
Abstract
Wound healing is an area of growing importance in the healthcare field, especially chronic wounds associated with comorbidities like diabetes mellitus (DM), hypoxic stress, obesity, and malnutrition. Chronic wounds significantly increase healthcare costs and reduce patients’ quality of life. Cytokines are a promising therapeutic target, as they regulate all stages of wound healing, and dysfunction in cytokine production can cause inflammatory non-healing wounds. Interleukin-1 (IL-1), IL-2, IL-6, IL-8, and tumour necrosis factor-α (TNF-α) facilitate leukocyte recruitment and clear dead cells during the initial inflammation stage while transforming growth factor-β (TGF-β), IL-4, and IL-13 inhibit inflammation and stimulate proliferation of fibroblasts to begin extracellular matrix (ECM) deposition. Given the complexity of cytokine interactions and their diverse cellular targets, a comprehensive understanding of these signaling pathways is crucial. This review examines the multifaceted roles of cytokines in wound healing and discusses recent advancements in the therapeutic application of cytokine modulation for improved wound care outcomes. Despite significant advancements in improving the specificity of cytokine therapies, further research is needed to focus on targeting downstream signaling pathways or specific receptors to minimize the adverse effects associated with these treatments.
Keywords
Wound healing, cytokines, wound regeneration, interleukins, growth factorsIntroduction
Wound healing is a complex and dynamic process which involves the coordinated actions of many different tissues and cell lineages [1]. It involves highly intricate and coordinated processes—namely hemostasis, inflammation, proliferation, and remodelling (Figure 1).
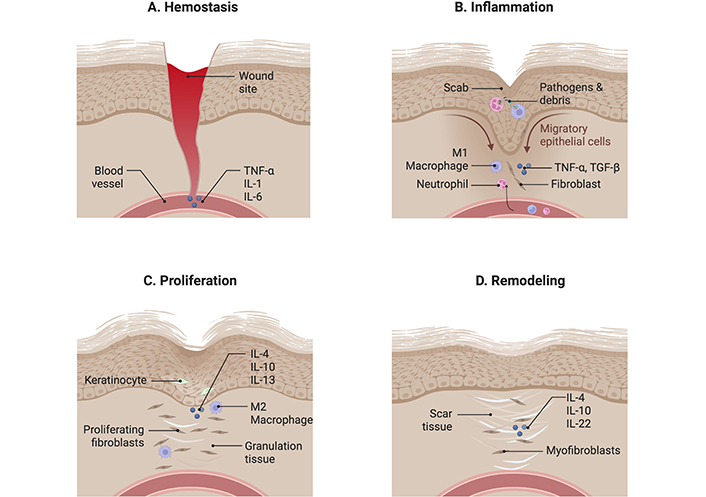
Wound healing is a complex and dynamic process comprising four distinct phases. (A) Hemostasis: This initial phase involves the body reducing blood loss by activating clotting factors to stem bleeding. Cytokines such as tumour necrosis factor-α (TNF-α), interleukin-1 (IL-1), and IL-6 are released to enhance clot formation. (B) Inflammation: Following hemostasis, cytokines like TNF-α and transforming growth factor-β (TGF-β) promote the chemotaxis of pro-inflammatory mediators, including neutrophils, macrophages, and fibroblasts, to the wound site. These cells work to clear cellular debris and pathogens. (C) Proliferation: This phase begins with the release of anti-inflammatory cytokines such as IL-4, IL-10, and IL-13. These cytokines stimulate granulation tissue formation and inhibit further release of inflammatory mediators, facilitating tissue repair. (D) Remodelling: During the final phase, IL-4, IL-10, and IL-22 contribute to the reorganization of collagen fibers, increasing the strength of the scar tissue and enhancing wound integrity. Created using BioRender (https://bioRender.com/o00w689)
The method includes extracellular matrix (ECM) formation, pathogen clearance through phagocytosis, and signaling coordination. The movement of these molecules to the wound site is driven by cytokines. Cytokines are soluble proteins that act as chemical signals facilitating communication between immune cells and are the primary mediators of an immune response [2]. They can be divided into 2 broad categories—pro-inflammatory and anti-inflammatory cytokines. Pro-inflammatory cytokines stimulate the immune system and cause vasodilation, to recruit more inflammatory mediators to the site of inflammation. These pro-inflammatory cytokines include tumour necrosis factor (TNF), interleukin-1 (IL-1), IL-6, and IL-8. Anti-inflammatory cytokines counteract the activity of pro-inflammatory cytokines, as they prevent the recruitment of inflammatory mediators to reduce inflammation and prevent an excessive immune response. Anti-inflammatory cytokines include IL-4, transforming growth factor-β (TGF-β), interleukin-1 receptor antagonist (IL-1Ra), and IL-13. The regulation of the balance between pro- and anti-inflammatory cytokines is crucial to ensuring that wound healing happens efficiently, with an imbalance of cytokines resulting in delayed wound healing or increased scarring, predisposing the patient to infections. Due to the pivotal role that cytokines play in immunity; they are attractive targets for therapeutic intervention in wound healing [3]. The biocompatible green zinc oxide nanoparticles, synthesized using Althaea officinalis chitosan gel, offer an excellent therapeutic approach for accelerating diabetic wound healing [4].
In our local context, wound healing is particularly significant not only due to surgical interventions but also as part of the broader ‘War on Diabetes’ initiative, addressing the high prevalence of diabetes-related chronic wounds. Singapore has the second highest prevalence of diabetes mellitus (DM) among developed nations, with the crude prevalence of DM increasing in recent years, now affecting nearly 10% of the population [5]. Complications of DM are common and include concerns such as nerve damage, which can cause wounds to go unnoticed, leading to a higher risk of infection and impaired healing. Further exploration into the use of cytokines in developing wound healing therapeutics holds significant potential to improve the quality of life and health outcomes for patients with conditions such as diabetes. This review will first outline the normal physiological process of wound healing, then examine the roles of specific cytokines and current therapeutics targeting them, and finally, discuss potential areas for future research and development.
Wound healing
Current approaches to wound healing
To aid wound healing and reduce the burden it places on the patient and healthcare systems; various therapeutic approaches have been developed [6]. The basic methods of cleansing, debridement, treatment with antimicrobials and dressing are universal across all wound healing approaches, but significant advancements have been developed to enhance each stage of the wound healing process [7]. The current approaches to wound healing are split into four categories, namely instrument-based therapy, wound dressings, cell therapy, and platelet therapy.
Instrument therapy is an approach that aids wound healing by altering the surrounding conditions of the wound to hasten healing, through methods such as negative wound pressure therapy, hyperbaric oxygen therapy, and low-level laser therapy. Negative wound pressure therapy uses suction to reduce wound oedema, improve tissue perfusion, stimulate granulation tissue and angiogenesis, and significantly increase the rate of granulation tissue formation and diabetic foot ulcer (DFU) healing in a randomized controlled trial [8, 9]. Alternatively, hyperbaric oxygen therapy involves the administration of pressurised oxygen to patients via hyperbaric chambers. It has been shown to improve the healing of chronic wounds through various factors such as reduced oedema, increased angiogenesis, and increased re-epithelization [10]. At a more topical level, low-level laser therapy shines diode lasers at different wavelengths directly onto wounds. This stimulates basic fibroblast growth factor (FGF) and vascular endothelial growth factor (VEGF) secretion and hence aids collagen production and wound closure [11].
Similarly, wound dressings provide a localized environment that promotes cellular migration and reduces infection risks [12]. Primarily, dressings aim to shield the exposed tissue from the surrounding environment, preventing pathogens from entering and infecting the wound. However, the advent of modern wound dressings involves the incorporation of biologically active materials or drugs into the dressing to enhance healing [13]. For example, hyaluronic acid (HA) wound dressings can moderate both inflammation and angiogenesis [14] and absorb exudate from the wound while maintaining wound hydration [15]. Collagen dressings can similarly absorb exudate and maintain wound hydration, but can also aid in hemostasis through platelet binding, as well as promoting angiogenesis, collagen deposition, and chemotaxis of fibroblasts, macrophages, and epithelial cells [16]. Although modern dressings incorporate diverse materials each suited for different applications, there are still limitations in their implementation.
Instead of using synthetic or foreign materials to shield the wound, cell therapies aim to directly replace the damaged tissue using materials similar to the patient’s normal skin. This can be done through therapies such as grafts, which can be further classified into autografts, allografts, and xenografts [17]. These allow for faster closure of the wound without the need for the body to regenerate all the damaged tissue, especially if the wound damage is extensive [18]. Stem cells are another form of cell therapy, which involves the transplantation of mesenchymal stem cells (MSCs) from adipose tissue, bone marrow, or other sources directly onto the wound site [19]. It offers alternatives to chronic wound healing due to its potential for angiogenesis and inflammation reduction [20], particularly in cases where interrupted blood supply to local tissue hinders chemotaxis and hence wound healing.
Platelet therapy involves autologous platelet-rich plasma (PRP) applied directly to the wound site. PRP therapy has been demonstrated effective in modulating inflammation and promoting re-epithelialization, angiogenesis, and wound closure by enhancing the expression of cytokines, including insulin-like growth factor-1 (IGF-1) and VEGF [21]. It has been utilised in the healing of both dermal wounds and deeper musculoskeletal conditions, such as osteoarthritis [22]. The effectiveness of administering concentrated platelets is promising, as it provides high levels of cytokines to the wound site to enhance healing [23] and opens up further avenues into streamlining this therapy to focus on individual cytokines in various stages of wound healing.
Many therapies are focused on topical treatment, especially different forms of wound dressing, to provide a more ideal environment for wound healing [24]. These approaches are effective, but only in specific situations, and lack coverage for all phases of wound healing. A novel area of research that targets all four phases of wound healing is cytokine therapy, which takes an alternative approach at a cellular level to enhance wound healing and reduce fibrosis [25].
Phases of wound healing
Understanding the process of wound healing is crucial in developing therapies targeted at specific mechanisms. The phases of wound healing are split into four parts: hemostasis, inflammation, proliferation, and maturation [26, 27]. The initial phase of wound healing begins with hemostasis, where platelet activation and aggregation occur around the site of injury. This is initiated by platelet contact with exposed collagen fibers, causing the release of adenosine diphosphate which stimulates platelet aggregation, as well as factor VIII, which accelerates clot formation [28]. Triggering of both the extrinsic and intrinsic clotting pathways causes the formation of thrombin and the subsequent conversion of fibrinogen into fibrin, forming a clot together with platelets. The release of cytokines TNF-α, IL-1, and IL-6 also causes endothelial cells to secrete more pro-inflammatory, procoagulant, and antifibrinolytic components, activates platelets, and promotes adhesion around the wound site. They also cause increased tissue factor expression on cell surface membranes for activation of factor VII and initiation of the coagulation cascade [29]. The shift in phase from hemostasis to inflammation is marked by the increase in cytokines that cause the recruitment and proliferation of fibroblasts [30].
The inflammatory phase is the result of lymphocyte recruitment, with the surrounding epithelial cells recognizing the danger-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) from the damaged skin. This activates the toll-like receptors (TLRs) located on the surface of the cell and causes the release of cytokines in response [31]. Cytokines such as TGF-β and TNF-α recruit neutrophils and macrophages to the injury, initiating phagocytosis and release of reactive oxygen species (ROS) [32] to destroy the cellular debris, pathogens, and foreign materials [33].
Following the inflammatory phase, the proliferative phase begins, involving re-epithelialization, angiogenesis, and the formation of granulation tissue. Re-epithelization refers to the recruitment and proliferation of keratinocytes to reform the basement membrane and the above dermis and epidermis [34]. Angiogenesis is initiated by the release of growth factors such as VEGF in response to hypoxic conditions, causing the migration and proliferation of endothelial cells to form new blood vessels [35]. Granulation tissue formation is a result of these two processes and is also a result of fibroblast activity. Fibroblasts are stimulated by growth factors to produce matrix metalloproteinases (MMPs) to degrade the fibrin clot, promoting cell migration, as well as ECM molecules like HA and collagen to replace the clot and better support re-epithelialization and keratinocyte migration [36]. In addition, the anti-inflammatory cytokines IL-4, IL-10, IL-13, and TGF-β are released by M2 anti-inflammatory macrophages to reduce the production of ROS and neutrophil recruitment so that the wound can begin to heal [37].
The final step of remodelling aims to improve the tensile strength of the wound site via reorganisation, degradation, and reformation of the ECM, converting the type III collagen into type I [38]. This process is mediated primarily by myofibroblasts, which not only secrete MMPs to degrade the ECM but also aid in wound contraction and ECM reorganisation with type I collagen and elastin [39]. In this stage, TGF-β, IL-4, IL-10, and IL-22 are secreted to regulate the balance between MMPs and tissue inhibitors of metalloproteinase (TIMPs), stimulate fibroblast migration, proliferation and differentiation into myofibroblasts and increase ECM deposition [32].
Throughout the 4 stages of wound healing, cytokines play a vital role in regulating the immune response, inflammation, and repair of the wound. Since the resolution of the inflammatory phase is essential for wound closure, this paper will focus on the cytokines involved in the inflammatory stage of wound healing and explore how they can be targeted to resolve inflammation in both acute and chronic wounds, facilitating progression to subsequent healing phases.
Factors affecting wound healing
Wound healing is a highly complex and dynamic process, involving coordinated efforts by multiple pathways, and is affected by many external and internal factors. Wounds are classified into two categories—acute and chronic wounds, each with different prognosis and treatment methods. Typical acute wounds heal within 4–6 weeks with minimal complications [40]. However, some wounds fail to progress through the stages of wound healing at the expected speed, eventually becoming chronic wounds. This can be attributed to local tissue hypoxia, bacterial colonisation of the wound, repetitive ischemia-reperfusion injury, and an altered cellular and systemic stress response in patients of older ages, all of which result in altered immune responses [41]. Multiple factors impact rates of wound healing and understanding and managing these factors aid in ensuring that acute wounds heal quickly and do not progress into chronic wounds. The factors affecting wound healing can be split into local and systemic factors (Figure 2).
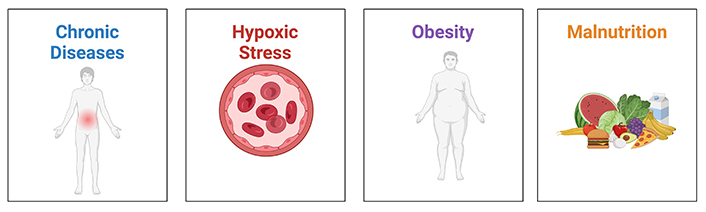
Factors affecting wound healing. These 4 factors—chronic diseases, hypoxic stress, obesity, and malnutrition contribute to poorer wound healing in an individual. Chronic diseases and obesity cause a poorer environment for wound healing, involving complications such as systemic inflammation. Hypoxic stress may result in worsened cellular function and poor angiogenesis, and malnutrition depletes the dietary sources of nutrients necessary for wound healing. Created using BioRender (https://bioRender.com/d25p430)
Chronic diseases
Chronic diseases such as DM can have complex interactions with wound-healing processes. DM can cause many complications with physiological wound healing, through vasculopathies, neuropathies, increased infection risks, cellular abnormalities severely lengthening healing times, and contributes to a major risk factor of DFUs [42, 43]. Similarly, atopic dermatitis arising from chronic type 2 inflammation mediated by IL-4, IL-5, and IL-13 resulted in eosinophil recruitment, chronic inflammation, and tissue damage, all of which slowed wound healing times and contributed to the formation of a chronic ulcer [44].
Hypoxic stress
Acute tissue hypoxia occurs after an injury, where the surrounding vessels are injured and cannot adequately supply the wound site with oxygen. This results in the increased expression of hypoxia-inducible factor-1 (HIF-1), which is involved in immune modulation, angiogenesis, and growth factors to generally aid wound healing [45]. However, chronic tissue hypoxia causes overexpression of HIF-1, resulting in inflammation, ROS production, and poor ECM deposition. Hypoxia additionally impaired angiogenesis within the wound, hindering cell migration to the wound site and thus slowing granulation tissue formation and wound healing [46]. Hence, ensuring adequate tissue perfusion is vital to prevent prolonged hypoxia and ensure timely healing.
Obesity
Obesity can contribute to impaired wound healing, as higher numbers of adipocytes contribute to increased production of pro-inflammatory cytokines such as TNF-α, IL-1, IL-6, and IL-8. This creates a persistent inflammatory state and suppresses immune responses in the inflammatory phase [47]. The large adipocytes grow faster than the vessels that supply them, leading to tissue hypoxia and nutritional deficiencies. Adipocytes also secrete inhibitors of angiogenesis [48], further reducing blood supply to surrounding tissue and creating an environment unconducive to wound healing. Hence, sustained inflammation and tissue damage, together with impaired immune responses and angiogenesis can lead to the formation of chronic wounds.
Malnutrition
Proper nutrition is vital for wound healing, with adequate levels of both micronutrients and macronutrients being necessary in all stages of healing. Proteins are necessary for collagen production and ECM deposition, with specific amino acids such as arginine aiding inflammation, collagen production, and macrophage function. Similarly, the consumption of carbohydrates and lipids provides energy for cellular proliferation, and lipids facilitate the absorption of fat-soluble vitamins A, D, E, and K which enhance immune responses, reduce oxidative stress, and aid coagulation [49]. Malnutrition has been linked to impaired cytokine release and immune response, placing patients at risk of infection and dysregulated wound healing [50]. Providing patients who are malnourished with preoperative nutritional supplements before surgical procedures are linked to reduced complications such as impaired wound healing, fewer postoperative infections, and shorter hospitalisations [51]. Hence, ensuring adequate nutrition is vital for wound healing, especially due to the prevalence of malnutrition among diabetic, elderly and obese patients, all of whom are at risk for chronic wounds [52].
Cytokines
TNF-α
TNF-α is an inflammatory cytokine released primarily from macrophages, T-lymphocytes and natural killer (NK) cells in response to pathogens or trauma. It has two forms, soluble-bound TNF-ɑ (sTNF-ɑ) and membrane-bound TNF-ɑ (mTNF-ɑ), of which the latter is processed by TNF-α-converting enzyme into the soluble form for release [53]. TNF-α signaling results are varied, playing a role in both therapeutic effects in immunomodulation, infectious pathogen resistance, and tumour resistance, as well as pathogenic effects by causing autoimmune diseases such as rheumatoid arthritis (RA) and worsening infections through excessive inflammation [54]. TNF-α has two main receptors, TNFR1 and TNFR2, with each receptor having different downstream effects.
TNFR1 is classified as a death receptor due to its death domain (DD) and is present in most cells. It binds to both sTNF-ɑ and mTNF-ɑ, after which it forms a trimer and triggers the formation of complex I. Complex I signals the downstream nuclear factor κB (NF-κB) and mitogen-activated protein kinase (MAPK) pathways, leading to inflammation, cell survival, proliferation, and immune activation [55]. Disruption of complex I formation can lead to complex IIa or IIb formation which results in apoptosis via caspase 8. If receptor-interacting protein kinase-1 (RIPK1), the kinase that forms complex I, cannot be degraded following the formation of complex IIa or IIb due to insufficient caspase, complex IIc is formed, and cell death occurs via necrolysis [56].
Similarly to TNFR1, TNFR2 also binds to both sTNF-ɑ and mTNF-ɑ but has a higher affinity for mTNF-ɑ. TNFR2 is mainly expressed on the surface of myeloid cells, T-lymphocytes, B-lymphocytes, endothelial cells, glial cells, and cardiomyocytes [57]. TNFR2 activation and effects are mainly through canonical or non-canonical NF-κB pathways. Canonical NF-κB pathways are similar to those activated in TNFR1 and result in the expression of increased inflammatory response, while non-canonical NF-κB pathways occur more slowly and result in cell proliferation and survival [56]. However, simultaneous activation of both TNFR1 and TNFR2 can contribute to cell death signaling, since TNFR2 canonical NF-κB pathway activation depletes the availability of TRAF2-cIAP1/2 in the cytoplasm, interfering with RIPK1 ubiquitination and causing complex IIa, IIb, and IIc formation, hence having an additive effect on TNFR1 stimulation [58].
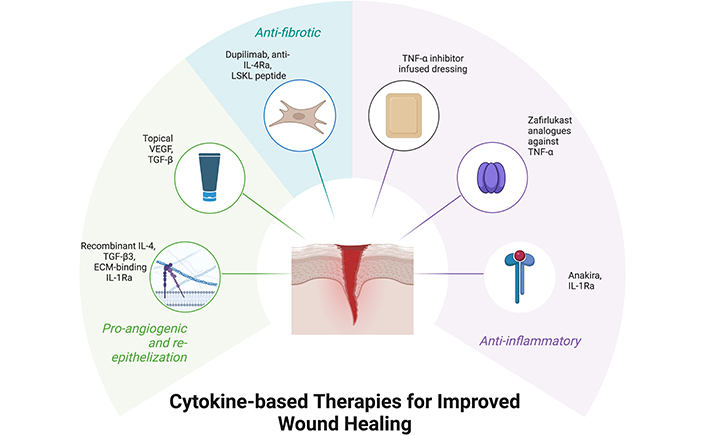
Due to the vital role TNF-α plays in inflammation, it has been linked to many different pathologies and is a target for therapeutic intervention. Excessive TNF-α production is associated with autoimmune diseases such as RA, psoriatic arthritis, and inflammatory bowel disease, and TNF-α inhibitors such as infliximab have been effective treatment options [53]. However, despite the utility of these TNF-α inhibitors, they are not without their risks, with some patients facing psoriatic cutaneous reactions [59, 60] and worsened granulomatous disease [61]. These adverse reactions are likely due to the non-specificity of current TNF-α inhibitors blocking both TNFR1 and TNFR2, causing impaired immune response. With TNFR2 playing a role in enhancing immunomodulation via regulatory T (Treg) cells and mesenchymal-derived suppressor cells, as well as having a protective effect against cardiac disease and multiple sclerosis [58], developing drugs targeting TNFR1 and TNFR2 is an avenue for reducing the side effects and enhancing the specificity of TNF-α therapy. Drugs such as zafirlukast analogues are in development that selectively inhibits TNFR1, targeting the inflammatory mechanisms of TNF-α while retaining the beneficial effects of TNFR2 stimulation [62].
Similar to the effects discussed earlier, TNF-α also has both normal physiological roles and pathological roles in wound healing. TNF-α is one of the first few types of inflammatory cytokines to be released upon trauma and tissue injury, stimulating neutrophil and macrophage recruitment, signaling for downstream release of inflammatory cytokines, and triggering the differentiation of macrophages to the M1 pro-inflammatory phenotype (Figure 3A) [63].
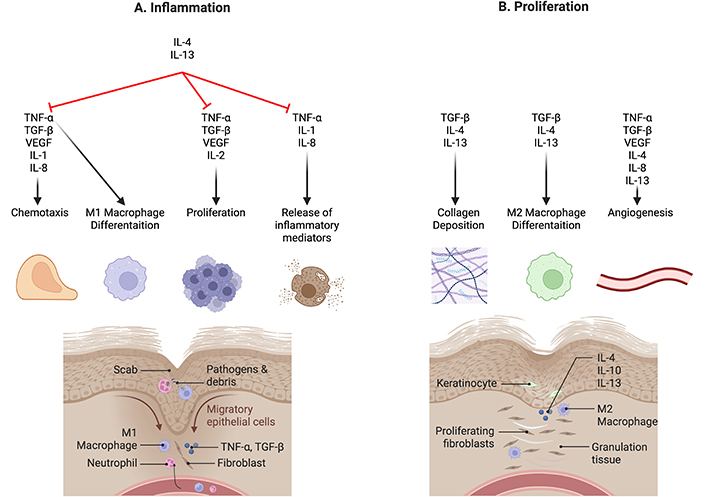
Cytokines in wound healing. (A) Inflammation: During the various stages of wound healing, cytokines are involved in fluctuating concentrations to regulate the process. Pro-inflammatory cytokines such as IL-1, IL-2, IL-8, and TNF-α drive inflammatory responses essential for clearing pathogens and cellular debris. (B) Proliferation: In contrast, anti-inflammatory cytokines like IL-4 and IL-13 suppress these inflammatory processes, promoting collagen deposition and M2 macrophage differentiation. This transition shifts the wound environment from a state of inflammation to one of proliferation, facilitating tissue repair and regeneration. IL-1: interleukin-1; TNF-α: tumour necrosis factor-α; TGF-β: transforming growth factor-β; VEGF: vascular endothelial growth factor. Created using BioRender (https://bioRender.com/v27x356)
Deficiencies in TNF-α levels can cause reduced leukocyte and fibroblast recruitment to the site of injury, causing delayed wound healing [64]. However, TNF-α overexpression has also been linked to injury complications, particularly those mediated by macrophage responses. TNF-α inhibition has been effective in cardiac and bone tissue regeneration, as well as aiding burn wound healing when delivered via HA dressing [65]. TNF-α is additionally associated with recurrent oral ulcers, and administering TNF-α inhibitors to oral ulcers resulted in reduced neutrophil recruitment and reduced inflammation, but also reduced fibroblast migration, maturation, and angiogenesis due to the lack of neutrophilic chemokines and growth factor release [66]. This is seen in patients with Behçets’s disease, who present with multiple non-healing ulcers which are most commonly located in the oral and ocular regions. Adalimumab, which is a monoclonal anti-TNF-α antibody, binds to TNF-α, preventing receptor binding and activation. This action reduces the pro-inflammatory effects of TNF-α and prevents further immune damage caused by excessive inflammatory mediators [67]. In a study involving 122 patients with Behçets disease, 60% exhibited oral ulcers prior to receiving treatment with adalimumab. After 56 weeks of treatment with adalimumab, the proportion of patients presenting with oral ulcers decreased to just over 20% [68]. Hence, TNF-α is a vital component of wound healing in a normal physiological state, and its levels must be maintained at these levels to prevent impaired wound healing.
TGF-β
TGF-β is a growth factor which regulates wound healing through inflammation, proliferation, and re-epithelization, and although each of the three isoforms is present throughout all phases of wound healing, each isoform exerts different effects on wounded tissue and has spikes in different phases [69]. On tissue injury, TGF-β is released by platelets, providing a rapid response and recruiting other immune cells like neutrophils to the site of the wound, which subsequently release more TGF-β. During the proliferative phase, TGF-β recruits fibroblasts, keratinocytes, and endothelial cells, stimulating collagen and ECM deposition, as well as angiogenesis via increased VEGF expression [70]. Aside from wound healing, TGF-β plays a significant role in immunomodulation by promoting the expansion of Treg cells, inhibiting T cells, B cells, and NK cells, and polarises pro-inflammatory M1 macrophages to anti-inflammatory M2 macrophages (Figure 3B; Figure 4) [71]. Hence, TGF-β has a significant role in all stages of wound healing and immune regulation.
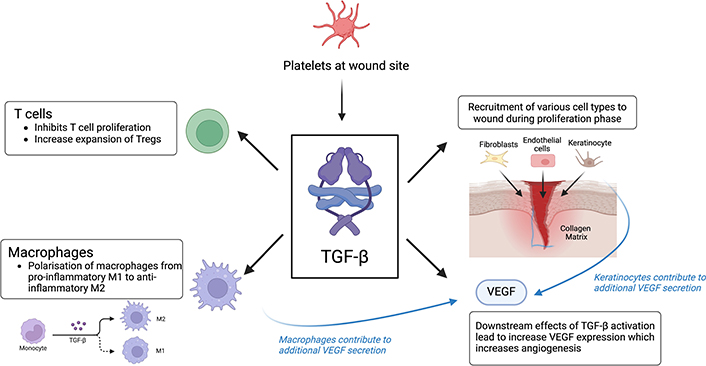
TGF-β signaling pathway. Platelets are the first responders at the wound site and release TGF-β upon activation. TGF-β then recruits various other cell types, such as fibroblasts, endothelial cells, and keratinocytes to the wound site. Additionally, TGF-β recruits other cytokines such as T cells, macrophages, and VEGF to the wound site. TGF-β polarises the production of pro-inflammatory M1 macrophages to anti-inflammatory M2 macrophages, as well as inhibiting the proliferation of T cells to prevent an excessive inflammation response. TGF-β results in an increased VEGF expression, which stimulates angiogenesis and encourages wound healing. IL-10: interleukin-10; TGF-β: transforming growth factor-β; VEGF: vascular endothelial growth factor. Created using BioRender (https://bioRender.com/r92p260)
TGF-β primarily binds to two receptor types found on all cell surfaces, transforming growth factor β receptor I (TGFβRI) and TGFβRII, and activation of the TGF-β receptors involves TGFβRII binding to the TGF-β ligand and recruiting TGFβRI, causing the phosphorylation of TGFβRI and subsequent phosphorylation of signal transduction proteins SMAD2 and SMAD3. They then form a complex with SMAD4 and translocate to the nucleus, altering DNA transcription of wound healing processes [72]. SMAD pathways are vital for signal transduction following the activation of TGF-β receptors and causing downstream changes to transcription and cellular function as a result of cytokine signaling. Alternative signaling pathways via PI3K/Akt, JAK/STAT, and MAPK are also present, and cause cellular processes such as inflammation, immune regulation, cytoskeletal changes, and differentiation, in comparison to cellular migration, proliferation, differentiation, and ECM deposition by classical SMAD signaling pathways [73].
With the pleiotropic effects of TGF-β playing multiple roles in wound healing and immune regulation, disruption in TGF-β release or signaling can lead to pathological states, and therapeutics targeting TGF-β must be specific or risk having severe adverse effects. Excessive levels of TGF-β can cause immunosuppression and promote fibrosis and cellular proliferation, leading to carcinogenesis [74] fibrotic diseases such as pulmonary fibrosis and systemic sclerosis [75], and infection [76]. However, TGF-β deficiency can also lead to inflammatory conditions, supporting carcinogenesis, fibrosis [77], and chronic wounds. TGF-β in chronic wounds was found to be decreased at the wound site, suggesting impaired chemotaxis and proliferation of fibroblasts, keratinocytes and endothelial cells, as well as reduced differentiation of fibroblasts into myofibroblasts, hence affecting collagen deposition and angiogenesis (Figure 3B; Figure 4). However, elevated levels of TGF-β at the epidermal edge of the chronic wound also result in hyperkeratosis and reduced keratinocyte proliferation [78], contributing to the chronicity of the wound possibly due to impaired keratinocyte migration and re-epithelization at the wound site.
TGF-β is a therapeutic target in the treatment of wound healing, and there are many different strategies towards inhibiting TGF-β signaling. Current developing treatments target the bioavailability of TGF-β, the interactions of TGF-β with its receptor, and other pathways TGF-β can stimulate intracellular signaling via neutralising antibodies, ligand traps, small molecule inhibitors or antisense oligonucleotides. However, many of these therapies have encountered challenges with adverse effects, owing to the broad range of physiological processes influenced by TGF-β [79]. Clinical trials of fresolimumab, an anti-TGF-β monoclonal antibody that targets all three TGF-β isoforms, show promise in reducing myofibroblast infiltration and fibrosis and is more well tolerated than many other anti-TGF-β therapies [75]. In the treatment of wound healing and fibrosis, reducing levels of TGF-β1 was shown to decrease the severity of fibrosis together with reducing ECM and collagen deposition, while administering TGF-β3 reduced scarring and improved the quality of dermis formation [80]. Leucine serine lysine leucine (LSKL) peptide therapy for blocking thrombospondin-1 (TSP-1), an activator of latent TGF-β, was also shown to effectively reduce fibrosis and the development of hypertrophic scars [81]. Conversely, recombinant TGF-β3 therapy like avotermin has found widespread success in reducing scarring following surgery with more organised ECM deposition [82]. Topical applications of TGF-β1 have also been shown to speed up wound closure, angiogenesis, inflammation, and ECM deposition in aged rats [83], and similar results were exhibited in human venous ulcer wounds using TGF-β2 [84]. As such, TGF-β is an enticing therapeutic target for fibrotic or chronic wound healing, but its pleiotropic effects can cause severe adverse reactions. Therapeutics must aim to maintain TGF-β at normal physiological levels, and further research should improve the mode of administration to localise treatment effects or target downstream signaling to enhance the specificity of treatment.
VEGF
VEGF is a proangiogenic mediator, which stimulates angiogenesis, and granulation tissue formation, influencing wound closure and epidermal repair [85]. The VEGF family consists of five members—VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factor (PlGF). VEGF production and recruitment are upregulated at wound sites when the body detects tissue injury, damaged blood vessels, or the presence of ROS. During the acute inflammatory phase, pro-inflammatory cytokines induce VEGF-C mRNA expression, resulting in more VEGF production [86]. During periods of hypoxia, smooth muscles, keratinocytes, endothelial cells, platelets, neutrophils, and macrophages produce increased amounts of VEGF-A, which stimulates wound healing. The expression of VEGF mRNA is further upregulated when there is an increase in HIF-1ɑ, which increases during periods of hypoxia or injury.
VEGF plays such a crucial role in angiogenesis and epidermal repair that the deletion of even a single VEGF allele result in embryonic lethality and causes increased mortality [87]. Patients with non-healing chronic wounds, such as diabetic nephropathy-associated wounds, have abnormally low levels of VEGF, which result in delayed angiogenesis and endothelial cell recruitment, leading to inefficient wound healing.
With the current findings of VEGF and its importance in angiogenesis and endothelial cell proliferation, there is a possibility that VEGF needs to be tapped into to influence wound healing. In chronic wounds, patients can be given supplementary VEGF to stimulate chemotaxis and angiogenesis. This treatment is currently being used on patients with DFUs [88] and has been shown to significantly reduce the time required for wound healing.
IL-1
IL-1 is a pro-inflammatory cytokine which has a crucial role in innate immunity by mediating the acute inflammatory response. In both local and systemic responses, IL-1 is involved in inducing pain sensitivity, fever, and hypotension, as well as allowing for the recruitment and migration of inflammatory and immunocompetent cells into the tissues through promoting vasodilation and increasing the number of adhesion molecules present on endothelial cells [89]. The 2 main isoforms of IL-1 are IL-1ɑ, and IL-1β, which are secreted by keratinocytes, macrophages, monocytes, and dendritic cells. IL-1ɑ is constitutively present in epithelial cells of healthy individuals, whereas IL-1β production is induced under disease or stress conditions [90]. Both forms of IL-1 bind to the interleukin-1 receptor-like 1 (IL-1R1), leading to the recruitment of IL-1 receptor accessory protein (IL-1RAcP), which forms a complex that includes IL-1R1, IL-1, and coreceptor [91]. This complex is a signaling-competent heterotrimeric complex, which recruits the myeloid differentiation primary response 88 (MyD88) [92]. This new complex continues to recruit other complexes, eventually leading to the NF-κB and MAPK pathway activation, as well as varying levels of pro-inflammatory gene transcription [93]. Inflammatory cytokines and pro-inflammatory mediators are then recruited predominantly during the acute reaction phase, which induces the classical signs of both local and systemic responses, such as pain, sensitivity, fever, and tachycardia (Figure 5).
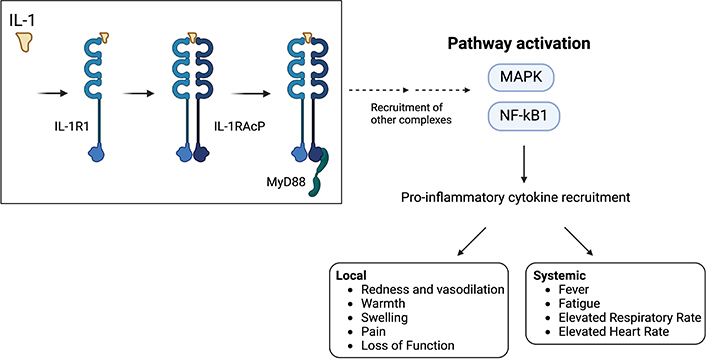
IL-1 signaling pathway. IL-1 is one of the first interleukins to be secreted during the acute inflammatory response. IL-1 recruits IL-1R1, which then forms a complex with IL-1RAcP. This complex binds to MyD88 which causes the recruitment of other complexes in a downward cascade, eventually resulting in the pathway activation of MAPK and NF-κB1. MAPK and NF-κB1 recruit pro-inflammatory cytokines. The pro-inflammatory cytokines cause both local and systemic inflammation, which manifests as redness and vasodilation, warmth, swelling, pain, loss of function at the wound site, and a systemic response including fever, fatigue, elevated respiratory rate, and elevated heart rate if the wound is of significant severity. IL-1: interleukin-1; IL-1R1: interleukin-1 receptor-like 1; IL-1RAcP: interleukin-1 receptor accessory protein; MAPK: mitogen-activated protein kinase; MyD88: myeloid differentiation primary response 88; NF-κB1: nuclear factor κB1. Created using BioRender (https://bioRender.com/c65v425)
While IL-1 is beneficial for triggering the initial immune response, a dysregulated IL-1 response would result in a prolonged inflammatory period during wound healing, increasing the likelihood of a chronic wound developing. The body regulates the activity of IL-1 by producing an IL-1Ra endogenously. IL-1Ra binds to the IL-1 receptors, hence preventing IL-1ɑ and IL-1β from binding, preventing signaling of pro-inflammatory mediators from occurring [94]. The discovery of IL-1Ra has significant implications for the therapeutic usages of IL-1Ra in the prevention of excessive inflammation. COVID-19 mRNA vaccines induce the production of IL-1 cytokines, but excessive inflammation is mitigated through the IL-1Ra axis [94]. These results suggest that IL-1Ra has significant potential to reduce inflammation, making it a promising candidate for application in wound healing—particularly for chronic wounds that remain in the inflammatory phase for prolonged periods [95].
IL-1 treatment has been found effective in treating wounds, with studies showing the effectiveness of ECM-binding IL-1Ra in treating diabetic wounds in rat models. The treatment reversed the senescence of fibroblasts at the wound site and increased the expression of growth factors, hence increasing angiogenesis, wound closure, and healing [96]. Additionally, recent research into the use of IL-1Ra such as anakinra in the treatment of infected wounds in zebrafish prevents the infection-induced inflammation and subsequent impaired wound healing and could be a possible therapeutic after infected wounds are cleared of pathogens [97]. With the relevance of raised levels of IL-1β, especially in diabetic patients, impairing healing through decreasing cell proliferation and migration [98], IL-1 is an exciting therapeutic target to enhance wound healing.
IL-2
IL-2 is a glycosylated globular protein and a pleiotropic cytokine. It has an important immunoregulatory role as it is a powerful growth factor and has strong mitogenic activities [99]. IL-2 is produced by a subpopulation of activated T lymphocytes (CD4+ and CD8+ T cells), as well as NK cells. Antigen-presenting cells (APC) stimulate CD4+ cells to produce the majority of IL-2. When the levels of IL-2 and IL-15 are raised in vivo, naive CD8+ T cells undergo rapid differentiation and expansion at a large scale into their effector cells. This observed behaviour is akin to that of the T cell response to foreign antigens and was seen to be driven predominantly by IL-2, signifying that IL-2 drives the homeostatic proliferation of naive CD8+ cells [100].
Additionally, IL-2 performs two crucial functions on Treg cells which express the IL-2 receptor (IL-2R). Treg cells are crucial in preventing excessive immune reactions as they preserve self-tolerance. Increased levels of IL-2 stimulate the production of IL-4, which then acts on antigen-activated CD4+ T cells to convert them into Treg cells. Beyond that, IL-2 binds directly to IL-2R on Treg cells, which causes the activation of the signal transducer and activator of transcription 5 (STAT5). STAT5 then intensifies the expansion of the fox head box protein 3 (FOXP3), which is the master regulator of the pathway involved in the production and development of Treg cells [101].
Apart from the roles that IL-2 plays in T lymphocytes, IL-2 activates macrophages and NK cells at wound sites to contribute to the proliferation of both B and T lymphocytes [102]. Identifying the role of IL-2 in wound healing would allow for greater advancements in the usage of IL-2 treatment in wound healing, as both the local and systemic IL-2 levels affect the levels of cell types in the local wound healing environment [103]. In a study carried out on adult burn patients, the systemic levels of IL-2 present in their blood were studied for a period of over 7 days. It was observed that the IL-2 amounts in the patient’s blood significantly increased during the first and fifth day since the burn, which is suggestive that differing levels of IL-2 are preferred at certain stages of wound healing [104]. Additionally, the same study also noted a direct correlation between the surface area and severity of burn wounds, with the amount of blood IL-2. A similar study done on child burn victims indicated that the amount of systemic IL-2 is higher than local IL-2, which is suggestive that IL-2 acts more systematically for patients with large wounds [105]. Applications of IL-2 in the treatment of such patients should be an area of future study, and the relationship between IL-2 levels present within the specific stages of wound healing should be studied to increase the efficiency of wound healing by potentially altering the systemic IL-2 levels.
Although IL-2 is a promising therapeutic target, limited research has been done into the efficacy of IL-2 inhibitors in wound healing, possibly due to its systemic effects in immunomodulation. IL-2 inhibitors have found success in immunotherapy for cancer and autoimmune diseases [106], and with increased interest in targeting IL-2 and its activity, its utility in wound healing warrants further investigation as well.
IL-4 and IL-13
IL-4 is an immunomodulatory cytokine and plays an important role in the activation of adaptive immune responses against helminths. IL-4 is primarily secreted by T helper 2 (Th2) cells to activate immunoglobulin production in B cells and initiates class switching to IgE, as well as by mast cells, basophils, and eosinophils at wound sites to facilitate activation of macrophages to M2 anti-inflammatory form to reduce inflammation [107]. IL-13 has a similar function to IL-4 and shares in one of its receptor targets but has other mechanisms such as promoting the dendritic cell migration to lymph nodes for antigen presentation and stimulating further activation of T lymphocytes [108].
IL-4 has two receptor targets, type 1 IL-4R which is expressed on hematopoietic cells, as well as type 2 IL-4R which is expressed on non-hematopoietic cells. Type 1 IL-4R is composed of IL-4Rα and γc subunits, while type 2 IL-4R is composed of IL-4Rα and IL-13Rα1 and can be stimulated by both IL-4 and IL-13 [109]. IL-4R activation and signaling occur via a few different signaling pathways, such as the JAK/STAT pathway. Type 1 IL-4 receptors activate JAK1 and JAK3, while type 2 IL-4 receptors activate JAK2 or TYK2. Both receptors target the downstream STAT6 to orchestrate changes in DNA transcription, specifically those for major histocompatibility complex class II and FcεRII expression, as well as IgE class switching. Other signaling pathways include the insulin receptor substrate-2 (IRS-2) pathway, which is highly similar to the pathway utilised by insulin or IGF-1. IRS-2 upregulates cellular proliferation and activates downstream PI3K and MAPK pathways to enhance cellular survival [110]. In comparison, IL-13 only binds to type 2 IL-4R, which also activates the JAK/STAT6 pathway, but only weakly stimulates IRS-2. Despite both cytokines having a shared receptor, differences in STAT6 signaling cause them to have differing roles and functions, with IL-4 having a greater effect on Th2 activation and B cell maturation, while IL-13 has a larger role in regulating allergic responses [111] and collagen remodelling [112].
With the importance of both IL-4 and IL-13 in modulating immune responses, they have come into focus as a therapeutic target, especially for chronic wounds which typically display ongoing inflammatory states. Typically, IL-4 and IL-13 fulfill immunosuppressive roles through inhibiting neutrophil migration, proliferation and production of pro-inflammatory mediators, and phenotype switching of macrophages from M1 pro-inflammatory to M2 anti-inflammatory [112]. Macrophages play a vital role in modulating inflammation via arginine metabolism, with M1 macrophages utilising nitric oxide synthase 2 (NOS2) to metabolise arginine and produce inflammatory and anti-microbial nitric oxide (NO) as a metabolite. This aids in the inflammatory phase to destroy pathogens and infected cells but can also cause damage to surrounding tissue and contribute to chronic inflammation. Conversely, M2 macrophages catabolize arginine via arginase Arg1, with the metabolites of proline and ornithine suppressing immune responses, inhibiting helminth growth and stimulating collagen production and ECM deposition. The competition for arginine use as a substrate also depletes the pool available for NOS2 use, inhibiting NO production and hence inflammation [113]. Additionally, M2 macrophages also release resistin-like molecule-α (RELMα), which activates the enzyme lysyl hydrolase 2 (LH2) to facilitate ideal collagen crosslinking and increases the synthesis of proteins for ECM remodelling, such as MMPs [114].
In chronic wounds such as DFUs or venous leg ulcers, these conditions exhibit increased NO synthesis produced by NOS2 of macrophages which have not completely switched from M1 to M2 phenotype, contributing to the persistent inflammation and tissue damage [115]. Recent studies on non-healing diabetic mouse wound models have found success in utilising a recombinant form of IL-4 bound to albumin with a collagen-binding domain and applied topically via HA-based gel. This treatment increased M2 phenotype switching and angiogenesis, and facilitated re-epithelialization and wound healing, posing a possible therapeutic to address chronic diabetic wounds [116]. However, overexpression of IL-4 and IL-13 can also lead to pathogenic wound-healing processes. IL-4 and IL-13 increase the expression of TGF-β, which induces fibrosis, as well as promoting fibroblast migration, proliferation and differentiation, and excessive production can result in fibrotic wound healing. These cytokines are linked to fibrotic pathologies such as pulmonary fibrosis, liver cirrhosis, and pathological cutaneous scars [117]. Dupilumab, a human monoclonal antibody against IL-4Ra, has been proven to effective in treating patients with atopic dermatitis. Results from a phase 3 clinical trial conducted on 155 patients revealed that 64% achieved a Physician Global Assessment (PGA) score of 0 or 1 out of 5 by week 52 of receiving dupilumab treatment [118]. Recent investigations have provided promising results on its use in treating hypertrophic scarring and keloids, with few major adverse reactions [119]. As such, the proper regulation of IL-4 and IL-13 in a normal physiological state is vital for wound healing without fibrosis and is an area of interest for the development of therapeutics.
IL-8
IL-8 is a pro-inflammatory cytokine produced by a variety of cells such as epithelial cells, fibroblasts, endothelial cells, lymphocytes, mast cells, and macrophages in response to inflammatory mediators [120]. It has two target chemokine c-x-c motif receptors (CXCRs), CXCR1 and CXCR2, which are expressed on neutrophils and monocytes to initiate chemotaxis, as well as on endothelial cells and neurons [121].
Upon binding to the receptors, CXCR2 is activated and the Gβγ subunit dissociates and undergoes signal transduction via the PI3K/Akt pathway, stimulating increased formation of actin filaments resulting in chemotaxis [122]. This process is vital during the initial stages of wound healing, where chemotaxis of leukocytes to the site of injury is essential to neutralise pathogens [123]. Once neutrophils reach the wound site, IL-8 continues to stimulate neutrophil degranulation, and formation of ROS and increases expression of adhesion molecules [124] contributing to local inflammation and immune response.
IL-8 has been identified as the causative mediator behind acute inflammatory conditions such as lung reperfusion injuries, acute immune complex-type glomerulonephritis, lipopolysaccharide-induced dermatitis and lipopolysaccharide-induced arthritis [125]. Elevated levels of IL-8 have also been linked to the development of acute respiratory syndrome in SARS CoV-2 patients [126], carcinogenesis [127], as well as chronic liver disease [128], posing a promising target for treatment of these inflammatory diseases via therapies such as tocilizumab.
However, IL-8 also has functions apart from inflammation, stimulating the chemotaxis of fibroblasts, endothelial cells, and keratinocytes via the JAK, Src/PI3K, FAK, and PLC pathways [129]. IL-8 plays a vital role in the proliferation phase of wound healing by promoting angiogenesis, lymphangiogenesis, and fibroblast migration [130], and chronic wounds deficient in IL-8 would have impaired wound healing. Applications of IL-8 through loading onto a poly-lactic-co-glycolic acid acellular dermal matrix dressing in combination with MSCs have been shown to promote diabetic wound healing [131], and further investigations for the use of IL-8 in therapeutic dressings or other modes of administration may advance treatment options for chronic wounds.
Conclusions
Many cytokines exhibit pleiotropic effects on the body and orchestrate the complex process of wound healing. Although the mechanisms of action of most cytokines have been extensively studied, few have been successfully developed into effective therapeutics for wound healing. With the large burden acute and chronic wounds place on healthcare systems, alongside the increase in chronic comorbidities for such wounds, wound healing therapeutics are becoming increasingly relevant.
Cytokines as the targets of therapeutics is not a new initiative, but recent studies have instead targeted cytokine receptors, such as dupilumab, reducing their adverse effects. Understanding when cytokines are produced, their normal physiological functions and the many downstream effectors they trigger can further enhance the specificity of treatment, and thus further the advancements in the development of effective wound healing therapeutics targeted at each phase of wound healing.
In this paper, we have highlighted the dynamic presence of specific cytokines during the various phases of wound healing, emphasizing their specialized roles. We also examined the localized and systemic effects of cytokines, such as their ability to trigger a systemic fever. Furthermore, we propose that future research should focus on determining the optimal levels of each cytokine required at different stages of wound healing and establishing a reference range that healthcare providers can use to assess and guide patients’ healing progress. Such data could also aid in identifying individuals at higher risk of developing chronic wounds or other complications. To achieve these goals, we recommend further studies to develop precise biomarkers capable of reliably monitoring cytokine levels at wound sites across the healing process, as current biomarkers remain relatively nonspecific. Additionally, research into optimizing cytokine delivery mechanisms—such as leveraging emerging nanotechnology—is essential to ensure adequate therapeutic cytokine concentrations at target sites while minimizing adverse side effects. While this field is still in its infancy and may encounter financial constraints and require a shift in clinical adoption mindsets, these challenges are anticipated to diminish over time as the field matures and demonstrates its clinical benefits.
In conclusion, the therapeutic application of cytokines in wound healing represents a rapidly advancing and promising field. By overcoming the key challenges discussed, the healthcare industry can move closer to fully harnessing the potential of cytokine-based therapies. This progress has the potential to significantly enhance patient care and improve outcomes, ultimately benefiting both individual patients and the broader medical community.
Abbreviations
| CXCRs: | chemokine c-x-c motif receptors |
| DFU: | diabetic foot ulcer |
| DM: | diabetes mellitus |
| ECM: | extracellular matrix |
| HA: | hyaluronic acid |
| HIF-1: | hypoxia-inducible factor-1 |
| IGF-1: | insulin-like growth factor-1 |
| IL-1: | interleukin-1 |
| IL-1R1: | interleukin-1 receptor-like 1 |
| IL-1Ra: | interleukin-1 receptor antagonist |
| IL-1RAcP: | IL-1 receptor accessory protein |
| IRS-2: | insulin receptor substrate-2 |
| MAPK: | mitogen-activated protein kinase |
| MMPs: | matrix metalloproteinases |
| MSCs: | mesenchymal stem cells |
| mTNF-ɑ: | membrane-bound tumour necrosis factor-ɑ |
| MyD88: | myeloid differentiation primary response 88 |
| NF-κB: | nuclear factor κB |
| NK: | natural killer |
| NO: | nitric oxide |
| NOS2: | nitric oxide synthase 2 |
| PRP: | platelet-rich plasma |
| RA: | rheumatoid arthritis |
| RIPK1: | receptor-interacting protein kinase-1 |
| ROS: | reactive oxygen species |
| STAT5: | signal transducer and activator of transcription 5 |
| sTNF-ɑ: | soluble-bound tumour necrosis factor-ɑ |
| TGF-β: | transforming growth factor-β |
| TGFβRI: | transforming growth factor β receptor I |
| Th2: | T helper 2 |
| TNF-ɑ: | tumour necrosis factor-ɑ |
| Treg: | regulatory T |
| VEGF: | vascular endothelial growth factor |
Declarations
Author contributions
RSYW and TT: Conceptualization, Data curation, Writing—original draft, Writing—review & editing. ASRP: Visualisation, Writing—review & editing. DKS: Conceptualization, Writing—review & editing. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2025.
Publisher’s note
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.