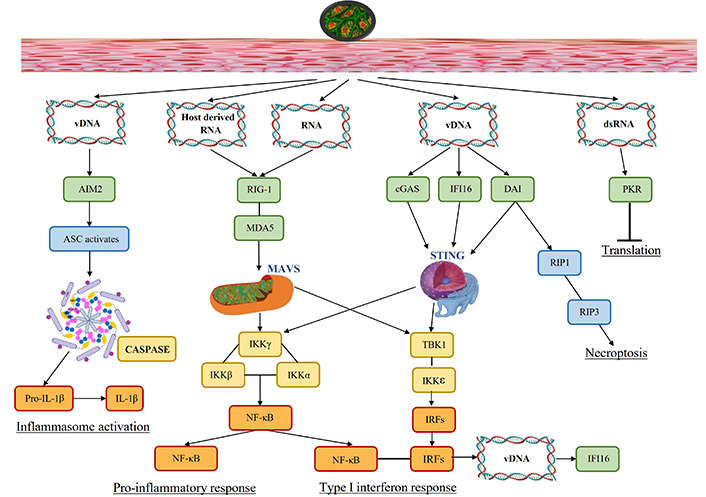
Signaling pathways activated due to viral presence. Viral infection results in molecular patterns (DNAs and RNAs) that activate pattern recognition receptors (green) and transmit innate immune signaling through various adaptor proteins (blue). This leads to the induction of antiviral responses, including the production of cytokines, activation of inflammasomes, inhibition of translation, and necroptosis. vDNA: viral DNA; dsRNA: double stranded RNA; AIM2: absent in melanoma 2; ASC: apoptosis-associated speck-like protein; RIG-1: retinoic acid-inducible gene-1; MDA5: melanoma differentiation-associated protein 5; cGAS: cyclic GMP-AMP synthase; DAI: DNA-dependent activator of interferon-regulatory factors; PKR: dsRNA-dependent protein kinase; IL-1β: interleukin-1β; IKKγ: inhibitor of kappa B kinase γ; NF-κB: nuclear factor kappa B; TBK1: TANK-binding kinase 1; IRFs: interferon-regulatory factors; IFI16: interferon-gamma-inducible protein-16; MAVS: mitochondrial antiviral signaling proteins; STING: stimulator of interferon genes; RIP1: receptor-interacting serine/threonine-protein
To detect and clear the invading pathogen, the innate immune system acts as the first line of defense. Human hosts have a wide variety of receptors including the PRRs to sense invading pathogens during infection by recognizing the pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). These receptors trigger intracellular signaling that converges on common transcription factors nuclear factor kappa B (NF-κB) and IFN-regulatory factors (IRFs), to release the infection regulating proinflammatory cytokines, chemokines, and IFN-1. The PRRs ensure infection surveillance; while transmembrane PRRs detect extracellular PAMPs and DAMPs including TLRs and C-type lectin receptors (CLRs). PRRs distributed in cytosol detect intracellular PAMPs and DAMPs via RIG-1 like receptors, such as nucleotide-binding leucine-rich repeat receptors (NLRs), and absent in melanoma 2 (AIM2)-like [24]. Figure 1 shows that the entry of HSV via skin induces diverse pathways, including molecular pattern recognition (viral genome signatures, host-derived RNA, and viral RNA) to trigger the PRRs. These activated PRRs (TLRs) recruit various adaptor proteins such as myeloid differentiation primary response protein (MyD) 88, which in turn activate various transcription factors namely NF-κB, activator protein (AP)-1, and IRF3/7 that culminates into the induction of various pro-inflammatory cytokines. This eventually leads to the activation of inflammasome resulting in death of the infected cell [25]. The vDNA activates AIM2 mediated inflammasome pathway activating apoptosis-associated speck-like protein (ASC) to release caspases and proinflammatory IL-1β. AIM2, senses the vDNA released by the incoming virion, or host-derived DNA from apoptotic cells [24]. In DNA virus infection, AIM2 inflammasome is vital for infected macrophages and dendritic cells (DCs) leading to caspase-1-dependent IL-1 responses. The viral and host-RNA jointly induce RIG-1 followed by MDA5 and mitochondrial antiviral signaling proteins (MAVS) that generate inhibitor of kappa B kinase (IKK) and NF-κB induced pro-inflammatory response. Moreover, vDNA releases cytosolic DNA sensors cyclic GMP-AMP synthase (cGAS), IFN-gamma-inducible protein-16 (IFI16), and DNA-dependent activator of IRFs (DAI) that activate stimulator of IFN genes (STING) which generate IFN-1 response as well as proinflammatory response. DAI contributes to necroptosis via receptor-interacting serine/threonine-protein (RIP) kinases 1 and 3. The dsRNA initiates translation via dsRNA-dependent protein kinase (PKR); while pro-inflammatory and IFN-1 response induce vDNA to produce antiviral IFI16.
Virus-induced cytokine production
Communication between immune cells takes place via cytokines, which not only regulate proliferation and differentiation, but are also implicated in other functions such as embryogenesis, neurogenesis, hematopoiesis, oncogenesis, and inflammation. Cytokines do not act as stored molecules but are synthesized rapidly and secreted usually upon stimulation. Moreover, a cytokine may influence the action of others by producing additive, antagonistic, or synergistic results. Cytokines have distinct pleiotropic or redundant functions, as they can antagonize or synergize to eliminate the virion through antiviral immune response regulation. The antiviral response is a multi-step process that includes (i) virion detection, (ii) signaling to neighboring cells, (iii) innate immune cell activation-differentiation, (iv) adhesion molecule production on endothelial cells, (v) release of molecules that are chemotactic to attract cells of the infection site, and (vi) increased phagocytosis along with cell activation. During viral infection, cytokine network is initiated by releasing some cytokines from the infected cell. The epithelial cells are known to produce IL-8, IL-6, IL-1, IFN, GM-CSF [26], TNF-α [27], IL-2 [28], IL-12 [29], IL-18 [30], and IL-23 [31, 32] with diverse roles. IFN is known to induce antiviral state, while IL-8 is a potent inflammatory molecule that targets phagocytes at the infection site. On the other hand, proinflammatory IL-1 is an apoptosis promoter, and chemotactic to neutrophils; but hematopoietic growth factor GM-CSF recruits diverse immune cells to help host defense [33].
Viral infections and IFN
IFN is produced during viral infection as a pleiotropic cytokine, classified into three types: type-I (α/β), II (γ), and III (λ). The production of IFN-α takes place in leucocytes infected by a virus, while IFN-β is found in virus-infected fibroblasts; but IFN-γ is produced by stimulation of antigen-sensitized/non-sensitized lymphocytes with mitogens. Type-I is critical in controlling early viral infections by interfering with viral progeny production through the activation of antiviral molecules. While type-III provides a major role as an antiviral defense at the viral contact site by acting as a first line of defense [34]. Once secreted, the IFN can act in an autocrine or paracrine manner. Interaction with IFN-receptor induces the antiviral state in the uninfected or virus-infected cells to inhibit one to several steps of the viral replication cycle. Secondly, IFN can promote the release of IL-6, IL-12, TNF-α, and IFN-γ in innate macrophages and natural killer (NK) cells [35]. Further, IFN may enhance DC differentiation [36] and promote the presentation of antigens [35] to stimulate B-cell and T cells [37], which is redundant with the function of IL-12 and -18 [38, 39]. The IL-12 and IFN-1 synergistically activate NK cells; while IL-3, -4, -6, -10, -13, and TGF-β suppress the IFN actions, and thus, these are functionally anti-inflammatory and immunomodulatory cytokines [40]. Another pleotropic cytokine is TNF-α, produced from activated nonhematopoietic infected cells, macrophages, DC, NK, B- and T-lymphocytes of the innate and adaptive immune systems [41]. Activation of TNF-α produces adhesion molecules in endothelial cells that promote the release of monocytes, neutrophils, and other immune cells to translocate to the infection site. Further, it participates in apoptosis by caspase activation. Moreover, TNF-α combined with IFN-γ induce macrophage to produce oxygen/nitrogen free radicals and superoxide anions [42]. Cytokines produced by macrophages include IL-1, -6, -12, -23, and TNF-α [40]. The first identified pleotropic IL-1 is known to act synergistically with IL-6 on the CNS to induce fever via activation of the hypothalamus-pituitary-adrenal axis [43]. Moreover, IL-1 induces histamine production by activating mast cells; and increases membrane permeability as a vasodilator [44]. Additionally, as a chemotactic factor, IL-1 induces neutrophils to travel to the infection site. This chemotactic ability is redundant with IL-8 action, [also known as chemokine C-X-C motif chemokine ligand 8 (CXCL8)] [33] produced by virus-infected cells. The cytokines that antagonize the functions of IL-1 are anti-inflammatory such as IL-4, -10, and -13 [45]. Another cytokine of IL-1 family, pleiotropic in nature, is IL-18, known as “IFN-γ-inducing factor”. IL-18 with IFN-1 is recognized by DCs to trigger signaling pathway via TNF receptor (TNFR)-associated factor 6 (TRAF6), and induce cluster of differentiation (CD) 11b+ expression on the cell surface [39]. The expressions of IL-6, -12, IFN-γ, TNF-α, and IFN-α, by activated cells, participate with other hematopoietic cells [46]. Additionally, IL-18 activates NK cells in combination with IL-12 [38], to stimulate CD25 and CD69 expression, proliferation, and cytotoxic ability. Activated NK cells then induce apoptosis of virus-infected cells and produce IL-6, -10, -12, IFN-γ, and TNF-α; while the function of IL-18 can be blocked by IL-10, -37, and TGF-β [47]. Genetic studies reveal that host-virus interactions induce the cellular antiviral state affecting HSV replication and latency. In fact, the viral modulation of the IFN system, particularly PKR, is responsible for neurovirulence and pathogenesis of HSV infection [31, 34, 38].
Immunology of the skin
Skin, being the largest and outermost organ, is exposed to thousands of pathogens. Thus, the skin protects the body via various defense mechanisms including physical, biochemical, and immunological. Anatomically the skin consists of three different layers namely epidermis, dermis, and subcutaneous fat. Epidermis, the outermost layer, is further subdivided into four layers: stratum corneum, stratum lucidum, stratum granulosum, and stratum basale. Stratus corneum consists of corneocytes, which are differentiated into skin-cell keratinocytes; while stratum lucidum contains dead keratinocytes; stratum granulosum is a thin layer between stratum lucidum and stratum basale; and stratum basale contains several immune cells including basal keratinocyte, Langerhans cell (LC), and T-cells [48, 49]. The dermis, below the epidermal layer, also contains several immune cells including macrophages, lymphocytes, mast cells, eosinophils, neutrophils, B cells, and fibroblast which synthesize extracellular matrix or ECM [50, 51]. The subcutaneous fat layers are rich in adipocyte and fibroblast, contain immune cells, and can produce cytokines [52]. Skin, particularly keratinocytes of skin produce hemopoietic colony stimulating factors and secrete several cytokines, including IL-1, -6, and -8. In early post-infection, cytokines are produced by innate immune cells that help to recruit circulating leucocytes, which in turn, produce more cytokines at the infection site resulting in enhanced tissue damage; along with the activation of DCs that move to the lymph nodes and activate T-cells. During primary HSV infection, a series of cytokines including IL-1β, IL-2, IL-6, IL-10, IL-12, IL-13, TNF-α, IFN-α/β, IFN-γ, and GM-CSF are released in skin [40–44].
Cytokine response in skin during HSV infection
Skin, specifically the skin keratinocytes is one of the major targets of HSV infection. Hence, keratinocytes develop many strategies to combat infection. Keratinocytes are one of the most superficial layers of the epidermis and consist of many layers with the oldest layer at the surface and newer at the bottom [53]. In-vitro infection of keratinocytes by HSV-1 initially induces the production of β-chemokine, followed by other cytokines such as IL-6, IL-10, IL-12, IL-1α, and IL-1β and thereby helps in the recruitment and release of IFN-γ by CD4 T-cell at the site of lesion, as presented in Figure 2 [54]. Skin biopsies taken from HSV-infected patients showed that T cells and monocytes are the predominant cells present in the lesion. Among T cells, CD4 T cells migrate first to the site, compared to CD8 T cells. The lesion site is associated with a high level of IFN-α, produced by keratinocytes, and IFN-γ produced by T cells [55]. It is suggested that HSV-1 first infects the LCs and epi-cDC2s (epidermal DCs type 2) in the skin. Infected LC migrates to the dermis and interacts with different dermal major basic proteins (MBPs) leading to the activation of T cells [56]. All these are directed by cytokines and chemokines, initially secreted by macrophages, LCs, DCs, and keratinocytes followed by infiltrating immune cells. Cytokines perform diverse functions from regulating inflammation to cellular proliferation, migration, and wound healing. Chemokines mainly control different immune cell migration by creating a chemotactic gradient [57–59].
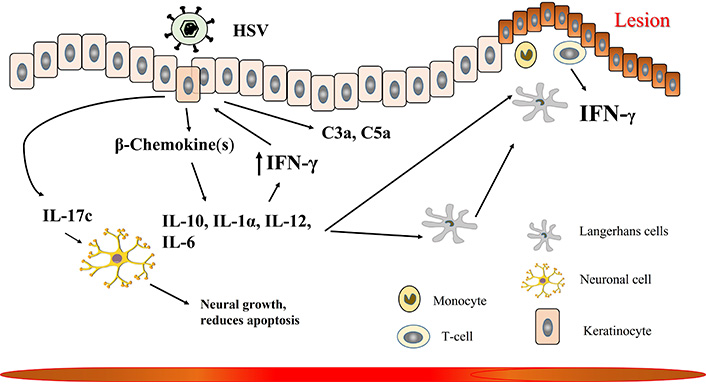
Cytokine response to herpes simplex virus type-1 (HSV-1) at the skin. The details are described in the text. IFN-γ: interferon-gamma; IL-10: interleukin-10; C3a: complement component 3a
HSV infections are associated with the upregulation of different pro-inflammatory cytokines and chemokines [60, 61]. Cytokine and chemokine type and levels change with days after recurrent HSV infection. On the first day, the lesion is associated with a high level of IL-1β, IL-6, macrophage inflammatory protein 1 beta (MIP-1β), and low levels of IL-12 and β-chemokine. However, at day three IL-1β, IL-6, and MIP-1β decreases while IL-10, IL-12, and MIP-1α remain same [54]. Keratinocytes, one of the most common cell types in the skin can recognize HSV through its TLR-2, TLR-3, and TLR-9, and activate the innate immune reaction [62, 63]. In response to HSV infection, keratinocytes produce different cytokines and chemokines to contain the virus. HSV-2 infection leads to the upregulation of antiviral cytokines IFN-α, IFN-β, TNF-α, GM-CSF, lymphocyte-activating factor (LAF), and IL-3. These infected keratinocytes stimulate the proliferation of lymphocytes, suggesting keratinocytes can combat HSV infection via two ways, either directly by producing cytokines or by recruiting immune cells at the site of the lesion [63]. After recognizing HSV, TLR-2 induces the production of pro-inflammatory cytokines TNF, IL-6, and IL-1, as well as antiviral cytokine IFN-1. This higher level of TNF increases the production of serum anaphylatoxins, complement component 3a (C3a) and C5a by keratinocytes which was otherwise secreted at a basal level, resulting in the recruitment of innate immune cells [63–65]. HSV-infected keratinocytes show cytopathic effect mediated by CD4+ and CD8+ T cells and in turn attract T cells and NK cells that further produce IFN-γ to increase TLR-3 production and thereby help in clearing the virus [66, 67]. HSV-infected keratinocyte produces IL-17c which binds to IL-17RE (receptor for IL-17c) on neuronal cells to promote neurite growth and reduce apoptosis. This suggests why, despite frequent reactivation of HSV, neuronal cells stay protected. Apart from cytokines, HSV infection in keratinocytes also induces the production of different chemokines. However, more studies are required to understand different chemokine induction in HSV-infected keratinocytes [54]. Recurrent HSV infection in later days recruits’ macrophages to the site of the lesion. Macrophages can either directly phagocytose virus-infected cells and/or produce TNF, IL-6, IL-12, and IFN-α [55, 68, 69]. HSV-infected keratinocytes secrete IL-1α as an alarm resulting in the recruitment of leukocytes, which helps in clearing off the virus [70]. The NK cells also migrate to the skin in response to HSV infection, to help in the recruitment and activation of CD8+ T-cells by producing IFN-γ [71]. Along with these cells, CD4+ T cells are also recruited to HSV-infected keratinocytes secreting IFN-γ. These CD4+ T-cells further secrete IFN-γ which induces keratinocytes to secrete CXCL9 and CXCL10 and thereby help in recruiting CD8+ T-cells. These CD4+ T cells remain at this location for 6 months after primary infection but can produce IFN-γ in response to future HSV reactivation [72, 73].
IFN-λ, another critical IFN involved in restricting pathogen activity in skin cells restricts CXCL9 mediated recruitment of neutrophils in keratinocytes and thereby reduces the severity of HSV-1 and HSV-2 infection [74]. HSV or any viral infection in general increases IFN production which stimulates IFN stimulated gene (ISG) expression that in turn suppresses viral spread and infection. One of such ISG induced by HSV-1 infection is IFI16, which maintains heterochromatin marks of HSV-1 by inhibiting viral infected cell protein 0 (ICP0) and thereby epigenetically silences it [75]. Induction of IFN on viral infection acts as a double sword, on one hand it restricts viral infection, while on the other, excessive IFN signaling might kill a cell, so a perfect balance is needed to maintain homeostasis. Sensing the HSV-1 DNA by cGAS and subsequent activation of STING induces IFN-1 expression, however, to inhibit permanent activation of IFN-1, myristic acid enhances myristoylation (lipid modification by adding 14-carbon myristate to N-terminal glycine protein) of ARF-1 (ADP-ribosylation factor-1) which promote autophagic degradation of STING [76]. NF-κB is a key transcription factor involved in the regulation of innate immune response. Activation of NF-κB leads to its translocation to the nucleus where it upregulates a number of genes related to the innate immune response including those of IFNs and ILs.
Inhibition of cytokine production: role of HSV proteins
The HSV develops several strategies to bypass and inhibit host immune reactions [77, 78]. One of the major responses of infected cells to combat viral infection is the upregulation of IFN. Similarly, viruses also develop many strategies to evade this. In response, HSV encodes several proteins namely, ICP0, ICP27, ICP34.5, and virion host shutoff (Vhs) which inhibit IFN signaling and help HSV to continue its lifecycle. The HSV mediated activation of immune responses is depicted in Figure 3, while the viral response to immune activation is presented in Figure 4 and Table 2.
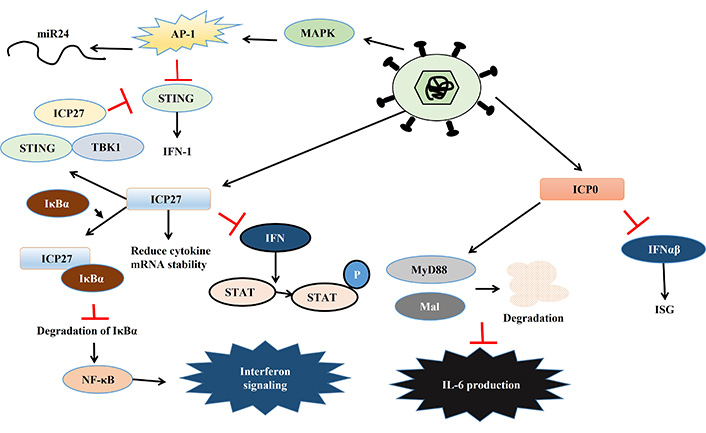
HSV mediated activation of immune responses. Details are explained in the text. Black arrows indicate activation, red bars indicate inhibitory actions. miR24: micro RNA-24; AP-1: activator protein-1; MAPK: mitogen-activated protein kinase; STING: stimulator of interferon genes; ICP27: infected cell protein 27; IFN-1: type-1 interferon; TBK1: TANK-binding kinase 1; IκBα: inhibitor of kappa B alpha; NF-κB: nuclear factor kappa B; STAT: signal transducer and activator of transcription; P: phosphorylation; MyD88: myeloid differentiation primary response protein 88; Mal: MyD88 adapter like; IL-6: interleukin-6; ISG: IFN stimulated gene
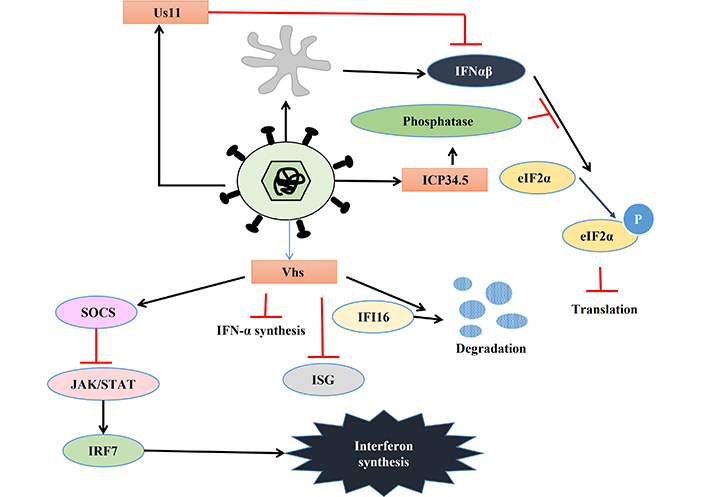
Viral response to immune activation. Details are described in the text. eIF2α: alpha subunit of eukaryotic initiation factor 2; IFN-α: interferon-alpha; ICP34.5: infected cell protein 34.5; Vhs: virion host shutoff; SOCS: suppressor of cytokine signaling; IFI16: IFN-gamma-inducible protein-16; ISG: IFN stimulated gene; STAT: signal transducer and activator of transcription; IRF7: IFN-regulatory factor 7; P: phosphorylation; JAK: Janus kinase
List of viral proteins inferring with chemokine-cytokine response
| Viral protein | Target | Effect | References |
|---|---|---|---|
| ICP34.5 or γ134.5 | Inhibits eIF2α | Continued translation in HSV | He et al. [81] 1997 |
| Binds to STING | Blocks STING’s transport from ER to Golgi, thereby induces IFN production | Christensen et al. [86] 2016 | |
| Us11 | Quenches dsRNA and binds PKR | Inhibits PKR-induced arrest of protein translation | Peters et al. [82] 2002 Poppers et al. [83] 2000 |
| ICP27 | Binds TBK-STING | Prevents IRF3 activation and thereby IFN production | Christensen et al. [86] 2016 |
| IκBα | Reduced cytokine production | Kim et al. [108] 2008 | |
| Suppresses IFN-induced STAT-1 phosphorylation | Dampen IFN signaling | Johnson et al. [95] 2008 Mogensen et al. [96] 2004 | |
| ICP0 | Interferes with IRF3 and IRF7 | Inhibits IFN-α/β mediated ISG expression | Mossman and Smiley [89] 2002 Mossman et al. [90] 2000 Shahnazaryan et al. [91] 2020 Härle et al. [92] 2002 Paladino et al. [93] 2010 |
| Binds MyD88, Mal | Interferes with IL-6 production | van Lint et al. [105] 2010 | |
| UsP7 induced by ICP0 | Deubquitinated TRAF6, IKKγ | Reduced cytokine production | Daubeuf et al. [106] 2009 |
| Vhs | Inhibits JAK-STAT pathway | Reduced IFN-1 production | Lin et al. [109] 2010 |
ICP34.5: infected cell protein 34.5; eIF2α: alpha subunit of eukaryotic initiation factor 2; HSV: herpes simplex virus; STING: stimulator of interferon genes; IFN: interferon; dsRNA: double stranded RNA; PKR: dsRNA-dependent protein kinase; IRF3: IFN-regulatory factor 3; TBK: TANK-binding kinase; IκBα: inhibitor of kappa B alpha; STAT-1: signal transducer and activator of transcription-1; ISG: IFN stimulated gene; MyD88: myeloid differentiation primary response protein 88; Mal: MyD88 adapter like; IL-6: interleukin-6; TRAF6: tumor necrosis factor receptor-associated factor 6; IKKγ: IκB kinase γ; Vhs: virion host shutoff; JAK: Janus kinase; ER: endoplasmic reticulum
PKR is one of the key players that activate IFN-α and β expression in macrophages during HSV-2 infection [79]. HSV encodes two different proteins which use different mechanisms to inhibit PKR-mediated IFN-induction. PKR activation leads to the phosphorylation of the alpha subunit of eukaryotic initiation factor 2 (eIF2α) via phosphoprotein p90 and thereby stops protein translation [80]. ICP34.5 binds to phosphatase and dephosphorylate eIF2α leading to continued protein translation so that HSV can translate its proteins [81]. Another HSV protein Us11 can also inhibit PKR activation in two ways, either by quenching dsRNA to prevent PKR activation or directly binding to PKR to make it inactive [82, 83]. Apart from PKR, STING also plays a key role in inhibiting viral replication by inducing IFN-1 expression. However, HSV-1 can inhibit the STING response by three different mechanisms. Firstly, HSV-1 activates the AP-1 pathway via mitogen-activated protein kinase (MAPK), which in turn activates micro RNA-24 (miR24), thereby degrading STING mRNA, enabling the virus to replicate. Secondly, ICP34.5 binds to STING and blocks its transport from the endoplasmic reticulum (ER) to the Golgi apparatus, further inhibiting its function. Thirdly, another viral protein ICP27 binds to TANK-binding kinase 1 (TBK1)-STING signalosome to prevent IRF3 activation leading to the suppression of IFN production [84–86]. HSV infection leads to dysregulation of micro (mi) RNA in host cells which might play an important role in viral immune evasion. One such miRNA is miR373, which is up-regulated in response to HSV infection leading to the degradation of IRF1 and thereby suppressing ISG expression [87]. UL-2 of HSV has been reported to bind to p65 and p50 subunit of NF-κB and prevent the phosphorylation of p65 by TNF-α and thereby rendering it inactive. This inactivation of NF-κB results in a decrease in IL-8 production and thereby also helps in viral evasion [88].
The IE gene transcribed ICP0 and ICP27 helps HSV to modulate IFN production and signaling in several ways. ICP0 inhibits IFN-α and β mediated ISG expression by interfering with IRF3 and IRF7 [89–93]. This protein interferes with MyD88, MyD88 adapter like (Mal), and NF-κB signaling leading to perturbation of cytokine signaling and thereby helping HSV to replicate [94]. The ICP27 could also suppress the phosphorylation of IFN-induced signal transducer and activator of transcription (STAT)-1, as well as its nuclear localization [95, 96]. Moreover, ICP27 hampers NF-κB signaling by binding and stabilizing inhibitor of kappa B (IκB) and thereby dampening IFN signaling [78]. However, cells also develop strategies to prevent ICP27-mediated immune evasion by down regulating transcription of ICP27, mediated by IL-29 and IFN-α [97].
Vhs, a ribonuclease tegument protein, is another important HSV-encoded protein playing a role in inhibiting host cytokine response. Animal and cell line-based studies suggest that Vhs inhibits IFN-1 synthesis as well as IFN-mediated antiviral response. However, it can’t inhibit IFN-β production in genital epithelial cells [98–100]. Vhs-induced inhibition of the Janus kinase (JAK)/STAT pathway via suppressor of cytokine signaling (SOCS)-3 in keratinocytes and decreased expression of IRF7 might lead to reduced IFN-1 production, but fails to inhibit IFN-β production, as it is JAK/STAT independent [101, 102]. Thus, Vhs null virus shows a higher level of ISG; and hyper-phosphorylated eIF2α indicating Vhs plays a role in inhibiting ISG production as well as dephosphorylation of eIF2α [103]. Vhs also increases the turnover of innate immune sensor IFI16, leading to a reduction in IFI16 in HSV-infected HeLa cells [104].
In response to HSV infection, skin produces a lot of cytokines which can directly inhibit HSV lifecycle and/or can attract immune cells to further inhibit HSV infection. To prevent this cytokine-mediated clearance, HSV codes for proteins that suppress cytokine production. HSV-encoded ICP0 targets MyD88 and Mal for degradation which are part of TLR-2 mediated NF-κB signaling and thereby reduce IL-6 production [105]. The ICP0 uses another mechanism to suppress the inflammatory response. It induces translocation of UsP7 from the nucleus to the cytoplasm where it deubiquitinates TRAF6 and IKKγ and thereby inhibits TLR mediated NF-κB and JNK activation, resulting in a reduction of cytokine production [106]. ICP27 blocks cytokine production in two ways, firstly by targeting NF-κB and IRF3 and secondly by reducing cytokine mRNA stability [96, 107]. The ICP27 binds to IκBα and prevents its degradation, thereby inactivating NF-κB signaling that results in a reduction of cytokine production [108]. Vhs being a tegument protein also helps in suppressing cytokine production in HSV-infected cells. Vhs-negative pseudovirus infection is associated with the upregulation of TNF-α indicating that Vhs plays a role in suppressing TNF-α, one of the key cytokines produced in response to many viral infections [109]. HSV-1 also uses its envelope protein gL to suppress NF-κB mediated expression of IFN-β and ISG15 by directly binding to p65 and thereby inhibits its translocation to the nucleus [110]. Another HSV-1 protein ICP8 also inhibits cGAS signaling by reducing the expression of cGAMP (2’3’-cyclic GMP-AMP), a downstream signaling molecule of cGAS pathway [111].
Conclusions
The cytokine response triggered by a pathogen invasion represents a highly intricate and dynamic interplay between the host immune system and the defensive mechanisms employed by the viral attacker. Among the primary defense barriers of human, the skin assumes a crucial role in thwarting the progression of pathogens, although its success is not always ensured. The skin encounters a diverse array of pathogens, as found in human HSV-1 (HHSV-1). Upon HSV-1 infection, the skin mounts a robust production of cytokines and IFNs as a protective response. However, the virus has evolved numerous strategies to evade this inflammatory response including the various innovative escape mechanisms to establish a successful infection, leading to an enduring evolutionary conflict between HSV-1 and the skin. While immune regulation and activation are complicated and extensive, we have tried to provide an overview of the various pathways and the interplay between them in skin infection. This becomes more complicated by the various mechanisms evolved in the virus to interfere with the immune response. Therefore, a better understanding of cytokine balance due to pathogen attack and an understanding of the various mechanisms by which a cytokine response is averted by the invader is key to unraveling preventives and producing a more balanced immune reaction. In this review, we have tried to highlight the critical role played by cytokines in mounting an effective immune response. Its importance is highlighted by the various mechanism viruses have evolved to interfere with the signaling pathways activating the cytokine response. Together, this leads to the imbalance known as a cytokine storm. Future research could delve into the intricate regulatory mechanisms governing a balanced IFN response in spite of the various mechanisms that viruses have developed to overcome it. Such research would need to be comprehensive in terms of a multi-level approach involving mechanisms related to gene transcription regulation at DNA level, RNA modification as well as protein signaling and expression regulation. Such an approach would help to understand the mechanisms of “cytokine storm” and ways to tame it. An elegant model has been suggested by Sun et al. [61] where they have prepared a vascularized skin-on-chip model where preclinical therapeutic testing and HSV disease modeling can be performed. Such models could help to unravel most of the mysteries of cytokine regulation [61].
Abbreviations
| AIM2: | absent in melanoma 2 |
| CD: | cluster of differentiation |
| cGAS: | cyclic GMP-AMP synthase |
| CXCL8: | C-X-C motif chemokine ligand 8 |
| DAMPs: | damage-associated molecular patterns |
| DCs: | dendritic cells |
| ds: | double stranded |
| eIF2α: | alpha subunit of eukaryotic initiation factor 2 |
| GM-CSF: | granulocyte macrophage colony stimulating factor |
| HSV: | herpes simplex virus |
| ICP0: | infected cell protein 0 |
| IFI16: | interferon-gamma-inducible protein-16 |
| IFN-1: | type-1 interferon |
| IL-17: | interleukin-17 |
| IRFs: | interferon-regulatory factors |
| ISG: | interferon stimulated gene |
| LC: | Langerhans cell |
| MC: | molluscum contagiosum |
| MIP-1β: | macrophage inflammatory protein 1 beta |
| MyD: | myeloid differentiation primary response protein |
| NF-κB: | nuclear factor kappa B |
| NK: | natural killer |
| PAMPs: | pathogen-associated molecular patterns |
| PKR: | double stranded RNA-dependent protein kinase |
| PRRs: | pattern recognition receptors |
| RIG: | retinoic acid-inducible gene |
| STAT: | signal transducer and activator of transcription |
| STING: | stimulator of interferon genes |
| TGF: | transforming growth factor |
| TLRs: | toll-like receptors |
| TNF: | tumor necrosis factor |
| vDNA: | viral DNA |
| Vhs: | virion host shutoff |
Supplementary materials
The supplementary table for this article is available at: https://www.explorationpub.com/uploads/Article/file/1003148_sup_1.pdf.
Declarations
Author contributions
AM and SI equally contributed to: Investigation, Data curation, Resources, Formal analysis, Writing—original draft, Writing—review & editing. JG: Writing—original draft, Visualization. SB: Data curation, Formal analysis, Writing—original draft. PP: Data curation, Formal analysis, Writing—review & editing. DC: Conceptualization, Project administration, Writing—original draft, Writing—review & editing. All authors read and approved the submitted version.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
AM is supported by Presidency University, which received funding from DBT-BUILDER; DBT-FIST; and WB-DST. PP received funding from ICMR. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2024.