
Abstract
Nociplastic pain is the fourth category of pain defined in recent years. It is a pain arising from altered nociception, despite the lack of clear evidence of actual or threatened tissue damage that causes activation of peripheral nociceptors nor evidence for disease or lesion of the somatosensory system causing the pain. This type of pain is usually multifocal, more diffuse or intense than expected and it is usually associated with other central nervous system-derived symptoms, such as fatigue, sleep, memory, and mood problems. It can occur in isolation or as part of a mixed-pain state in combination with ongoing nociceptive or neuropathic pain. It is associated with increased social and sanitary costs due to the difficulty of adequately treating it. Its pathogenesis is still poorly understood, even if a mounting body of evidence suggests a pivotal role in inflammation and immunity, which may be triggered by an infection and/or a trauma. This narrative review aims to summarise the current knowledge about the interplay of the immune system and nociplastic pathways activation and amplification. The challenge for the future will be to identify the exact role of inflammation and immunity, the cause of this activation, and its link to other pathogenetic factors of nociplastic pain, such as diet or microbiota alteration, social and phycological factors, together with a genetic and epigenetic predisposition.
Keywords
Nociplastic pain, fibromyalgia, immunity, inflammationIntroduction
According to the International Association for the Study of Pain (IASP), pain is defined as “an unpleasant sensory and emotional experience associated with, or resembling that associated with, actual or potential tissue damage” [1] that involves biological, social, environmental, and psychological aspects. According to its pathogenesis, it can be categorized in different ways, such as nociceptive, neuropathic, and inflammatory. Nociceptive pain always originates from peripheral nociceptor activation in response to a noxious stimulus and has a protective role [2]. Neuropathic pain results from a lesion or a disease of the somatosensory system, causing its abnormal function [2]. Recent evidence suggests that acute pain can evolve into chronic pain if inflammation persists, leading to ongoing activation of pain networks [3].
In recent years, a third category of pain has emerged, i.e., nociplastic pain (NcP) that was defined as “a pain that arises from altered nociception, despite the fact that there is no clear evidence of actual or threatened tissue damage that causes activation of peripheral nociceptors, nor evidence for disease or lesion of the somatosensory system causing the pain”, according to the most recent IASP definition [4]. This kind of pain is usually reported as multifocal, diffuse, intense, or both, that expected and it is quite often associated with other central nervous system (CNS)-derived symptoms, such as fatigue, sleep, memory, and mood disorders. It can occur in isolation or as part of a mixed-pain state in combination with ongoing nociceptive or neuropathic pain. Examples of NcP conditions are chronic widespread pain and fibromyalgia (FM), chronic temporomandibular disorders, chronic pelvic pain syndromes, irritable bowel syndromes (IBSs), complex regional pain syndrome (CRPS), chronic low back pain of unknown causes (non-specific low back pain) [4, 5].
The diagnosis of NcP can be very challenging, since it is based on description of subjective symptoms, with no known clinical laboratory test that can confirm or exclude it [6]. Symptoms are commonly multiple, fluctuant, and may mimic other medical conditions, so that diagnosis is sometimes late since several exclusion steps are performed [6]. A recent paper suggests that NcP can be both under and over-diagnosed, and even misdiagnosed [7]. Finally, NcP can overlap with other pain syndromes and mental health disorders [7]. Recent evidence suggests a relationship between some form of NcP such as chronic temporomandibular disorders with other life factors such as lower levels of sleep quality [8], while an anti-inflammatory, fiber-rich diet or specific vitamin or probiotic supplementation could reduce the burden of FM by altering the gut microbiota [9].
The incidence of NcP is therefore difficult to estimate. It has been reported that FM alone has a prevalence of 2–4% in the entire population, which increases with age, and it ranked as the third most frequent musculoskeletal condition after back pain and osteoarthritis [10]. Chronic pelvic pain has an incidence of 12–27% in developed countries in women [11] and 2–10% in men [12]. About 10% of the population has IBS at any one time [13] while burning mouth syndrome and CRPS are relatively rare, with a respective incidence of 1.73% [14] and 0.07% [15].
The mechanisms that underlie these types of pain syndrome are not entirely clear, but proposed pathogenetic pathways are the amplified processing of or decreased inhibition of pain stimuli at multiple levels in the nervous system, or both [4]. Several spinal and supraspinal mechanisms have been identified as responsible for pain amplification or perpetuation [5]. The first is supraspinal hyper-responsiveness to pain stimuli, mediated by increased connectivity between brain regions such as the anterior cingulate cortex, the thalamus, and the medial prefrontal cortex [16, 17]. The second mechanism includes spinal pathways such as temporal summation or increasing pain perception after repeated noxious stimulus secondary to a so-called wind-up (i.e., an enhanced spinal neuron response after C-fibre or, less commonly, A-δ stimulation) and the expansion of receptive fields and hyperalgesia or allodynia, or both, secondary to central sensitization [18, 19]. Moreover, diminished descending modulation has been widely involved in NcP pathogenesis [20], while others proposed a central role of microglia in NcP development [21]. Recent evidence also suggests that dorsal root ganglia could be the primary NcP source, at least in some form, due to dorsal root ganglion’s ability to amplify and modulate painful transmission [22, 23]. Finally, also peripheral nerves have been involved in NcP, as a higher cross-sectional area of sural and vagus nerves in NcP patients than in healthy subjects was retrieved [24] and may be linked to neuropathy of the small A-delta and C fibers [25] or an activation of an immune-mediated neuro-inflammation [26]. All these mechanisms could interact each other, contributing to pain sensitization, both in bottom-up way (i.e., neurogenic peripheral inflammation) and in up-down direction (i.e., increased matrix connectivity or diminished descending modulation) [5].
The causes of NcP are still unknown. It was traditionally linked to emotional stress and psychological and social trauma. In recent years, the biopsychosocial model was proposed to explain NcP development: in this model, several triggers (such as infection, trauma, but also history of abuse, psychological trauma, environmental exposure, and genetic and epigenetic alterations of the genome) are advocated to promote NcP [4, 5, 27]. Others have proposed a significant association between immunity and inflammation and some form of NcP [28].
Strong evidence suggests a key role of a pro-inflammatory state and immune activation in all types of chronic pain [29, 30], and a mounting body of reports supports the role of inflammation, innate and adaptive immunity also in some form of NcP.
The present review aims to summarise the current knowledge about the interplay of the immune system and nociplastic pathways activation and amplification.
Materials and methods
The review was developed following the principles of the Scale for the Assessment of Narrative Review Articles (SANRA) [31]. A literature search was performed on the MEDLINE/PubMed (National Center for Biotechnology Information, NCBI) database; the search string contained free-text and/or Medical Subject Headings (MeSH) related to the following keywords: “chronic pain”, “diffuse pain”, “chronic pain disorder”, “nociplastic pain”, “functional pain”, “somatoform disorder”, “somatization”, “somatic symptom disorder”, and “widespread pain”, along with keywords adapted for individual conditions (e.g., “fibromyalgia”, “fibrositis”, “temporomandibular disorder”, “temporomandibular joint pain”, “chronic pelvic pain”, “irritable bowel syndromes”, “wide-spread pain”, “non-specific low back pain”, “complex regional pain syndrome”, “chronic fatigue syndrome”, “burning mouth syndrome”), combined with other keywords such as “immunity”, “immune system”, “neutrophils”, “natural killers”, “macrophage”, “cytokines”, “T cells”, “inflammation”, “inflammatory”, “immunological”, “antibodies”, “mast cells”, “B cells”, “glial cells”. No language or time restrictions were imposed. Papers were selected by the authors based on the relevance for the topic and using the following inclusion criteria: articles approved by an ethics committee or institutional review board if needed, articles, reviews, and systematic reviews were accepted. Finally, selected papers from the authors’ personal library were included. Case reports, commentary, and letters, as well as papers not relevant to the topic, were excluded. Data were independently collected by two investigators (AC, MG) with any discrepancy resolved by re-inspection of the original article by a third investigator (FP). To avoid transcription errors, the data were rechecked by a third investigator (AP).
Results
A total of 15,228 results were retrieved. After excluding duplicates, non-relevant articles (i.e., papers focusing on neuropathic pain), a subset of 67 articles [5, 20, 21, 26–28, 32–92] were included in the present narrative review. For schematic purposes, the literature retrieved was divided into two main blocks, the innate and the adaptive immune system. Figure 1 and Tables 1 and 2 summarise the main mechanisms involved and NcP condition studied.
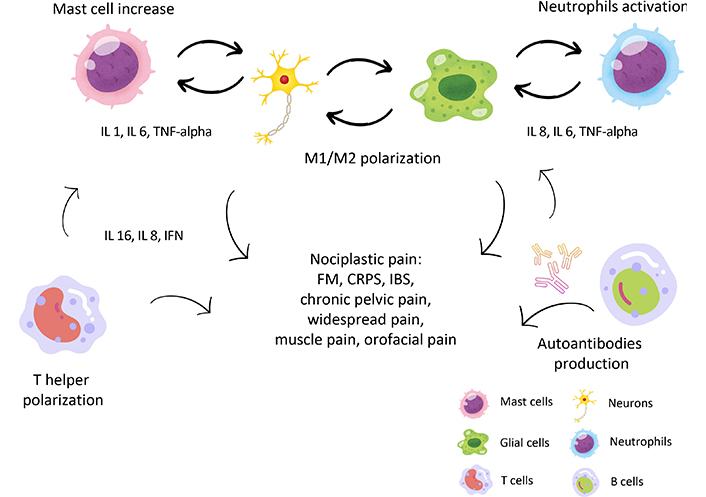
Different mechanisms of immune activation in nociplastic pain (NcP). The most important immune cells involved in NcP are shown: mast cells promote a pro-inflammatory state (through secretion, among others, of IL 1, IL 6, and TNF-alpha) and support bidirectional activation between neurons and glial cells. Also, neutrophils and NK cells contribute to releasing cytokines and sensitizing neurons and glial cells. Once stimulated, the latter undergoes both proliferation and shift to M1 phenotype, which causes further increased pro-inflammatory cytokine secretion. Also, the T cell phenotype switch is demonstrated in many forms of NcP, with an increase in helper (Th1 and Th17) and a reduction of regulatory T cells (Tregs), with an increase on the inflammatory milieu. Finally, an increase in B cells and autoimmune immunoglobulin G (IgG) production against glial cells and other antigens is shown in NcP, which supports inflammation. See text for details. IL 1: interleukin 1; TNF-alpha: tumor necrosis factor-alpha; IFN: interferon; FM: fibromyalgia; CRPS: complex regional pain syndrome; IBS: irritable bowel syndrome. Part of the icons made by Amethyst prime from www.freepik.com
Studies that explore the role of innate immunity and NcP
| Study and reference | Mechanism involved | NcP condition |
|---|---|---|
| Mast cells involvement | ||
| Blanco et al. [32] | Increased mast cells correlate with pain, fatigue | FM patients |
| Salemi et al. [33] | Increased mast cells and mediators such as IL 1, IL 6, TNF-alpha, IL 17 | FM patients |
| Murphy et al. [34] | Increased mast cells and mediators such as IL 1, IL 6, TNF-alpha, IL 17 | Chronic pelvic pain patients |
| Tsilioni et al. [36] | Increase in serum substance P which increases mast cell activation | FM patients |
| Liu et al. [41] | Higher levels of mucosal leptin, linked to increased levels of mast cells through the induction of IFN and the suppression of IL 4 | IBS patients |
| Li et al. [43] | Mast cell accumulation, activation, and degranulation in proximity to peptidergic nerve fibers were mediated by substance P | CRPS patients |
| Neutrophils involvement | ||
| Caxaria et al. [46] | Neutrophils invade sensory ganglia, increasing mechanical hyperalgesia and favouring sensitization of spinal cord neurons | FM patients |
| Microglia and astrocyte activation | ||
| Atta et al. [21] | M1/M2 polarization with an augmented pro-inflammatory signature (mediated by the M1 phenotype, which secretes TNF-alpha, IL 1β, and IL 6) | FM patients |
| Ye et al. [54] | Glial activation with an increase of pro-inflammatory cytokines and overexpression of purinergic receptors | Orofacial pain patients |
| Linan-Rico et al. [55] | Enteric glia activation, linked to toll-like receptor 4 signaling | IBS patient |
| Dodds et al. [56] | Glial activation by toll-like receptors, purinergic receptors, and chemokines | Female patients with pelvic pain |
| Lao et al. [57] | Microglia activation is mediated by purinergic receptors, TNF-alpha, and IL 1β | Male patient with pelvic pain |
| Tian et al. [58] | Astrocytes activation via metalloproteinase pathway | CRPS patients |
| NK recruitment | ||
| Verma et al. [60] | NK cells recruitment and pro-inflammatory cytokines spill over | FM patients |
NcP: nociplastic pain; FM: fibromyalgia; IL: interleukin; TNF: tumor necrosis factor; IBS: irritable bowel syndrome; IFN: interferon; CRPS: complex regional pain syndrome; NK: natural killers. MI/M2 microglia phenotypes (see text for details)
Studies that explore the role of adaptive immunity and NcP
| Study and reference | Mechanism involved | NcP condition |
|---|---|---|
| T helper involvement | ||
| Guggino et al. [65] | Th activation with an increase of a pro-inflammatory state, in particular by TNF-alpha | FM patients |
| Dolcino et al. [66] | Th17 polarization, associated with higher levels of CD4+ T cells and of sTGF-beta, IL 6, IL 21 | FM patients |
| Koike et al. [68] | High CD4/CD8 ratio identified as an independent variable for pain development | Burning mouth syndrome patients |
| Quick et al. [69] | T helper polarization with increased expression of interferon-gamma and IL 17A | Mouse model of pelvic pain |
| Chen et al. [70] | Th1, Th17, and Th22 cells increase in the peripheral blood with augmented pro-inflammatory state | Male patient with pelvic pain |
| Lenert et al. [73] | Reduction of peripheral Th2-cells that was linked to sustained inflammation | Mice model of chronic muscle pain |
| Chen et al. [74] | Th1 hyper-expression that supports persisting inflammation with increased levels of interferon-gamma associated with reduced IL 10 | IBS patients |
| B cell involvement | ||
| Fineschi et al. [76] | Elevated B cell levels and a heightened interferon-pro-inflammatory signature | FM patients |
| Goebel et al. [77] | IgG against satellite glial cells, neurons, myelin fibers, macrophages, and endothelial cells induces sensory hypersensitivity | Mouse model of FM |
| Ryabkova et al. [80] | Autoantibodies against collagen, gastric, pulmonary cells, and beta 2 glycoprotein | Chronic fatigue syndrome patients |
| Cuhadar et al. [81] | Pathological auto-IgG transferring enhances hyperalgesia | Mouse model of CRPS |
| Ohman et al. [82] | Augmented expression of B cells, IgG, and the co-stimulatory molecules CD80 and CD86 | IBS patients |
| Dunphy [83] | Auto-IgG against nucleolar autoantigen | Pelvic pain patients |
NcP: nociplastic pain; Th: T helper cells; TNF: tumor necrosis factor; FM: fibromyalgia; IL: interleukin; IgG: immunoglobulin G; CRPS: complex regional pain syndrome; IBS: irritable bowel syndrome
Innate immune system
The innate immune system is represented by mast cells, macrophages, neutrophils, natural killers (NK), and glial cells, which are immediately activated in response to an infection or a trauma, promoting a pro-inflammatory state, driven by the secretion of several cytokines and interleukins (ILs) such as IL 1, IL 6 and tumor necrosis factor-alpha (TNF-alpha), to defend the body from a pathogen or a lesion.
The role of the innate immune system in NcP pathogenesis is under investigation, with several reports that propose a key role of mast cells, neutrophils, microglia, and NK cells.
For example, the role of mast cells is supported by their significant increases in the skin of FM patients as compared to control [32]. This study also suggests that FM symptoms such as fatigue, headache, flushing, abdominal discomfort, hypotension, and tachycardia could be associated with mastocyte degranulation [32]. The release of active mediators by mast cells and their involvement in promoting a pro-inflammatory state (through secretion, among others, of IL 1, IL 6, TNF-alpha, IL 17) is also well established in FM [33]. This pro-inflammatory signature is, in turn, linked to central sensitization in several forms of NcP, such as FM and pelvic pain [34, 35]. Mast cell activation may also suppress Tregs activation and can activate neural sensitization, since they express tyrosin kinase (Trk) A receptor on their cell membrane and, therefore, nerve growth factor binding might cause their degranulation [34]. Furthermore, an increase in serum substance P was found in FM patients, which further increases mast cell activation, suggesting a two-way activation between the nervous system and mast cells [36]. In addition, mast cells can influence microglia activation, thus promoting NcP (see below) [37]. Elevated mast cell levels were also retrieved in chronic pelvic pain and colitis models [38–40]: for example, men with chronic pelvic pain syndrome as well as mice with an experimental model of chronic prostatitis showed increased mast cell tryptase and nerve growth factor [39]. Moreover, treatment of experimental autoimmune prostatitis with a mast cell stabilizer combined with a histamine 1 receptor antagonist resulted in a synergistic decrease in chronic pelvic pain [39]. Also in IBS, the role of mast cells is under investigation, since their activation has been associated with mucosal changes in the intestine of IBS, even if other factors such as diet and microbiota alteration could intervene to modulate mast cell density [40].
Recently, in IBS with diarrhoea a higher level of mucosal leptin was retrieved [41] that has been linked to the generation of a pro-inflammatory phenotype of mast cells through the induction of interferon (IFN) and the suppression of IL 4 [41]. Increased activation of mast cells has been consistently reported both in the ileum and in the colon of IBS patients and has been associated with increased gut permeability and increased severity of symptoms [42]. The pathogenetic role of mast cells is also recognized in a rat model of CRPS, in which mast cell accumulation, activation, and degranulation in proximity to peptidergic nerve fibers was mediated by substance P [43], while in burning mouth syndrome an increase of mast cell density was not consistently reported [44]. Also, persistent facial pain was linked to plastic changes in nociceptive signaling pathways involving various cells such as satellite glial cells, astrocytes, microglia, mast cells, and nociceptive neurons [45].
Among other cells of adaptive immunity, neutrophils are implicated in widespread pain pathogenesis in FM, invading sensory ganglia, increasing mechanical hyperalgesia, and favouring sensitization of spinal cord neurons [46]. Moreover, the transfer of neutrophils from primed mice and from patients with FM syndrome causes neutrophil infiltration into the dorsal root ganglia and favours mechanical hypersensitivity to recipient naïve mice [46]. Several cytokines secreted by neutrophils, such as IL 8 and IL 6, have been extensively identified in FM and correlated to symptom severity [47] while higher levels of TNF were identified in skin biopsies of CRPS patients but not in blood samples, suggesting a local pro-inflammatory activation [48]. Moreover, in FM patients with high neutrophil/lymphocyte ratio levels, the severity of pain seems higher [49].
The microglia activation and its M1/M2 polarization are well demonstrated in neuroinflammation and NcP such as FM with an augmented pro-inflammatory signature (mediated by the M1 phenotype, that secretes TNF-alpha, IL 1β, and IL 6) and a decreased anti-inflammatory state (mediated by the M2 phenotype that produces IL 10, IL 4, and IL 13) [21, 50]. At the spinal cord level, these modifications are modulated by purinergic receptors [51], toll-like receptors activation [52], chemokines, and substance P secretion [21]. Activated microglia express purinergic receptors, which, in turn, stimulate the release of proinflammatory cytokines and enhance neural depolarization [51], and express also toll-like receptors, which can promote an inflammatory response associated with persistent pain and with increased neurons firing rate [52]. Finally, high levels of substance P (as retrieved in cerebrospinal fluid of patients with FM) stimulate M1 phenotype switch and favour central inflammation [21]. Interestingly, treatment with naltrexone at low doses seems to reduce microglia activation, inducing a shift from an activated pro-inflammatory phenotype to an anti-inflammatory M2 phenotype. This action is mediated by the antagonism of toll-like receptor 4 and a metabolic shift of microglial cells into a suppressed phenotype, suggesting a role of low-dose naltrexone as a mediator of immuno-neuro-metabolic pathway in NcP [53]. The role of glial activation with an increased secretion of pro-inflammatory cytokines and overexpression of purinergic receptors was also well reported in orofacial pain [54] and in several forms of IBS, where enteric glia seems to be activated [55], linked to toll-like receptor 4 signaling, but also to increased production of macrophage colony-stimulating factor 1. The role of glial cells is also well-known in female pelvic pain [56], where glial activation by toll-like receptors, purinergic receptors, and chemokines enhance a pro-inflammatory milieu that facilitates neuronal transmission via increased glutamate release, reduced uptake, and suppression of inhibitory influences within the spinal cord [56].
Recent evidence suggested a pivotal role of astrocytes and microglia in both pain and psychosocial symptoms in chronic pelvic pain in males [57], mediated by purinergic receptors, TNF-alpha, IL 1β, chemokines among others Astrocyte activation contributes to the pathogenesis of pain hypersensitivity in a CRPS model, mainly by an increased activation of the phosphorylated c-jun N-terminal kinase 1/2 pathway, contributing to the pathogenesis of pain hypersensitivity [58].
NK cells are a subtype of cells derived from a lymphoid precursor, which are rapidly activated by major histocompatibility complexes in response to viral infection or cancer cell invasion [59]. Their expression was increased around peripheral nerves in FM patients, while circulating levels decreased, suggesting that, in persistent pain, peripheral nerves chronically recruit NK cells, leading to their extravasation and, in turn, promoting a long-lasting pro-inflammatory condition and perpetuating neural degeneration [60]. However, in women with symptomatic IBS, the activation of NK was significantly lower as compared to control [61].
The link between chronic inflammation and NcP is further corroborated by some reports that underline the role of the tryptophan (TRP)—kynurenine pathway in pain modulation. This pathway is activated during inflammatory state, resulting in elevation of oxidative compounds and neurotoxic compounds, such as 3-hydroxykynurenine and quinolinic acid, favouring the resulting neuroinflammation [62]. Higher levels of this axis activation were correlated with pain in temporomandibular disorders [63] and also with chronic fatigue syndrome and depressive symptoms in long COVID [64], suggesting an involvement of this metabolic pathway in central sensitization.
The adaptive immune system
The adaptive immune system involves both cell-mediated and humoral responses, activated after days or weeks following a trauma or an infection.
Cell-mediated responses are mainly represented by helper and suppressor T cells. Both these subtypes of T cells have been advocated in NcP pathogenesis. For example, in FM patients, an increase of Th was shown, and this pattern was associated with an increase in a pro-inflammatory state, mediated in particular by TNF-alpha [65]. Moreover, gene expression profiling in peripheral blood mononuclear cells obtained from FM patients showed a Th17 polarization [66], associated with higher levels of CD4+ T cells and of sTGF-beta, IL 6, IL 21, and IL 23 that promote Th17 differentiation, survival, and expansion in fibromyalgic patients were noted, suggesting an autoimmune pathogenesis for FM development. Finally, a very recent paper supports the role of Th1 polarization in FM and chronic fatigue symptoms development in patients with major depressive disorder, further suggesting an association between increased Th1 helper and macrophage activation, increased release of IL 16 and IL 8, and neurotoxicity [67].
In patients with burning mouth syndrome, low CD8+ cell count and high CD4/CD8 ratio were identified as independent variables for pain development, suggesting that the modulation of adaptive immunity could have a key role in its pathogenesis [68]. Experimental evidence suggests that T cells can mediate pelvic pain by activating T helper response [69]: In mice with an experimental model of induced chronic pelvic pain, CD4+ T cells expressed IFN-γ and IL-17A but not IL 4, promoting a Th1/Th17 immune signature. Th17 switch and IL-17A expression could, in turn, favour disease/inflammation. These findings were further corroborated by the observation that naive mice in which CD4+ T cells from experimental animals were injected, developed visceral hyperalgesia [69]. Other groups evidenced that Th1, Th17, and Th22 cells increase in the peripheral blood of men with chronic pelvic pain [70]. This abnormal auto-regulation of Th1 and Th17 drives inflammation, recruiting macrophages, promoting IL secretion, and favouring cell damage [71]. A Treg alteration with increased macrophages has been found in women with endometriosis, linked with infertility and chronic pain. Alteration of the regulatory function of T cells and an imbalance between T helper cells of the Th1 and Th2 types have been reported in women with endometriosis and chronic pain [72].
In a mice model of chronic muscle pain, an alteration of T regs and T suppressor away from immuno-regulation was noted, with a reduction of peripheral Th2-cells that was linked to sustained inflammation. Treatment with IL 5 successfully relieved persistent pain, suggesting a key role of IL 5, even if the effects of IL 5 treatment on T-cell populations, anti-inflammatory cytokine production, and neurons activation are not so evident [73].
Increased levels of IFN-gamma associated with reduced IL 10 were also retrieved in the intestinal mucosa of IBS patients, suggesting an imbalance between Th1 and Th2, toward an immunodominance pattern with Th1 hyper-expression that, in turn, supports persisting inflammation [74]. This pro-inflammatory status would influence intestinal mucosal permeability and the subsequent immunological response, further eliciting the Th1 response by microbiotic antigens. In addition, this Th1/Th2 shift may lead to abnormal sensation through the neuro-immune cross-talk [74]. Indeed, even if the hypothesis of NcP as an autoimmune disease mediated by hyperactive T cells is very stimulating, further investigation is needed to clarify their effective pathogenetic role.
The involvement of B cells in the pathogenesis of various NcP conditions has been increasingly recognised [75]. In FM patients, elevated B cell levels and a heightened IFN-pro-inflammatory signature have been observed [76]. A compelling study demonstrated that transferring serum IgG from FM patients—targeting satellite glial cells, neurons, myelin fibers, macrophages, and endothelial cells in the dorsal root ganglia—induces sensory hypersensitivity in mice by activating these cells [77]. These same antibodies were also found in the spinal ganglia of FM patients, supporting the hypothesis that FM is an autoimmune disease driven by antibodies against neurons and glial cells [77]. High levels of IgG against satellite glial cells have been linked to increased pain in FM patients, even though a specific autoantibody has not yet been identified [78]. Other researchers suggest that autoantibodies against myofascial-derived antigens could trigger neuronal hyperexcitability in the dorsal root ganglion, leading to pain hypersensitivity and central sensitization in FM [79]. Similar dysregulated autoantibodies have been found in chronic fatigue syndrome, with increased levels of autoantibodies against collagen, but also against gastric and pulmonary cells, and beta 2 glycoproteins [80]. Additionally, in CRPS, pathological autoantibodies were identified, and patients with more severe pain have higher autoantibody titers [81]. Moreover, transferring IgG from patients with CRPS to naive mice caused prolonged postsurgical hypersensitivity, sustained by A and C nociceptors sensitization [81]. Augmented expression of B cells, IgG, and the co-stimulatory molecules CD80 and CD86 may also contribute to inflammation in the enteric mucosa of IBS patients, either promoting or inhibiting inflammation [82]. In some cases of chronic pelvic pain syndrome, serum autoantibodies against specific nucleolar autoantigen have been identified [83], and anti-endometrial IgG has been detected in about 50% of women with endometriosis and pelvic pain [84].
All these results suggest a pathogenetic role of autoantibodies in NcP development, even if little is still known about the specific autoantibody responsible for NcP and why some patients present this increase contrary to others.
Conclusions
NcP is a real challenge for patients and physicians who are facing it due to its devastating impact on quality of life, its closed link to other physical symptoms (such as fatigue and insomnia), the psychological involvement (i.e., anxiety and depression), the increased social cost related to medication overuse and social burden such use increased divorces and suicide rates [85]. Understanding its pathophysiology is paramount to establishing a correct multimodal treatment strategy.
The biopsychosocial model is the most advocated one to explain NcP development: in this model, several triggers (such as infection, trauma, but also history of abuse, psychological trauma, environmental exposure, and genetic and epigenetic alterations of the genome) are advocated to promote NcP [4, 5]. Other authors have proposed a key role for inflammation and immunity which can be triggered by an infection, since symptoms associated with NcP have often been retrieved after hepatitis C infection, HIV infection, Lyme disease, Epstein-Barr viral infection, and, more recently, during the so-called long COVID syndrome [86–90]. Moreover, the bidirectional interaction between immune and nervous systems (i.e., immune cells can modulate nerve fibers function, and neurotransmitters and neuropeptides can influence immune cell activation) is well recognized in both acute and chronic pain [91], supporting the role of a pro-inflammatory state both peripherally and centrally in the pathogenesis of chronic pain [92]. In the peripheral nervous system, the activation of glial cells and immune cells (i.e., neutrophils and macrophages) with the subsequent release of pro-inflammatory mediators (such as TNF, ILs, prostaglandins) contribute to the hypersensitivity and hyperexcitability of nociceptor neurons (peripheral sensitization), through modulation of various ion channels of nociceptors, leading to transition to chronic pain [92]. Nociceptors, once activated, produce cytokines, and chemokines but also substance P that further promote neurogenic inflammation, favouring increased vascular permeability, leukocytes infiltration, and glial cell activation [93]. Moreover, neuroinflammation occurs also in CNS, sustained by glial, astrocytes, and neurons activation, resulting in the release of multiple pro-inflammatory mediators that finally induce hyperalgesia and allodynia (central sensitization) [92].
The link between neuroinflammation and chronic pain conditions such as FM, headache, temporomandibular disorder, back pain, IBS, primary headaches, pelvic pain, and vestibulodynia is emerging [93], even if the exact pathogenetic mechanism is yet to be understood. The challenge for the future will be the exact role of inflammation and immunity, their cause, and their link to other pathogenetic factors of NcP, such as diet or microbiota alteration, social and psychological factors, and genetic and epigenetic predisposition. The development of chronic widespread pain and FM is strictly linked with gender and age, with older women showing higher prevalence, but also other factors have been involved, such as genetic predisposition linked to specific nucleotide polymorphisms or particular lifestyle stress by mental and/or physical auto-overexertion, perfectionism traits and self-sacrificing behaviour [94]. The exact mechanism explaining the link between gender as well as psychological habits on inflammation and NcP could represent a novel area of research for the future.
Several evidence reporting a strict link between NcP condition and dietary influence are emerging [9], suggesting that specific supplementation with probiotics [95] or vitamins such as vitamin D [96] or coenzyme Q10 [97] could reduce inflammation in FM patients, as well as a plant-based or a gluten-free diet [98]. Also, poor sleep quality has been involved in NcP, as it suppresses pathways of descending pain inhibition [99]. Moreover, manual therapy has been demonstrated to improve mechanical hyperalgesia, as well as temporal summation and conditioned pain modulation in chronic musculoskeletal pain, even if the effects seem limited in a short time [100]. Future trials should focus on the effect of lifestyle changes such as exercise, manual therapies, and dietary and microbiota modification on NcP and immune-inflammatory mechanisms.
Key conclusions
NcP is a real challenge for patients and physicians who are facing it due to its devastating closed link to other physical symptoms (such as fatigue and insomnia), the psychological involvement (i.e., anxiety and depression), the increased social cost related to medication overuse and social burden such use increased divorces and suicide rates.
The biopsychosocial model is the most advocated one to explain NcP development: in this model, several triggers (such as infection, trauma, but also history of abuse, psychological trauma, environmental exposure, and genetic and epigenetic alterations of the genome) are advocated to promote NcP.
The link between neuroinflammation and chronic pain conditions such as FM, headache, temporomandibular disorder, back pain, IBS, primary headaches, pelvic pain, and vestibulodynia is emerging, even if the exact pathogenetic mechanism is yet to be understood.
This narrative review summarizes the current knowledge about the interplay of the immune system and nociplastic pathways activation and amplification.
Future research directions should focus on the exact role of inflammation and immunity, their cause, and their link to other pathogenetic factors of NcP, such as diet or microbiota alteration, social and psychological factors, and genetic and epigenetic predisposition.
Abbreviations
| CNS: | central nervous system |
| CRPS: | complex regional pain syndrome |
| IASP: | International Association for the Study of Pain |
| IBSs: | irritable bowel syndromes |
| IFN: | interferon |
| ILs: | interleukins |
| NcP: | nociplastic pain |
| NK: | natural killers |
| TNF-alpha: | tumor necrosis factor-alpha |
| TRP: | tryptophan |
Declarations
Author contributions
MG and AC: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. AP: Investigation, Writing—original draft, Writing—review & editing. FP: Conceptualization, Validation, Writing—review & editing, Supervision. GV: Validation, Writing—review & editing, Supervision. All authors read and approved the submitted version.
Conflicts of interest
Giustino Varrassi, the Editorial Board Member and a Guest Editor of Exploration of Immunology, had no involvement in the decision-making or the review process of this manuscript. The other authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2025.
Publisher’s note
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.