 Review
Review

Affiliation:
ITC Limited-Corporate Division, Life Science and Technology Centre, Bangalore 560 058, India
ORCID: https://orcid.org/0000-0002-1590-7313
Affiliation:
ITC Limited-Corporate Division, Life Science and Technology Centre, Bangalore 560 058, India
Email: ganesh.gopal@itc.in
ORCID: https://orcid.org/0000-0002-9408-211X
Explor Immunol. 2024;4:744–769 DOI: https://doi.org/10.37349/ei.2024.00170
Received: March 12, 2024 Accepted: October 27, 2024 Published: November 15, 2024
Academic Editor: Calogero Caruso, University of Palermo, Italy
The article belongs to the special issue The Complement System in Health and Disease
The complement system is a key component of the innate immune system that mediates the clearance of pathogens, apoptotic cells, and cellular debris. However, the complement system also has diverse roles in the central nervous system (CNS), where it regulates synaptic pruning, neural plasticity, and neuroinflammation. Dysregulation of the complement system has been implicated in various neurodegenerative disorders such as Alzheimer’s disease, multiple sclerosis, epilepsy, stroke, and traumatic brain injury. In these conditions, excessive or chronic activation of the complement system may lead to synaptic loss, neuronal damage, immune dysregulation, and inflammation, which leads to exacerbating the disease’s progression and severity. Moreover, the complement system may interact with infectious agents that invade the CNS, such as bacteria, viruses, fungi, and parasites, and modulate their pathogenicity and host response. Therefore, understanding the complex interplay between the complement system and the CNS is crucial for developing novel therapeutic strategies to prevent or treat neurodegenerative and neuroimmune disorders. Natural compounds, such as plant extracts, phytochemicals, and nutraceuticals, have emerged as promising candidates for modulating the complement system and its effects on the CNS. These compounds may exert anti-inflammatory, antioxidant, neuroprotective, and immunomodulatory effects by regulating the expression of various complement components and pathways. In this review, we summarized the current knowledge on the roles of the complement system in human neurodegenerative disorders and the benefits of natural compounds for complement-targeted therapy.
The complement system is a part of the immune system that helps to fight against infections and remove damaged cells from the body. It comprises approximately 50 proteins that are found in the fluid phase of the blood or bound to cells where they function as receptors or regulators of complement activation [1]. These proteins can be activated by different triggers, such as bacteria, viruses, or antibodies, and start a chain reaction that leads to inflammation, phagocytosis, and cell lysis. The complement system has three main pathways: the classical pathway (CP), the alternative pathway (AP), and the lectin pathway (LP) [2, 3].
The CP is triggered when IgM or IgG antigen/antibody complexes bind to C1q, the first protein in the cascade. This binding activates C1r, which subsequently cleaves C1s. The activated C1s then cleaves C4 and C2, resulting in the formation of the C3 convertase, C4b2a. This convertase cleaves C3 into C3a and C3b [4]. C3a functions as an anaphylatoxin, recruiting inflammatory cells, while C3b binds to the C4b2a complex to form the C5 convertase, C4b2a3b. The C5 convertase initiates the assembly of the membrane attack complex (MAC), which creates pores in bacterial membranes, leading to cell lysis [5]. Additionally, the CP can be activated by other danger signals such as C-reactive protein, viral proteins, polyanions, apoptotic cells, and amyloid, indicating that it can be activated independently of antibodies [6].
The AP is activated by various molecules such as carbohydrates, lipids, and proteins present on foreign and non-self surfaces [7]. In this pathway, C3 undergoes continuous low-level hydrolysis, producing C3b, which attaches to targets like bacteria. This bound C3b then recruits factor B, which is subsequently cleaved by factor D to form the C3 convertase, C3bBb. This convertase is stabilized by plasma properdin [8]. Properdin, a protein released by activated neutrophils, as well as macrophages and T cells, binds to C3b, preventing its degradation by factors H and I. Recent research indicated that properdin can also directly bind to apoptotic and necrotic cells, initiating complement activation [8].
The LP is initiated when mannose-binding lectin (MBL) or ficolin binds to carbohydrate moieties on the surfaces of pathogens such as yeast, bacteria, parasites, and viruses. Both MBL and ficolin circulate in the bloodstream as complexes with MBL-associated serine proteases (MASPs) [9, 10]. There are four MASPs: MASP-1, MASP-2, MASP-3, and a truncated form of MASP-2 known as MAP19 [10]. Upon binding to pathogens, conformational changes occur, leading to the autoactivation of MASP-2, which then cleaves C4 into C4a and C4b. C4b attaches to the pathogen surface, promoting the binding of C2, which is subsequently cleaved by MASP-2 into C2a and C2b. The complex of C4b and C2a forms the LP C3 convertase, C4bC2a. While the specific roles of the other MASPs are not fully understood, MASP-1 can cleave C2 but not C4, thereby aiding in the amplification of complement activation by the bound complexes [9].
The complement system plays a crucial role in maintaining health by enhancing the immune system’s ability to eliminate harmful microbes and foreign substances from the body. Additionally, it supports wound healing and prevents excessive bleeding. However, problems can arise from an overactive or underactive complement system. Research indicated that an overactive complement system can lead to cellular and tissue damage, potentially triggering autoimmune conditions like lupus or rheumatoid arthritis (RA). Conversely, an underactive complement system can increase susceptibility to infections such as meningitis or pneumonia [11]. The effects of complement components and activation products can have both positive and negative impacts on various neurodegenerative disorders, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) which have been reported [12].
The neuroimmune system is a network of immune cells and molecules that can sense and respond to harmful stimuli in the central nervous system (CNS). Classical complement proteins mediate many physiological and pathological processes both in the periphery and in the CNS. An established function of these proteins is in mediating microglial phagocytosis of synapses during CNS development and diseases. CNS injury resulting from both neurodegeneration and viral infections is associated with increased complement expression and activation. Activation of classical complement proteins alters microglial responses in a context-specific manner and synaptic elimination occurs via similar complement-mediated pathways in neurodegeneration versus viral infection. Various viruses are neutralized in the periphery via mechanisms involving classical complement proteins to limit viral spread. Some of these proteins are upregulated in the CNS after neurotropic viral infection [13]. Therefore, it is important to maintain a balanced complement system for good health.
Natural compounds, such as plant extracts, phytochemicals, and nutraceuticals have emerged as promising candidates for modulating the complement system and its effects on the CNS. These compounds may exert anti-inflammatory, antioxidant, neuroprotective, and immunomodulatory effects by inhibiting or activating the function of various complement components and pathways [14–16].
Therefore, understanding the complex interplay between the complement system and CNS is crucial for developing novel therapeutic strategies to prevent or treat neurodegenerative and neuroimmune disorders. We also provided: (a) an overview of the structure, function, and regulation of the complement system; and (b) the mechanisms of complement-mediated neurodegeneration and neuroprotection. Finally, we discussed the challenges and opportunities for the development of natural compounds as complement inhibitors or modulators for the treatment of CNS diseases.
Complementing the host’s defense machinery plays a vital role in maintaining barrier function and protecting against microbial invasion after an injury, along with the contact and coagulation systems. This complement system also modulates different branches of innate and adaptive immunity [17]. The purpose of complement is to identify, tag, and eliminate microbial intruders with immediate reactivity and sufficient specificity in order to avoid damage to host cells. This reactivity and specificity occur via a series of circulating pattern-recognition proteins (PRPs) that sense pathogen-associated molecular patterns (PAMPs) and initiate the complement cascade. A strong coordination network of interactions occurs through three canonical pathways, which include, the CP, AP, and LP. It is essential for the surveillance, immunomodulatory, and effector functions of the complement system. All activation pathways result in the formation of C3 and C5 convertase enzyme complexes that cleave C3 to anaphylatoxin C3a and opsonin C3b, and C5 to anaphylatoxin C5a and C5b respectively. The release of C5b into a target initiates MAC formation and targeting lysis. Opsonins and anaphylatoxins are also promoted by scavenger cells to promote phagocytic uptake of pathogens, as well as activation of monocytes, neutrophils, or mast cells [18].
The complement system plays a critical role in modulating immune responses and tissue homeostasis in peripheral organs which influences various physiological processes, including metabolism, inflammation, and tissue repair. Peripheral organs, such as the liver, adipose tissue, gastrointestinal tract, and kidney express complement components and receptors allowing for local complement activation and modulation of organ function [19]. Complement activation products, such as C3a and C5a, serve as an important mediator of inflammation and tissue injury in peripheral organs contributing to the pathogenesis of various diseases [20]. In the cardiovascular system, complement activation can contribute to the pathogenesis of cardiovascular diseases such as myocardial infarction, atherosclerosis, and heart failure. Complement activation products such as C3a and C5a can promote endothelial dysfunction, leukocyte recruitment, and vascular inflammation thereby exacerbating cardiovascular pathology [21].
Moreover, dysregulation of the complement system has been implicated in the pathogenesis of metabolic disorders including obesity, diabetes, and non-alcoholic fatty liver disease (NAFLD). Complement components and regulators are dysregulated in these conditions, leading to chronic low-grade inflammation, insulin resistance, and tissue damage [22]. Additionally, complement activation has been implicated in the pathogenesis of gastrointestinal disorders, such as inflammatory bowel disease (IBD) and celiac disease, where it contributes to mucosal inflammation and tissue damage [23].
Similarly, in the lungs, complement system components are involved in the regulation of pulmonary inflammation and injury. Dysregulation of the complement system has been implicated in the pathogenesis of inflammatory lung diseases such as acute respiratory distress syndrome (ARDS) and chronic obstructive pulmonary disease (COPD). Complement activation products, particularly C5a, can mediate pulmonary inflammation, neutrophil recruitment, and tissue damage in these conditions [24].
Beyond the peripheral organs, the complement system, plays a multifaceted role in CNS, influencing various physiological processes and contributing to both homeostasis and pathology. Complement proteins such as C1q, C3, and C4 are abundantly expressed in the CNS and are involved in synaptic refinement during development and synaptic plasticity in adulthood [25]. However, dysregulated complement activation in the CNS can lead to excessive neuroinflammation and neuronal damage, contributing to the pathogenesis of neurological disorders [26]. The CP has been implicated in synaptic pruning and neuroinflammation in the CNS [27]. The LP contributes to immune surveillance and inflammation in the CNS, particularly in response to microbial infections [28]. The AP amplifies complement activation and contributes to neuroinflammation and tissue damage in CNS disorders [29]. Excessive complement activation and the generation of complement fragments, including C3a, C4a, and C5a, can contribute to neuroinflammation, synaptic dysfunction, and neuronal damage in AD and PD [30]. Complement activation products, including C3a and C5a, exert neurotoxic effects and promote microglial activation and cytokine release. Moreover, the deposition of complement proteins, such as C3d and C5b-9, has been observed in the brains of patients with neurodegenerative diseases [25].
The complement system interacts with neuroimmune cells to modulate immune responses, synaptic pruning, and neuronal function in the CNS. Neuroimmune cells, including microglia, astrocytes, and neurons, play essential roles in maintaining CNS homeostasis and responding to pathological insults (Figure 1). Microglia, the resident immune cells of the CNS, express complement receptors (CRs) and actively participate in complement-mediated immune responses. Complement activation products, such as C3a and C5a, can modulate microglial activation and migration contributing to neuroinflammation and synaptic remodeling [31]. Dysregulated complement signaling in microglia has been implicated in the pathogenesis of neurodegenerative diseases, including AD and PD [29]. Astrocytes, the most abundant glial cells in the CNS, also express CRs and can respond to complement activation products. Complement-mediated signaling in astrocytes regulates their activation, cytokine secretion, and neuroprotective functions [32]. Dysregulated complement signaling in astrocytes has been implicated in various CNS disorders, including multiple sclerosis (MS) and ALS [33]. Neurons, traditionally viewed as non-immune cells also express complement components and receptors and can respond to complement activation [34]. Complement-mediated signaling in neurons influences synaptic pruning, neuronal survival, and synaptic plasticity [30]. Dysregulated complement signaling in neurons has been linked to synaptic dysfunction and neuronal loss in neurodegenerative diseases [25]. The interactions between complement system pathways and neuroimmune cells are complex and bidirectional. Neuroimmune cells can regulate complement activation through the expression of complement regulators and inhibitors, such as CD46, CD55, and CD59 [35]. Conversely, complement activation in neuroimmune cells can modulate their functions and influence CNS homeostasis and pathology.
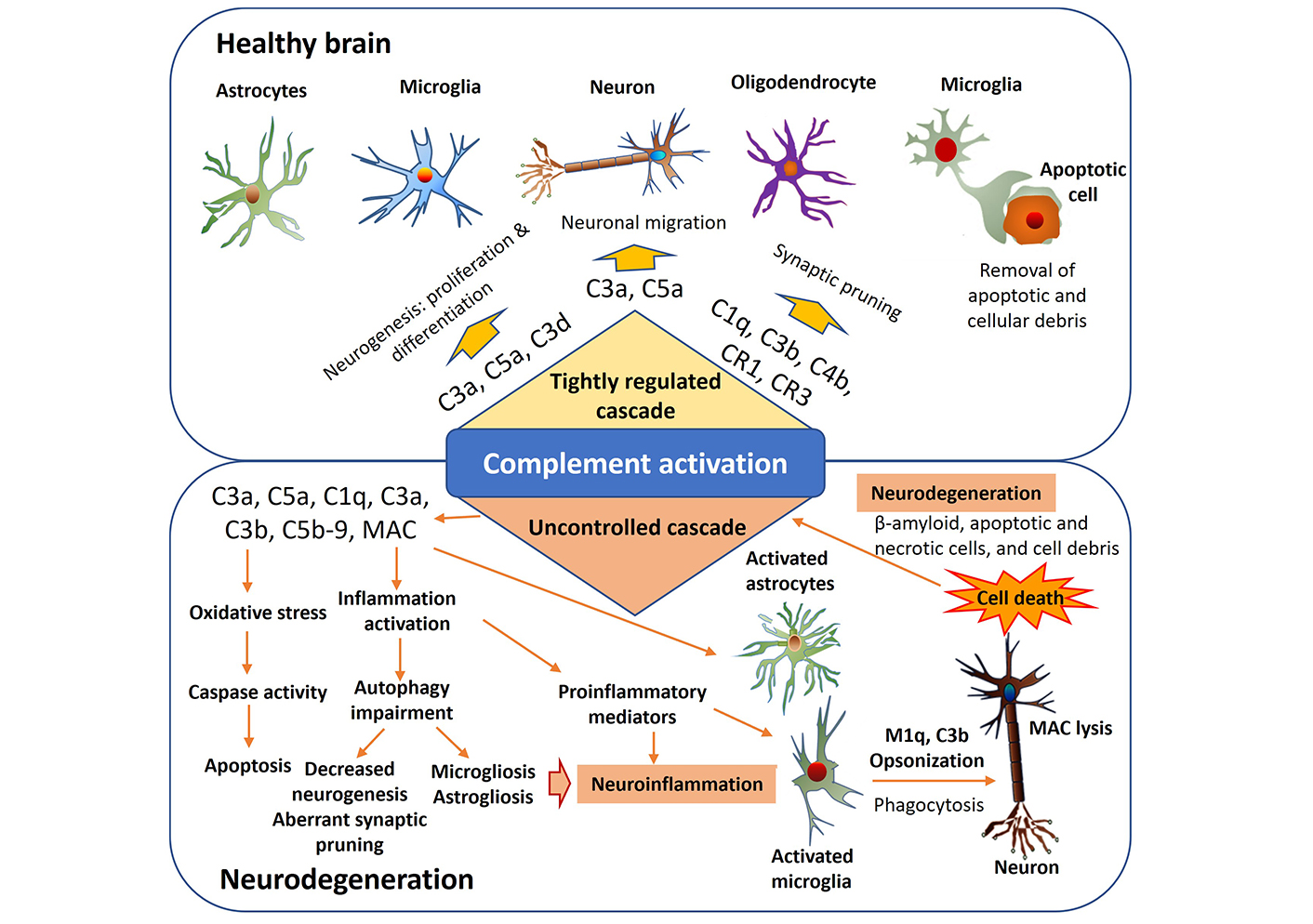
The role of the complement system in both the physiological and pathophysiological functioning of the central nervous system. Various pathogenic factors like β-amyloid, apoptotic and necrotic cells, and cellular debris can initiate an overactivation of the complement cascade, resulting in persistent inflammation, increased oxidative stress, and impaired autophagy. Consequently, this process worsens neuroinflammation and contributes to the neurodegeneration. MAC: membrane attack complex
Understanding the intricate relationship between the complement system, aging, and aging-related diseases is crucial for elucidating the underlying mechanisms of age-related pathologies and identifying potential therapeutic interventions. Aging is a multifaceted biological process characterized by a progressive decline in physiological functions and increased susceptibility to various age-related diseases [36]. As individuals age, they experience alterations in immune function, including dysregulation of the complement system [37, 38]. Age-related changes in the complement system include alterations in complement component expression, activation patterns, and regulation, which can lead to chronic low-grade inflammation, tissue damage, and increased vulnerability to infections [38]. The dysregulation of the complement system in aging has been implicated in the pathogenesis of several aging-related diseases, including cardiovascular diseases, neurodegenerative disorders, and age-related macular degeneration (AMD) [38, 39]. Inflammation, oxidative stress, and complement dysregulation contribute to the development and progression of these age-related diseases [40, 41].
Brain aging is a multifaceted process characterized by structural and functional changes in the CNS, often leading to cognitive decline and increased susceptibility to neurodegenerative diseases. The complement system, a crucial component of innate immunity, has recently emerged as a key player in the pathogenesis of age-related neuroinflammation and neurodegeneration. In the aging brain, dysregulation of complement system components has been implicated in the pathogenesis of neurodegenerative diseases such as AD and PD. Complement proteins such as C1q, C3, and C4 are abundantly expressed in the CNS and play crucial roles in synaptic refinement, neurogenesis, and neuroinflammatory responses [26]. However, aberrant complement activation and the generation of complement fragments, including C3a, C4a, and C5a, can contribute to neuroinflammation, synaptic dysfunction, and neuronal damage in aging and neurodegenerative diseases [31]. The dysregulation of complement system components in brain aging can trigger neuroinflammatory responses through various mechanisms. Complement activation products such as C3a and C5a can promote microglial activation, induce the release of pro-inflammatory cytokines, and exacerbate synaptic loss and neuronal damage [31]. Additionally, complement-mediated neuro-inflammation can lead to the recruitment of peripheral immune cells, further amplifying the inflammatory cascade, and contribute to neurodegeneration [25].
The nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway and the complement system are two critical components of the innate immune response that play key roles in regulating immune responses and inflammation. Recent research has revealed intricate interactions between NF-κB signaling and the complement system, influencing immune regulation and disease pathogenesis. NF-κB is a transcription factor that regulates the expression of genes involved in inflammation, immunity, cell survival, and proliferation. In response to various stimuli, such as pro-inflammatory cytokines, pathogens, and cellular stress, NF-κB is activated and translocates to the nucleus, where it induces the expression of target genes [42]. Dysregulated NF-κB signaling has been implicated in the pathogenesis of inflammatory and autoimmune diseases, as well as cancer [43]. The complement system can activate NF-κB signaling through multiple pathways. Complement activation products, such as C5a and C3a, can stimulate NF-κB activation directly by binding to their respective receptors on immune cells [44]. Additionally, complement components, including C3 and C4, can induce NF-κB activation through the engagement of pattern recognition receptors (PRRs) and the formation of signaling complexes [45]. This crosstalk between the complement system and NF-κB signaling amplifies immune responses and promotes inflammation. Additionally, NF-κB can regulate the expression of complement regulatory proteins, such as CD46 and CD55, in order to finely control complement activation and prevent excessive tissue damage [46]. The interaction between NF-κB signaling and the complement system has significant implications for immune modulation and the development of various diseases. The dysregulation of NF-κB signaling and complement activation has been implicated in the pathogenesis of numerous diseases, including IBD, RA, and systemic lupus erythematosus (SLE) [42, 44]. The aberrant activation of NF-κB and the complement system can perpetuate chronic inflammation and contribute to tissue damage and organ dysfunction in these diseases.
Modulating NF-κB signaling and complement activation presents potential therapeutic strategies for immune-related diseases. Inhibitors of NF-κB signaling, such as small molecule inhibitors and biologics targeting NF-κB components, have been developed and tested in preclinical and clinical studies for various inflammatory conditions [43]. Similarly, targeting complement components or receptors has shown efficacy in preclinical models and clinical trials of inflammatory and autoimmune diseases [47]. Combined therapies that target both NF-κB signaling and the complement system may offer synergistic effects and improve therapeutic outcomes in immune-related disorders.
MicroRNAs (miRNAs) are small, non-coding RNA, molecules that play a pivotal role in post-transcriptional gene regulation. They fine-tune gene expression by binding to the 3’ untranslated region (UTR) of target mRNAs, leading to mRNA degradation or translational repression. Recent research has discovered the intricate interactions between miRNAs and the complement system, a crucial component of the innate immune system. Several miRNAs have been identified as regulators of complement components, receptors, and regulators. miR-223 has been implicated in modulating the expression of the complement component C3, influencing the activation of the complement cascade [48]. Additionally, miR-146a has been shown to target complement factor H (CFH), a critical regulator of the alternative complement pathway [49]. These findings emphasized the role of miRNAs in finely tuning the expression of complement-related genes to maintain immune homeostasis. The crosstalk between miRNAs and the complement system holds significant implications in immune modulation. Dysregulation of miRNA-mediated complement regulation may contribute to chronic inflammation, autoimmunity, and other immune-related disorders.
In the CNS, where complement dysregulation is implicated in neuroinflammatory disorders, the interplay with miRNAs is of particular interest. miRNAs have been shown to modulate the expression of complement factors in microglia, astrocytes, and neurons, influencing neuroinflammation and synaptic plasticity [50]. Understanding the complex connections between miRNAs and the complement system in the CNS may offer new avenues for therapeutic interventions in neurodegenerative diseases. The dysregulation of miRNA-complement interactions has been implicated in various diseases. In SLE, aberrant miRNA expression profiles have been linked to complement activation and immune dysregulation [51]. Similarly, in AMD, alterations in miRNA-mediated complement regulations are associated with disease progression [52]. Harnessing the regulatory potential of miRNAs in the complement system opens avenues for therapeutic intervention. miRNA-based therapeutics, such as miRNA mimics or inhibitors, could be designed to modulate complement activity in a targeted manner, providing precision in immune modulation.
Genetic variations in complement genes have been linked to neurodegenerative disorders, as mutations in the CFH gene are associated with an increased risk of AMD and AD [53]. Chronic inflammation plays a significant role in dysregulation of the complement system in neurodegenerative disorders. In conditions like AD, chronic neuroinflammation perpetuates complement activation, exacerbating neuronal damage [25]. Oxidative stress-induced activation of the complement system contributes to neuroinflammation and neurodegeneration. Studies have shown that oxidative stress markers are elevated in neurodegenerative disorders such as PD and AD, correlating with complement dysregulation [54, 55]. Dysfunctional microglia can aberrantly activate the complement system, leading to synaptic loss and neuronal injury. Recent research highlighted the role of microglial complement signaling in neuroinflammation and neuro-degeneration, implicating it in diseases like AD and PD [56, 57]. Impairment of the blood-brain barrier (BBB) allows peripheral immune cells and complement proteins to infiltrate into the CNS, exacerbating neuroinflammation. Studies have demonstrated BBB dysfunction in neurodegenerative disorders such as AD and MS, contributing to complement dysregulation [58, 59]. Environmental factors like air pollution and heavy metal exposure can trigger neuroinflammation and complement activation, contributing to neurodegenerative diseases. Recent studies have highlighted the impact of environmental toxins on complement dysregulation in disorders such as PD and ALS [60, 61]. Metabolic dysregulation, including obesity and insulin resistance, can promote complement activation and neuroinflammation. Studies have shown a link between metabolic syndrome and increased risk of neurodegenerative disorders, partly mediated by complement dysregulation [62, 63]. Emerging evidence suggested a bidirectional communication between the gut microbiota and the brain, influencing neuroinflammation and complement activation. Dysbiosis of gut microbiota has been implicated in neurodegenerative disorders, potentially affecting complement regulation [64, 65]. Epigenetic changes can modulate complement gene expression and contribute to neuroinflammation in neurodegenerative diseases. Studies have identified epigenetic alterations in complement-related genes in AD and PD, highlighting their role in disease pathogenesis [66, 67].
Dysregulation of the complement system can lead to various pathophysiological conditions, including autoimmune diseases, inflammatory disorders, and complement-mediated tissue damage. Understanding the molecular mechanisms underlying complement system-related pathophysiology is essential for the development of targeted therapeutic interventions.
CRs, including CR1 (CD35), CR2 (CD21), and CR3 (CD11b/CD18), are integral to immune cell function and antigen presentation. Dysregulated CR signaling has been implicated in autoimmune diseases such as SLE and RA [68]. Activation of CRs can initiate inflammatory responses, perpetuating tissue damage in autoimmune pathologies. Targeting CRs presents a promising avenue for therapeutic development [69].
Various regulatory proteins play a crucial role in maintaining a delicate equilibrium between complement activation and regulation. Among these proteins, CD55 (decay-accelerating factor) and CD59 (protectin) act as inhibitors of MAC formation, effectively protecting host cells from complement-mediated lysis [70]. However, deficiencies in these regulatory proteins, as seen in conditions like atypical hemolytic uremic syndrome (aHUS) and paroxysmal nocturnal hemoglobinuria (PNH), increase the susceptibility of individuals to complement-mediated disorders [71]. Therefore, exploring strategies to enhance the function of regulatory proteins holds promising therapeutic potential in mitigating complement-driven pathology.
Genetic polymorphisms in complement components and regulatory proteins influence disease susceptibility and phenotype manifestation. Genome-wide association studies (GWAS) have identified variants in genes encoding CFH and CR3 associated with autoimmune disorders [72]. Unraveling the genetic architecture of complement dysregulation aids in personalized risk assessment and therapeutic targeting in affected individuals.
Advancements in deciphering the molecular intricacies of complement system-related pathophysiology offer promising avenues for therapeutic intervention. Understanding the crosstalk between complement and inflammatory pathways, the role of CRs, the regulation of complement activation by regulatory proteins, and genetic determinants of disease susceptibility provide a comprehensive framework for targeted therapeutic strategies. Harnessing this knowledge may pave the way for precision medicine approaches in managing complement-mediated disorders.
Neurodegenerative and neuroimmune disorders pose significant challenges to human health. It is characterized by progressive neuronal loss, cognitive decline, and motor dysfunction. Dysregulated complement system activation contributes to neurodegenerative disorders including Guillain-Barré syndrome variants, chronic inflammatory demyelinating polyneuropathy (CIDP), genetic demyelinating neuropathy, IgM neuropathy, neuropathic pain, myasthenia gravis, Lambert-Eaton myasthenic syndrome, dermatomyositis, necrotizing autoimmune myositis, MS, neuromyelitis optica spectrum disorders, autoimmune encephalopathies, Alzheimer disease, traumatic brain injury, ALS, Huntington disease, and schizophrenia are among others [73–76].
AD is the most common cause of dementia, characterized by progressive cognitive decline and memory impairment. The pathological hallmarks of AD include the accumulation of amyloid-beta (Aβ) plaques and neurofibrillary tangles (NFTs), synaptic dysfunction, and neuronal loss. While the exact etiology of AD remains elusive, growing evidence suggests the role of neuroinflammation in disease progression. The complement system, a crucial component of innate immunity, has emerged as a key regulator in AD pathogenesis, contributing to neuroinflammation and neurodegeneration [75]. The complement system comprises a complex network of proteins involved in immune surveillance, host defense, and tissue homeostasis. In the context of AD, dysregulated complement activation leads to the increased deposition of complement proteins and activation products in the brain parenchyma and around Aβ plaques. Studies have shown elevated levels of complement components, including C1q, C3, and C4, in post-mortem AD patient’s brains [77]. Complement activation products, such as C3b and C5b-9, are also detected in AD brain tissues, suggesting ongoing complement-mediated inflammation [78]. Synaptic dysfunction is an early pathological feature of AD, preceding neuronal loss and cognitive decline. The complement system plays a critical role in synaptic pruning and elimination during development and synaptic plasticity in the adult brain [79]. However, dysregulated complement activation in AD leads to excessive synaptic loss and dysfunction. Complement proteins, including C1q and C3, localize to synapses in AD patient’s brains and mediate synapse elimination through microglial phagocytosis [80]. Targeting complement activation has emerged as a potential therapeutic strategy for AD [76]. Preclinical studies using complement inhibitors or genetic deletion of complement components have demonstrated neuroprotective effects in AD mouse models [81]. A recent study showed that genetic deletion of C3 or treatment with complement inhibitors reduces Aβ deposition, neuroinflammation, and synaptic loss in transgenic AD mice [82].
PD is the second most common neurodegenerative disorder, characterized by motor symptoms such as bradykinesia, rigidity, and tremor as well as non-motor symptoms including cognitive impairment and autonomic dysfunction. The complement system, a crucial part of innate immunity, has emerged as a key player in PD pathogenesis, contributing to neuroinflammation and neurodegeneration. Dysregulated complement activation occurs in PD, leading to increased deposition of complement proteins and their activation products in the brain parenchyma and around Lewy bodies [83]. Studies have shown elevated levels of complement components, including C1q, C3, and C5b-9 in post-mortem PD patient’s brains [54]. Complement activation products, such as C3b and C5b-9 are also detected in PD brain tissues, suggesting ongoing complement-mediated inflammation [54]. Activated complement proteins contribute to neuroinflammation in PD through various mechanisms. Complement activation promotes microglial activation and recruitment of peripheral immune cells, leading to the release of pro-inflammatory cytokines and chemokines, such as interleukin-1β (IL-1β), tumor necrosis factor-alpha (TNF-α), and IL-6 [84]. Additionally, complement-mediated synaptic dysfunction and neuronal damage exacerbate neuro-inflammatory responses, further contributing to disease progression [13].
Targeting the astrocyte-neuron C3/C3ar (C3a receptor) pathway could be a promising therapeutic strategy for PD [85]. Preclinical studies using complement inhibitors or genetic deletion of complement components have demonstrated neuroprotective effects in PD animal models [86–88]. For instance, genetic deletion of C3 or treatment with complement inhibitors reduces neuroinflammation, synaptic loss, and motor deficits in PD mice [89]. Translating complement-targeted therapies to clinical applications holds promise for slowing disease progression and preserving motor function in PD patients.
Huntington’s disease (HD) is a progressive neurodegenerative disorder characterized by motor dysfunction, cognitive impairment, and psychiatric symptoms. Emerging evidence suggests that dysregulated complement system activation contributes to the pathogenesis of HD [90]. Complement proteins and their activation products are found in HD patients’ brain tissues, where they promote neuroinflammation, synaptic dysfunction, and neuronal damage [90]. Histopathological studies have revealed the presence of complement proteins and activation products in the brains of HD patients [91]. Complement components such as C1q, C3, and C5b-9 are detected in HD brain tissues, where they colocalize with areas of neuronal loss and gliosis [92]. Complement activation products, including C3b and C5b-9, contribute to neuroinflammatory responses and exacerbate neuronal damage in HD. Activated complement proteins promote neuroinflammation in HD by activating microglia and astrocytes, leading to the release of pro-inflammatory cytokines and chemokines [90]. Complement-mediated inflammation further exacerbates neuronal damage and synaptic dysfunction, contributing to disease progression [90]. Additionally, dysregulated complement activation disrupts the BBB, allowing peripheral immune cells to infiltrate the CNS and exacerbate neuroinflammatory responses [91]. Targeting complement system has emerged as a potential therapeutic strategy for HD [29, 93]. Preclinical studies using complement inhibitors or genetic deletion of complement components have demonstrated neuroprotective effects in HD animal models, showing that genetic deletion of C3 or treatment with complement inhibitors reduces neuroinflammation, synaptic dysfunction, and motor deficits in HD mice [94].
Autoimmune encephalitis (AE) is a group of disorders characterized by inflammation of the brain mediated by autoantibodies against neuronal surfaces or synaptic proteins. Emerging evidence suggests that dysregulated complement system activation contributes to the pathogenesis of AE [95]. Histopathological studies have revealed the presence of complement proteins and activation products in AE brain tissues [96]. Complement components such as C1q, C3, and C5b-9 are detected in AE brain lesions, particularly in areas of neuroinflammation and neuronal injury [97]. Complement activation products, including C3b and C5b-9, contribute to neuroinflammatory responses and exacerbate neuronal damage in AE. Synaptic dysfunction is a prominent feature of AE, associated with cognitive deficits and psychiatric symptoms [75, 98]. Dysregulated complement activation contributes to synaptic dysfunction by promoting the elimination of synapses through microglial phagocytosis. Complement-mediated synaptic pruning disrupts neural circuits and impairs neuronal communication, further exacerbating cognitive impairments in AE [99]. Targeting complement activation has emerged as a potential therapeutic strategy for AE [100]. Preclinical studies using complement inhibitors or genetic deletion of complement components have demonstrated neuroprotective effects in AE animal models [101]. Treatment with complement inhibitors reduces neuroinflammation, synaptic dysfunction, and behavioral abnormalities in AE-like rodents [102].
MS is an autoimmune disorder characterized by demyelination, neuroinflammation, and axonal damage in the CNS. Emerging evidence implicates dysregulated complement system activation in the pathogenesis of MS [103]. Complement proteins and activation products are found in MS lesions and cerebrospinal fluid, contributing to myelin destruction and oligodendrocyte injury [104–107]. Recent research has implicated dysregulated complement system activation in the pathogenesis of MS, suggesting a key role for innate immunity in driving disease progression [108]. Evidence from histopathological studies demonstrated the presence of complement proteins and activation products in MS lesions, particularly in active demye-linating lesions [109, 110]. Complement components such as C1q, C3, and C5b-9 are detected in MS plaques, where they colocalize with areas of myelin destruction and axonal injury [111]. The deposition of complement proteins in MS lesions contributes to the recruitment of immune cells, activation of microglia and astrocytes, and promotion of pro-inflammatory cytokine production, exacerbating neuroinflammation and tissue damage [112, 113]. Oligodendrocytes, the myelin-producing cells of the CNS, are targets of complement-mediated injury in MS [114]. Complement activation products, such as C3b and C5b-9, directly damage oligodendrocytes and disrupt myelin integrity [115]. Additionally, complement-mediated inflammation leads to the recruitment of immune cells, including macrophages and microglia, which further contribute to oligodendrocyte death and demyelination [116]. Dysregulated complement activation in MS contributes to BBB dysfunction, facilitating the entry of immune cells and inflammatory mediators into the CNS [117]. Complement components, particularly C5a and C5b-9, disrupt tight junction proteins and endothelial cell integrity, leading to increased BBB permeability and leukocyte infiltration into the CNS [118]. Given the central role of complement dysregulation in MS pathogenesis, targeting complement components or inhibiting complement activation has emerged as a promising therapeutic strategy for disease intervention [119]. Several complement inhibitors and modulators are currently under investigation in preclinical and clinical studies for their efficacy in attenuating neuroinflammation, preserving myelin integrity, and slowing disease progression in MS patients [70].
CIDP is an autoimmune disorder characterized by chronic inflammation and progressive demyelination of peripheral nerves, leading to motor and sensory dysfunction [120]. Despite extensive research, the underlying mechanisms driving CIDP pathogenesis remain elusive. However, growing evidence suggests a role for the complement system in mediating immune-mediated nerve damage in CIDP [121].
The complement system, a crucial component of the innate immune response, has been implicated in driving inflammation and tissue injury in CIDP [122]. Dysregulated complement activation leads to the generation of complement components and activation products, which contribute to immune cell recruitment, demyelination and axonal damage in peripheral nerves [123]. Genetic studies have identified associations between complement gene polymorphisms and susceptibility to CIDP, highlighting the genetic predisposition to complement dysregulation in CIDP [124].
Complement dysregulation contributes to the diverse clinical manifestations of CIDP, including weakness, sensory deficits, and gait disturbances [124]. Deposition of complement components and complement activation products at sites of nerve injury exacerbates inflammation and amplifies tissue damage in CIDP patients [75, 125]. Moreover, complement-mediated mechanisms may underlie treatment-resistant forms of CIDP, suggesting a potential role for complement-targeted therapies in disease management [126].
The complement system interacts with other immune mediators implicated in CIDP pathogenesis, including autoantibodies and pro-inflammatory cytokines [127]. Dysregulated complement activation may amplify autoimmune responses and promote tissue inflammation, contributing to nerve damage and clinical symptoms in CIDP [124].
Targeting the complement system represents a promising therapeutic strategy for CIDP management [126]. Several complement inhibitors, including monoclonal antibodies and small molecule inhibitors, are under investigation as potential treatments for CIDP [122]. These novel therapies aim to attenuate complement-mediated nerve damage and inflammation, offering potential benefits for CIDP patients refractory to conventional immunosuppressive agents.
Neurodegenerative and neuroimmune disorders represent a significant burden on global healthcare systems, characterized by progressive neuronal dysfunction and immune dysregulation. The complement system, a critical component of the innate immune response, has emerged as a key player in the pathogenesis of these disorders, offering potential targets for therapeutic intervention [128]. Complement-targeted therapies hold promise for modulating neuroinflammation, preserving neuronal integrity, and improving clinical outcomes in affected patients [129].
Complement-targeted therapies encompass a diverse array of interventions aimed at inhibiting specific complement components or pathways implicated in neurodegeneration and neuroinflammation [128]. Monoclonal antibodies targeting complement proteins or receptors block complement activation or promote complement clearance, thereby reducing neuroinflammatory responses and neuronal damage [130]. Small molecule inhibitors disrupt key enzymatic steps in the complement cascade, offering potent inhibitory effects on complement-mediated neurotoxicity [131].
Complement-targeted therapies have demonstrated efficacy across a spectrum of neurodegenerative and neuroimmune disorders including AD, PD, MS, and neuromyelitis optica spectrum disorder [132]. These therapies offer targeted approaches to suppress neuroinflammation, reduce neuronal damage, and preserve cognitive and motor function in affected patients [128]. Furthermore, complement-targeted therapies hold promise for combination therapies with existing neuroprotective agents, offering synergistic effects and enhanced treatment outcomes [133]. Complement-targeted therapies represent a paradigm shift in the treatment of neurodegenerative and neuroimmune disorders, offering personalized and precision medicine approaches tailored to individual disease phenotypes and patient characteristics [134]. Aim to precisely modulate specific components of the complement system, reducing unwanted side effects and improving therapeutic outcomes [135]. By targeting different parts of the complement cascade, these therapies can potentially treat a wide range of diseases, from autoimmune disorders to age-related conditions [135]. The development of complement-targeted therapies is grounded in extensive research and clinical trials, replacing long-held assumptions with evidence-based practices [135]. These therapies work by either inhibiting or modulating specific proteins within the complement system, which plays a key role in immune surveillance and tissue homeostasis. This targeted approach can prevent the cascade of inflammatory responses that contribute to disease progression [135]. Overall, complement-targeted therapies exemplify a shift towards more precise, effective, and personalized medical treatments, marking a significant advancement in the field of immunology and therapeutic development.
Despite existing challenges, the field of complement-targeted therapies in neurology continues to evolve rapidly, with ongoing efforts to develop novel therapeutic agents, refine treatment strategies, and explore combinatorial approaches [134]. Advances in biomarker discovery, patient stratification, and CNS drug delivery technologies hold promise for enhancing treatment efficacy and improving patient outcomes in neurodegenerative and neuroimmune disorders [131].
Complement-targeted therapies have emerged as promising treatment modalities for neurodegenerative and neuroimmune disorders, offering targeted approaches to modulate dysregulated immune responses and preserve neuronal integrity. However, the clinical translation of complement-targeted therapies faces several challenges, including issues related to drug specificity, treatment efficacy, safety profiles, and translational barriers. Understanding the limitations of current complement-targeted therapies is crucial for overcoming these challenges and improving treatment outcomes in neurologic disorders.
Lack of target specificity: Many current complement-targeted therapies lack specificity for the desired complement component or pathway, leading to off-target effects and potential adverse events [136].
Limited treatment efficacy: Despite their potential benefits, some complement-targeted therapies have shown limited efficacy in clinical trials, failing to achieve significant improvements in disease outcomes [25].
Risk of immunogenicity: Certain complement-targeted therapies may elicit immune responses, leading to the development of anti-drug antibodies and reduced treatment efficacy over time [130].
BBB penetration: Many complement-targeted therapies face challenges in crossing the BBB, limiting their access to the CNS and efficacy in treating neurologic disorders [102].
Complexity of complement regulation: The complement system is tightly regulated. Disrupting its balance with therapeutic interventions may lead to unintended consequences, exacerbating inflammation and tissue damage [137].
High treatment costs: Some complement-targeted therapies are associated with high treatment costs, limiting their accessibility to patients and healthcare systems [130].
Risk of thrombotic events: Certain complement inhibitors, particularly those targeting the terminal complement pathway, may increase the risk of thrombotic events, posing safety concerns in patients with underlying cardiovascular or thrombotic conditions [138].
Complex disease pathophysiology: Neurodegenerative and neuroimmune disorders are multifactorial diseases with complex pathophysiology, making it challenging to develop targeted therapies that effectively modulate complement-mediated mechanisms [139].
Heterogeneity of patient populations: Variability in disease presentation, progression, and treatment response among patient populations complicates the evaluation and optimization of complement-targeted therapies in clinical practice [134].
Future research should focus on developing more specific and efficacious complement inhibitors, optimizing treatment regimens, and identifying novel drug delivery strategies to enhance BBB penetration. Furthermore, the discovery of reliable biomarkers for patient stratification and treatment response monitoring will facilitate the personalized implementation of complement-targeted therapies in neurologic disorders.
Herbal bioactives have gained increasing attention as potential therapeutic agents for complement-targeted therapies, offering novel approaches to modulate dysregulated immune responses and mitigate inflammatory processes. This section provided a comprehensive review of herbal bioactives with complement-modulating properties, elucidating their mechanisms of action, therapeutic potentials, and clinical applications. We discussed the recent advancements in understanding the molecular interactions between herbal bioactives and the complement system, highlighting their efficacy in preclinical models of various diseases. Furthermore, we analyzed the challenges and opportunities associated with the development of herbal bioactives as complement-targeted therapies, underscoring their potential to complement existing treatment modalities and improve patient outcomes.
Herbal bioactives, derived from medicinal plants, have emerged as promising candidates for complement-targeted therapies due to their diverse pharmacological activities and favorable safety profiles [139]. Several herbal bioactives have been identified for their ability to modulate complement activation and regulation. Curcumin, a polyphenolic compound derived from turmeric, exhibits potent inhibitory effects on the complement cascade by targeting multiple complement components and pathways [140]. Similarly, resveratrol, a natural stilbene found in grapes and red wine, has been shown to attenuate complement-mediated inflammation and tissue injury in various disease models [141].
Herbal bioactives exert their complement-modulating effects through various mechanisms, including inhibition of complement activation pathways, modulation of CR expression, and enhancement of complement regulatory protein activity [139, 142]. By targeting different points of the complement cascade, herbal bioactives can effectively suppress excessive inflammation, tissue damage, and autoimmune responses associated with complement dysregulation [139, 142]. Furthermore, herbal bioactives exhibit pleiotropic effects on various cellular signaling pathways involved in immune regulation, oxidative stress, and inflammatory responses contributing to their therapeutic potential in diverse disease conditions [143].
Despite of existing challenges, herbal bioactives hold great promise as complement-targeted therapies for various diseases, offering potential benefits for patients as adjunctive or alternative treatment options. Future research endeavors should focus on elucidating the molecular mechanisms underlying the complement-modulating effects of herbal bioactives, optimizing their pharmacological properties, and conducting well-designed clinical trials to evaluate their safety and efficacy in human subjects.
Recent progress in the field of neurodegenerative and neuroimmune disorders has led to a growing interest in the potential therapeutic benefits of plant-derived bioactives. These natural compounds have shown promise as complement-targeted therapies, with their molecular interactions with the complement system being a key focus of research [130, 139, 144]. In particular, their effectiveness in preclinical models of neurodegenerative diseases like AD, PD, and MS has been highlighted [132]. The plant-derived bioactives that are enriched with a variety of phytochemicals have been found to possess complement-modulating properties, making them attractive candidates for the development of novel therapies for neurological conditions [75].
Numerous plant-derived bioactives have been identified for their ability to modulate complement activation and regulation (Table 1). Flavonoids such as quercetin, kaempferol rosmarinic acid, and apigenin exhibit potent inhibitory effects on the complement cascade by targeting key components of the CP and AP [145]. Targeting complement-mediated neuroinflammation and neuronal damage through plant-derived bioactives offers promising avenues for disease modification and symptom management in affected patients. Furthermore, standardization of herbal extracts, quality control measures, and regulatory considerations pose additional challenges to their clinical development and commercialization as complement-targeted therapies.
Complement inhibitory active ingredients from medicinal plants
| Sl No | Medicinal plants | Active ingredients | Actions on complement system | Reference(s) |
| 1 | Wedelia chinensis | Wedelosin | Inhibits CP and AP | [146] |
| 2 | Persicaria lapathifolia | Kaempferol glycoside and acylated quercetin glycosides | Inhibits CP | [147] |
| 3 | Rosemarinus officinalis | Rosmarinic acid | Inhibits CP and AP; binds C3; inhibits C5 convertase | [142, 148, 149] |
| 4 | Petasites hybridus | Sesquiterpin ester petasin | Inhibits C5a-induced calcium concentration | [150] |
| 5 | Jatropha multifida | Proanthocyanidin | Inhibits CP | [151] |
| 6 | Aloe vera | Polysaccharide | Inhibits AP | [152, 153] |
| 7 | Jatropha curcas | Curacycline A | Moderately inhibits CP | [154] |
| 8 | Centaurium spicatum | Quercetin | Complement inhibition | [155] |
| 9 | Piper kadsura | Piperlactam | Inhibits C5a-induced migration | [156] |
| 10 | Lithospermum euchromum | Polysaccharide | Inhibits CP and AP | [157] |
| 11 | Olea europaea | Apigenin and otherflavonoids | Inhibits CP | [158] |
| 12 | Crataegus sinaica | Whole extract | Inhibits CP and AP | [159] |
| 13 | Angelica acutiloba | Fucans | Inhibits CP and AP | [160] |
| 14 | Ascophyllum nodosum | Aqueous leaf extract | Complement inhibition | [161] |
| 15 | Trichilia glabra | Sesquiterpenes, quinines, coumarins, and flavonoids | Inhibits of CP than AP | [162] |
| 16 | Arnebia euchroma | Crude polysaccharides | Modulation of the complement system | [163] |
| 17 | Bupleurum chinense | Crude and acidic polysaccharides | Inhibits of the excessive activation of complement | [164] |
| 18 | Houttuynia cordata Thunb | Crude polysaccharides | Inhibits inappropriate activation of the complement system | [165] |
| 19 | Juniperus pingiivar | Polysaccharides | Inhibits the activation of complement | [166] |
| 20 | Scutellaria baicalensis | Flavonoids-enriched extract | Reduce complement deposition and decrease complement activation | [167] |
| 21 | Areca catechu | Areca nut extract | Inhibits the production of complement receptors (CR1, CR3, and CR4) | [168] |
| 22 | Curcuma longa | Curcumin | Upregulates the expression of factor H and complement regulatory proteins | [169, 170] |
| 23 | Boswellia serrata | β-Boswellic acid | Inhibits CP and AP and upregulation of conversion factor B | [171] |
| 24 | Essential oils from Myristica fragrans, Melaleuca quinquenervia, Syzygium aromaticum, Artemisia dracunculus; Coriandrum sativum, Juniperus communis, Melaleuca alternifolia, Zingiber officinale; Rosmarinus officinalis, Laurus nobilis, Cymbopogon martini, Melaleuca cajuputil, and Citrus limon; Thymus zygis, Coridothymus capitatus, and Thymbra capitata | (α/β)-Pinene, α-thujene, camphene, δ-3-carene, myrcene, 1,8-cineole, γ-terpinene, p-cymene, camphor, linalool, bornyl acetate, β-caryophyllene, terpinen-4-ol, α-humulene, α-terpineol, and borneol | Inhibits CP | [172] |
Sl No: serial number; CP: classical pathway; AP: alternative pathway; CR: complement receptor
This section provided an in-depth review of the mechanisms of action by which plant-derived compounds modulate the complement system. We discussed how these compounds influence complement activation, regulation, and effector functions through diverse molecular pathways. Furthermore, we highlighted recent advancements in understanding the therapeutic implications of plant-derived compounds in complement-related disorders, offering insights into the development of novel therapeutic strategies.
Inhibition of complement activation pathways: Many plant-derived compounds inhibit the activation of complement pathways, including the CP, LP, and AP [173]. Recent studies showed that kaempferol, quercetin, rosmarinic acid, and piperlactam can directly inhibit complementary pathways thereby preventing the activation of downstream complement components [142, 147, 156].
Modulation of CR expression: Plant-derived compounds can modulate the expression and function of CRs on immune cells, influencing their responsiveness to complement activation products. For instance, Areca catechu has been reported to downregulate the expression of CRs such as CR1, CR3, and CR4 on neutrophil responses and tissue damage [168].
Enhancement of complement regulatory proteins’ activity: Some plant-derived compounds can enhance the activity of complement regulatory proteins, such as factor H and factor I, which play crucial roles in controlling complement activation and preventing excessive tissue damage. Curcumin has been shown to upregulate the expression of factor H, leading to enhanced complement regulation and reduced inflammatory responses [169].
Induction of complement-inhibitory proteins: Certain plant-derived compounds can stimulate the production or secretion of complement-inhibitory proteins by immune and tissue cells, providing additional mechanisms for complement modulation. For instance, curcumin has been demonstrated to induce the expression of membrane-bound complement regulatory proteins like CD59, and B cell non-Hodgkin’s lymphoma cells, thereby protecting them from complement-mediated inflammation [171].
Antioxidant and anti-inflammatory effects: Plant-derived compounds have been found to possess both antioxidant and anti-inflammatory effects. These effects indirectly regulate complement activation and downstream effector functions. The C3 promoter has many oxidative stress and inflammation-related factors such as activator protein-1, IL-1β, p38, and CCAAT/enhancer-binding protein delta (C/EBPδ) [174–176]. Oxidative stress is closely associated with C3 activation. Mice with lower ROS levels showed a better neurological outcome with lower C3 levels [177]. As a result, antioxidant and anti-inflammatory potential phytochemicals can help reduce complement-mediated neuroinflammation and tissue damage.
Overall, plant-derived compounds exert their effects on the complement system through multifaceted mechanisms, offering potential therapeutic benefits for neurodegenerative and neuroimmune disorders (Figure 2). Further elucidation of these mechanisms will facilitate the development of novel complement-targeted therapies from natural sources.
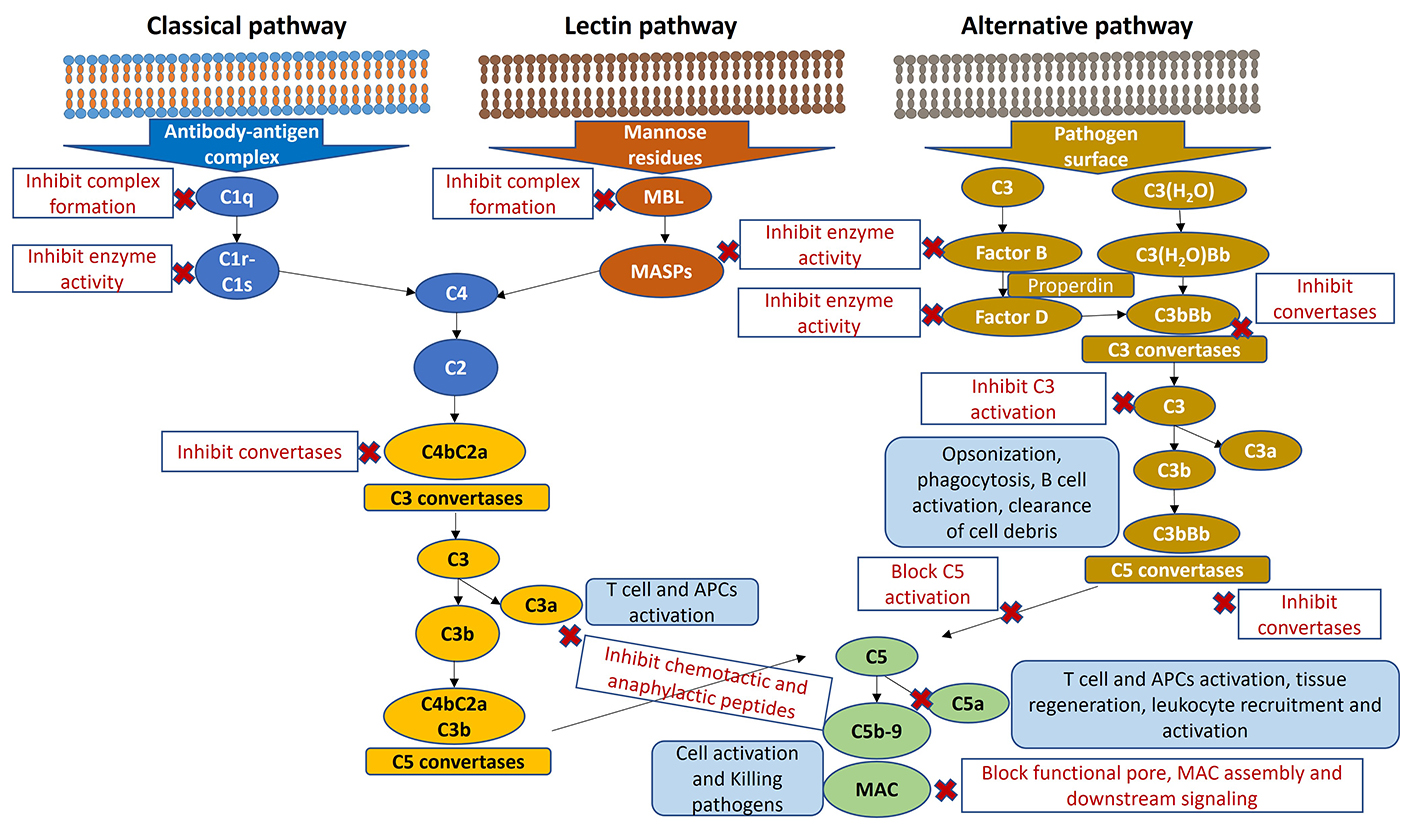
The complement system and herbal compounds therapeutic targets. MBL: mannose-binding lectin; MASPs: mannose-binding lectin-associated serine proteases; MAC: membrane attack complex; APCs: antigen-presenting cells
Understanding the mechanisms of action of plant-derived compounds on the complement system has important therapeutic implications for various diseases, including autoimmune disorders, inflammatory conditions, and neurodegenerative diseases. By targeting complement-mediated inflammation, tissue damage, and immune dysregulation, these compounds offer potential benefits for disease management and symptom relief.
In conclusion, the utilization of plant-derived compounds for targeting the complement system associated with neurodegenerative and neuroimmune disorders holds immense potential. The diverse array of bioactive molecules found in plants offers a rich source of therapeutic agents with multifaceted pharmacological properties, including anti-inflammatory, antioxidant, and neuroprotective effects. Through their ability to modulate complement activity, these plant-derived compounds offer a novel approach to addressing the underlying pathophysiological mechanisms of neurodegenerative diseases such as AD, PD, and MS. However, despite the considerable therapeutic potential of plant-derived compounds, several challenges and considerations must be addressed. These include issues related to bioavailability, pharmacokinetics, formulation optimization, and standardization of plant extracts. Furthermore, the translation of preclinical findings to clinical applications requires rigorous evaluation through well-designed clinical trials to establish safety, efficacy, and dosage regimens.
Aβ: amyloid-beta
AD: Alzheimer’s disease
AE: autoimmune encephalitis
ALS: amyotrophic lateral sclerosis
AMD: age-related macular degeneration
AP: alternative pathway
BBB: blood-brain barrier
CFH: complement factor H
CIDP: chronic inflammatory demyelinating polyneuropathy
CNS: central nervous system
CP: classical pathway
CRs: complement receptors
HD: Huntington’s disease
IBD: inflammatory bowel disease
IL-1β: interleukin-1β
LP: lectin pathway
MAC: membrane attack complex
MASPs: mannose-binding lectin-associated serine proteases
MBL: mannose-binding lectin
miRNAs: microRNAs
MS: multiple sclerosis
NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells
PD: Parkinson’s disease
RA: rheumatoid arthritis
SLE: systemic lupus erythematosus
JN: Conceptualization, Methodology, Writing—original draft. GG: Conceptualization, Writing—review & editing, Supervision, Funding acquisition. Both authors read and approved the submitted version.
Both authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
The study was financially supported by the ITC Limited, Kolkata, India. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 5331
Download: 59
Times Cited: 0
Martin Kolev ... Pascal Deschatelets
Célia Dos Santos ... Analía Sánchez-Luceros
Mariagiulia Spazzapan ... Roberta Bulla
