 Review
Review

Affiliation:
Rheumatology Unit, Sheba Medical Center, Ramat-Gan 52621, Israel
Email: ibank@tauex.tau.ac.il
ORCID: https://orcid.org/0000-0001-9866-7548
Explor Immunol. 2022;2:320–333 DOI: https://doi.org/10.37349/ei.2022.00053
Received: November 30, 2021 Accepted: February 28, 2022 Published: May 31, 2022
Academic Editor: Dominique J. Charron, Hospital Saint Louis, France
The article belongs to the special issue Interplay of γδ T cells and Tumor Cells
Obesity has become a worldwide scourge, affecting more than 10% of adults worldwide. While widely recognized to be associated with increased incidence of medical conditions such as diabetes mellitus and atherosclerosis, obesity also accounts for 9% of the cancer burden in some populations. This is due in part to perturbation of protective immune mechanisms involving natural killer cells, macrophages, and neutrophils. Recent studies indicate that γδ T cells play a prominent protective role against cancer, but in some circumstances are detrimental and pro tumorogenic. In this review, the current scientific literature was explored to determine whether and how obesity affects the anti- and pro-tumoral functions of γδ T cells. Considerable perturbations of γδ T cells by obesity were revealed, suggesting that the “obesity-γδ T cell axis” may profoundly impact the increased incidence of cancer in obese individuals and is worthy of further study.
Obesity, i.e. the abnormal or excessive accumulation of body fat that presents a risk to health wherein the body mass index (BMI) is > 30, is a condition with increasing prevalence, which affected 10.8% of men and 14.9% of women worldwide in 2014 [1]. Obesity accounted for 9% of the cancer burden in women in Europe, America, and the Middle East. The risk of cancers in obese individuals is increased, with relative risks (RRs) ranging from 1.5 to 1.8 for cancers of the colon, gastric cardia, liver, gallbladder, pancreas, and kidney [1–3]. Moreover, the RR for esophageal adenocarcinoma was up to 4.8 for a BMI of 40 or more [1].
Effects of obesity on the functions of the immune system are thought to be an important mechanism responsible for the increased risk of cancer. A review of pre-clinical murine studies by Farag et al. [4] showed that obesity induces lymph node atrophy, impedes lymphatic transport, and reduces T cell receptor (TCR) diversity. Other effects included accumulation of tumor-promoting myeloid cells, e.g., macrophages, neutrophils, and myeloid-derived suppressor cells (MDSCs) within the tumor microenvironment [4]. In humans, the function of type 1 innate lymphoid cells (ILC-1) in adipose tissue (AT) that regulate macrophages via cytotoxicity is impaired in obesity, thus contributing to a pro-inflammatory phenotype [5]. Obesity impairs the function of anti-cancer natural killer (NK) cells by impeding their ability to direct lytic granules to the tumor cell and degranulate [6]. Interestingly, impaired NK activity characteristic of obesity can be improved by weight loss [7].
γδ T cells, i.e. T cells expressing TCR encoded by the γ and δ genes, were discovered in the 1980s, and constitute one of the three arms of the immune system (the other two being αβ T cells and B cells) characterized by surface membrane expression of somatically rearranging immunoreceptors [8–10]. γδ T cells emerged evolutionarily about 450 million years ago, along with B cells and αβ T cells, and remain the least well understood [11]. Some outstanding unique features of γδ T cells include their characteristic lack (on the majority of cells) of expression of cluster of differentiation 4 (CD4) and CD8 (unlike αβ T cells), their early establishment of residence in surface epithelial tissues, and their independence from major histocompatibility complex (MHC) molecules for antigen recognition [12–14]. Indeed, the manner wherein the γδ TCR recognizes antigens is unique although differing between subsets of γδ T according to the variable (V) regions of the TCR. Thus, the majority of γδ T cells expressing Vγ9 and Vδ2 in the TCR (Vγ9δ2 cells), which form the major subset in human peripheral blood (PB), innately recognize cell surface membrane expressed butyrophilin (BTN) 3A1 and 2A1 molecules when they assume a novel conformation induced by intracellular binding of phosphoantigens, upregulation of which occurs during oncogenic transformation or intracellular infections [15]. On the other hand, Vδ1+ γδ T cells, which are chiefly located in tissues along epithelial surfaces, recognize MHC-like molecules including CD1d, a, b, and c, either directly, or when presenting lipid antigens including sulfatides [15]. Furthermore, Vδ1+ or Vδ3+ clones express TCR that directly recognizes sites on cell surface membrane proteins including MHC class I-related 1 (MR1), endothelial receptor C protein, annexin, phycoerythrin, and ephrin A. γδ T cells also express NK like receptors, e.g., NK receptor group 2 member D (NKG2D) that can mediate cytotoxicity by interactions with MHC class I polypeptide-related sequence A (MICA) or MICB [15]. Thus, γδ T cells appear to exhibit a functional repertoire straddling innate and adaptive immunity [13, 14, 16].
The very first paper describing the discovery of human thymus-derived T cells expressing a γδ TCR already described the cytotoxic potential of γδ T cells and the potential role of the CD3–TCR complex in mediating killing against hematological tumor cells [8]. This anti-tumoral cytotoxic potential has been confirmed by numerous studies suggesting that these cells may play a strong anti-tumoral role in humans further stimulated by the revelation that, relative to other leukocytes and immunocytes, the presence of γδ T cells in the tumor stroma of a large cohort of cancer patients was the best positive prognostic indicator [17, 18]. The mechanisms include Vγ9δ2 TCR interactions with BTNs bound to intracellular tumor phospho-antigens, as well as recognition of stress-related molecules, expressed on tumor cell membranes, which result in perforin and granzyme B mediated killing of tumor cells [17]. In addition, NK cell-like NKG2D mediated killing by recognition of MICA and MICB has been implicated [17]. Cytotoxic molecules such as tumor necrosis factor α (TNFα) may act directly on tumor cells and type I cytokines interferon γ (IFNγ) that enhance the anti-tumoral response of adaptive immune cells, both of which are highly secreted by γδ T cells, are additional important mechanisms of anti-tumoral effects of γδ T cells [17]. Finally, activated Vγ9δ2 cells may function as tumor antigen-presenting cells, activating tumor antigen-specific CD8+ and CD4+ αβ T cells [19].
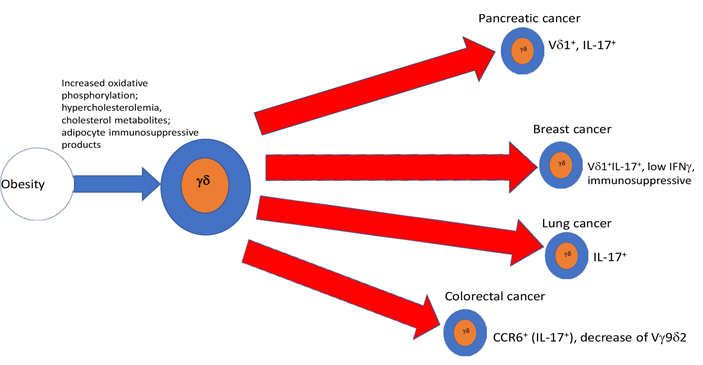
Despite these potent anti-tumoral mechanisms, there are strong indications that γδ T cells may also critically play pro-tumoral roles [20]. For example, immunosuppressive γδ T cells have been shown in breast cancer and pancreatic cancer [21–23]. Another important mechanism involves γδ T cells that secrete interleukin-17 (IL-17). Thus, IL-17 secreting T cells were shown to enhance breast cancer growth and metastasis in mice [24]. In pancreatic cancer, IL-17 producing γδ T cells are a poor prognostic factor and were shown in murine models to directly enhance tumor growth [25, 26]. In murine models of lung cancer, IL-17 producing γδ T cells triggered by the microbiota were shown to mediate cancer growth [27]. The mechanism may involve IL-17 mediated promotion of pro-tumoral neutrophils, promotion of vascular endothelial growth factor (VEGF) to enhance tumor angiogenesis, or enhancement of pro-tumoral mesenchymal stem cells [3, 28, 29].
Despite growing interest in the role of γδ T cells in cancer, there are no published reviews of how obesity impacts this important arm of the immune system. Critically, although there exists considerable functional overlap (e.g., cytokine repertoire, cytotoxic, helper and regulatory) of γδ T cells and other T cells of either “adaptive” (classical MHC restricted CD4+ and CD8+ αβ T cells) or of “innate” type [including NKT and mucosal associated invariant T (MAIT) cells] their highly unique antigenic repertoire, which includes phospho-antigen metabolites in the cholesterol synthetic pathway, as well as lipid antigens, strongly suggests that they could play a pivotal role in the effect of obesity on cancer [13, 14, 16]. Here, we will present the available evidence addressing the impact of obesity on the distribution and functions of γδ T cells in animal models and in humans, and evaluate how obesity-induced changes might influence the immune effects of γδ T cell in the setting of cancer.
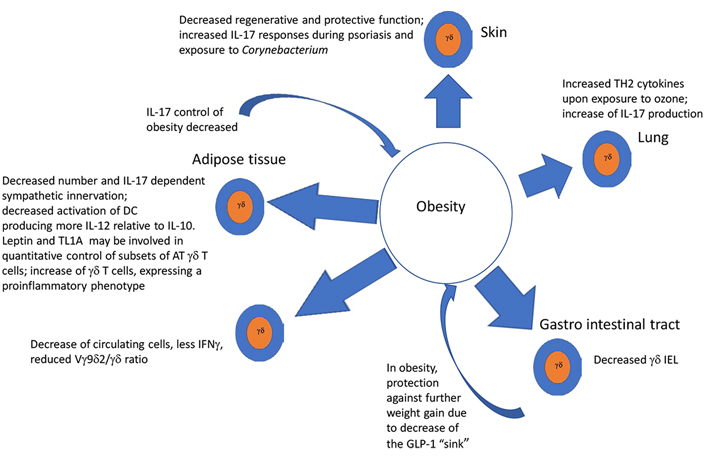
γδ T cells, similarly to other classical and innate like T cells, are present in AT in mice, humans, and cows. In cows, the frequency of both γδ T cells and CD8+ non-γδ T cells was found to be higher in mesenteric than in subcutaneous AT and expressed a CD45RO+CD62L– phenotype [30]. Importantly, in murine models, it was found that the predominant cells secreting IL-17 in AT are γδ T cells. Moreover, IL-17 produced by these cells was shown to inhibit adipogenesis, to moderate AT accumulation, and to regulate glucose metabolism [31]. Adipose T cells promyelocytic leukemia zinc finger (PLZF)+ γδ T cells producing both TNFα and IL-17 induced IL-33 production by platelet-derived growth factor receptor α (PDGFRα) expressing stromal cells, which by its effects on suppression of tumorigenicity 2 (ST2)+ regulatory T (Treg) cells controlled non-shivering thermogenesis. Thus, γδ knock out mice exhibited loss of temperature regulation, in particular in cold conditions [32]. The IL-17 produced mainly by γδ T cells in AT binds to the IL-17 receptor C (IL-17RC). Ablation of IL-17RC reduced transforming growth factor β1 (TGFβ1) expression which in turn reduced sympathetic innervation in AT, induced obesity and reduced thermogenicity. Accordingly, reduction of IL-17 producing γδ T cells had similar effects. Interestingly, ablation of IL-17RC or γδ T cells also reduced sympathetic innervation in the lung and salivary glands [33]. Others confirmed that mice lacking γδ T cells have impaired glucose homeostasis. Interestingly, a ketogenic diet (KD) expanded metabolically protective γδ T cells that restrain inflammation. However, long term ad libitum KD induces obesity and impairs metabolic health while depleting AT fat tissue γδ T cells, suggesting that γδ T cells mediate protective immunometabolic response and thus decrease AT inflammation [34]. On the other hand, γδ TCR expressing intraepithelial lymphocytes (IEL) in the gut, may have an opposite effect in terms of immunometabolic control. Thus, by expressing receptors for glucagon-like peptide-1 (GLP-1), γδ, as well as αβ IEL, serve as a “sink” for this important metabolic regulator by decreasing its bioavailability, thus contributing to insulin resistance (IR) and obesity. Indeed, mice lacking IEL by virtue of ablation of α4β7 and αeβ7 integrins, are resistant to the effects of a high fat diet (HFD) [35].
On the other hand, previously, it had been shown that an HFD which induced obesity in mice, also increased γδ cells in inguinal fat [36] and that the γδ T cells increase in fatty tissue in obese mice was less pronounced in leptin-deficient mice, suggesting a role for leptin in the control of AT γδ T cells [37]. An additional factor controlling the homeostasis of γδ T cells in obesity may be TNF-like cytokine 1A (TL1A), a TNFα-related molecule [38]. Thus, TL1A deficient mice were both less obese and had fewer γδ T cells in epididymal white fat, concomitant with a reduction of mitochondria regulatory genes. Interestingly, in humans, a non-coding polymorphism close to the TL1A locus was associated with body mass, suggesting that studies of AT γδ cells in these individuals would be of interest [38]. Others have reported that HFD may specifically induce an increase of CD44+CD62lo γδ T cells in epididymal fat of mice. In this study, although Vγ4 and 6 deficient mice or TCRδ–/– mice were no different in weight relative to wild type (WT), they had less inflammatory macrophages producing IL-6 in their AT—suggesting a proinflammatory role of Vγ4 and 6 expressing γδ T cells, which are the main IL-17 producing γδ T cells, in visceral AT (VAT). Interestingly, however, in this study, TCRδ–/– mice were less insulin resistant and had reduced levels of inflammation in the liver and skeletal muscle [39]. In another study, it was shown that decreasing αβ/γδ T cell ratio in white AT (WAT) depots alters their inflammatory state and mass repartition, which might be involved in the improvement of insulin sensitivity. Thus, a mouse model overexpressing peroxisome proliferator-activated receptor β (PPAR-β) specifically in T cells [transgenic (Tg) T-PPAR-β] exhibits a partial depletion in αβ T cells and no change in γδ T cell number resulting in an increased γδ/αβ T cell ratio in lymphoid organs. Tg T-PPAR-β mice were partially protected against HFD-induced weight gain and exhibited decreased IR and liver steatosis independently of animal weight. Thus, in this model, a relative increase of γδ T cells was associated with beneficial metabolic effects [40]. Overall, it appears that while γδ T cells are fundamental for AT innervation and thermogenesis, the expansion of visceral WAT promotes dysregulation of its immune cell composition and likely promotes low-grade chronic inflammation [41]. The highlights of these studies described in this paragraph are presented in Table 1.
Murine models: γδ T cells in AT and adiposity
| Study | IL-17+ γδ T cells | IFNγ+ γδ T cells | Number/functions | Reference |
|---|---|---|---|---|
| γδ T cells in AT | Present | Inhibition of adipogenesis | [31] | |
| γδ T cells in AT | Present | Induction of IL-33 production; γδ T cells are responsible for non-shivering thermogenesis | [32] | |
| γδ T cells in AT | Present | Binds to the IL-17RC, increases sympathetic innervation | [33] | |
| γδ T cells in AT, effect of KD | Short term KD increases anti-inflammatory γδ T cells, long term-induces obesity and depletes γδ T cells in AT | [34] | ||
| IEL γδ T cells | Express GLP-1R, forming a “depleting sink” for GLP-1R, thus possibly contributing to obesity | [35] | ||
| γδ T cells in AT-effect of HFD induced obesity | HFD increased γδ T cells in inguinal fat | [36] | ||
| γδ T cells in AT-effect of HFD induced obesity | HFD increased γδ T cells. AT was decreased in the absence of leptin | [37] | ||
| γδ T cells in AT-effect of HFD in TL1A deficient mice | TL1A deficient mice were less obese and had fewer γδ T cells in AT | [38] | ||
| Subsets of γδ T cells in AT, effect of HFD | Present, mainly Vγ4 and 6+ | HFD induces an increase of CD44+CD62lo γδ T cells in epididymal fat which appear to play a proinflammatory role | [39] | |
| Effect of a transgene PPAR-β expressed in T cells on HFD and IR | Mice bearing Tg T-PPAR-β in T cells have depleted αβ T cells and no change in γδ T cell number resulting in an increased γδ/αβ T cell ratio in lymphoid organs and preserved γδ T cells in WAT. These mice have less IR and are less obese | [40] | ||
| Hyperglycemic obese mice | Reduced skin γδ cells dysfunctional in keratinocyte repair | [42–44] | ||
| Effect of HFD on imiquimod induced psoriasis | More γδ low cells expressing PD1 in the skin | [45] | ||
| Obesity-related to adiponectin deficiency in psoriasis | Elevated in the skin by obesity | Reduced by adiponectin | [46] | |
| Effect of HFD on Corynebacterium activated skin γδ T cells | Elevated in the skin by HFD | [47] | ||
| Effect of obesity on ozone induces lung changes | Increase of IL-13+ and ST2 related IL-33 responsive γδ T cells in the lung | [48] | ||
| Effect of obesity on γδ T cells in the lung | Elevated by HFD in the lung | Increased airway hyperresponsiveness | [49] | |
| Effect of HFD on gut T cells | IEL, which express GLP-1R, CD69, and CD103 are decreased by HFD, leading to increased sensitivity to dextran sulfate-induced colitis | [50–52] | ||
| Deficiency of integrin β7-effect on HFD | Decreased GLP-1R+ γδ and αβ IEL, leading to resistance to HFD causing obesity, due to decrease of GLP-1R | [35] | ||
| Effect of obesity on ocular T cells in NRF2–/– mice (model for AMD) | Increased in the eye | Induction of retinal lesions | [53] | |
| A high cholesterol diet in mice | Reduced number of γδ T cells in the thymus, colonic submucosa, and early tumorogenesis | [54] | ||
| Effect of 27-hydroxycholesterol | An increase in the number of polymorphonuclear neutrophils and pro tumoral γδ T cells at distal metastatic sites | [55] | ||
| Diet-induced obesity | CCR6+ pro-tumoral γδ T cells attracted to chemically induced colon adenocarcinoma | [56] | ||
| Metabolic characteristics of pro and anti-tumoral γδ T cells and effect of the obesity | IL-17 producing cells are dependent upon oxidative phosphorylation, exhibit high lipid uptake and increase in obesity | IFNγ producing γδ T cells are dependent upon glycolysis | IL-17+ γδ T cells were pro-tumoral, whereas IFNγ+ cells were anti-tumoral | [57] |
GLP-1R: glucagon-like peptide-1 receptor; PD1: programmed death 1; NRF2: nuclear erythroid 2-related factor 2; AMD: age-related macular degeneration; CCR6: C-C motif chemokine receptor 6
Obese adult humans have a decrease of γδ cells in the PB, which are less capable of IFNγ production and express less IL-2R and IL-7R than those of non-obese individuals but can still be activated with (E)-4- hydroxy-3-methyl-but-2-enyl pyrophosphate (HMBPP), a known potent activator of human Vγ9δ2 T cells, indicating retention of functional responses of the Vγ9δ2 subset [58]. Likewise, in a study of 10 obese women who also had diabetes, 10 obese women without diabetes and 12 controls, it was found that both PB invariant NKT (iNKT) and γδ T cells of obese patients were characterized by diminished IL-2 and IFNγ production [59]. γδ T cells are also reduced in obese patients with obstructive sleep apnea (OSA) compared to healthy controls [60]. Another study revealed that the decline may be specific for a subset of γδ T cells since obese as well as patients with colorectal carcinoma (CRC) shared a significant reduction of the Vγ9δ2/γδ T cell ratio in PB [61]. Interestingly, obese people treated with aged garlic extract (AGE) that was added to their diet, increased their γδ T cells in the PB, along with a decrease of NKT cells [62]. However, a study of 18 women with morbid obesity, revealed that only IFNγ producing NK cells but not γδ (or CD4 or CD8) T cells were positively associated with obesity, IR and hemoglobin A1C (HBA1C) levels [63]. The effect of obesity resulting in the reduction of γδ T cells in the PB may moreover, be age-dependent, since an analysis of PB of 890 children using magnetic resonance imaging (MRI) and dual-energy X-Ray absorptiometry, revealed that higher Vγ9δ2 cells were found in patients with higher fat mass index, visceral fat index, and android to gynoid ratio [64].
Similar to mice and cows, human γδ T cells may also reside in AT. For example, γδ T cells expressing Vδ1 and Vδ3 and producing IFNγ and IL-17 were increased in omental fat of patients with oesophageal cancer relative to their PB [65]. In one study, analysis of NK, NKT-like, γδ T, and Treg cell populations in VAT and blood of healthy lean subjects revealed that CD56hi NK and OX40+ Treg cells were more abundant in VAT with respect to blood. Conversely, CD56dim NK and total Treg cells are mostly present in the circulation, while γδ T lymphocytes were uniformly distributed in the two compartments [61]. Furthermore, analysis of AT in 120 obese and 275 controls revealed that macrophages were the source of inflammation in obese fat, whereas the relatively reduced γδ T cells that were present in AT accounted for reduced sympathetic response pathways in the AT [66]. Moreover, adiposity affects the functions of the tissue-resident γδ T cells. Thus, adipocyte-conditioned media (ACM) from obese and CRC subjects released higher levels of pro-inflammatory/immunoregulatory factors as compared to ACM from healthy lean subjects. Dendritic cells (DC), differentiated in the presence of ACM from obese (and CRC) subjects, expressed elevated levels of the inhibitory molecules PD-ligand 1 (PD-L1) and PD-L2, and showed a reduced IL-12/IL-10 ratio in response to both toll-like receptor (TLR) ligand and γδ T lymphocyte-induced maturation [67]. The highlights of these findings, pertaining to the effects of obesity on human γδ T cells, are summarized in Table 2.
γδ T cells in obese humans
| Subjects studied | IL-17 production by γδ T cells | IFNγ production by γδ T cells | Number and function of γδ T cells | Reference |
|---|---|---|---|---|
| Obese adults | PB decrease | Decrease of γδ cells in the PB, which are less capable of IFNγ production and express less of IL-2R and IL-7R | [58] | |
| Obese women | PB decrease | Decrease, less IL-2 and IFNγ production | [59] | |
| Obese adults | Decrease Vγ9δ2/γδ ratio in PB | [61] | ||
| Morbid Obesity adults | PB IFNγ+ γδ T cells are not correlated with obesity | [63] | ||
| Obese children | Increase of Vγ9δ2 cells in PB | [64] | ||
| Obese adults with OSA | Decrease of γδ T cells in PB | [60] | ||
| Adults with esophageal cancer | Increase in omental fat | Increase in omental fat | Increase relative to PB of Vδ1 and Vδ3+ cells in omental fat, producing IL-17 | [65] |
| Lean adults | Equally represented in VAT and PB | [61] | ||
| AT of 120 obese and 275 controls | Decreased γδ T cells in the AT of obese people, leading to decrease sympathetic activity | [66] | ||
| In vitro study of effects of adipocyte conditioned medium on immune functions | Adipocyte conditioned medium decreases responses of DC to γδ T cell activation signals, with decreased IL-1/IL-10 ratio | [67] |
It is well known that obesity negatively affects the clinical course of a number of non-malignant diseases, including psoriasis and asthma as well as that of cancer. While the primary goal of this review is to examine how obesity impacts γδ T cells in relation to their involvement in malignancy, we considered it likely that additional insights into the possible mechanisms of this three-way interaction could be gleaned by examining how γδ T cells are impacted by in the course of chronic inflammatory diseases, which are often linked to cancer [68]. Here, and in the highlighted form in Table 1, we summarize studies that have addressed the role of γδ T cells in mediating the deleterious effects of obesity in disease states in specific organs, and, finally, in cancer.
In hyperglycemic obese mice, γδ cells in the skin are reduced by half and are dysfunctional in maintaining epithelial repair of keratinocytes [42, 43]. In the absence of growth factors, cytokines, and chemokines produced by γδ T cells, wound repair is negatively impacted. This disruption in γδ T cell function is apparent in metabolic diseases such as obesity and type 2 diabetes [44].
Importantly, in the context of studies exploring the effect of a western diet, known to be associated with obesity, impacting imiquimod induced psoriasis, Yu et al. [45] found that there were more γδ low cells expressing PD1 in the skin than in mice fed a normal diet and that anti PD1 increased their activation, worsening psoriasis. In obesity-related to adiponectin deficiency in mice, γδ T cells in psoriasiform infiltrate in the skin produced more IL-17 and were increased, whereas adiponectin suppressed IL-17 via AdipoR1 suggesting a regulatory anti-inflammatory role for adiponectin in this context [46]. Furthermore, it was found that facultative Corynebacterium serves to promote and maintain IL-23 dependent γδ T cells in the skin. In mice placed on an HFD, these cells became proinflammatory in an IL-23 dependent manner in the presence of Corynebacterium [47]. These data suggest promotion of IL-17 producing γδ T cells in the skin by diet associated with obesity at the expense of γδ T cells that function in a protective role in health.
In obese mice, ozone increased lung IL-13+ ILC2 and IL-13+ γδ T cells. Ozone increased ST2+ γδ T cells, indicating that these cells can be targets of IL-33, and γδ T cell deficiency reduced the obesity-related increases in the response to ozone, including increases in type 2 cytokines induced by obesity. Thus, obesity and ozone interacted to promote type 2 cytokine production in γδ T cells and ILC2 in the lungs perhaps due to the effects of IL-33 [48]. In the absence of obesity, however, IL-33 appeared to suppress γδ T cells producing IL-17 in a Treg-dependent manner [69]. Interestingly, IL-33 was also recently shown to reduce the cytotoxic functions of γδ T cells by inducing the release of matrix metallopeptidase 9 (MMP9) from macrophages, which cleaves NKG2D, a molecule that mediates anti-tumoral cytotoxicity of γδ T cells, suggesting an IL-33 related decrease of the potential anti-tumoral mechanism by γδ T cells in lung tumors [70]. HFD fed obese mice also develop airway hyperresponsiveness, which was associated with a larger increase IL-17 RNA in γδ T cells in the airways [49].
Feeding mice an HFD reduced IEL in the jejunum, duodenum, and ileum [50]. Furthermore, it was shown that treating mice with HFD to induce obesity resulted in a reduction of both αβ and γδ IEL after 7 weeks. Concomitantly, a down-regulation of CD103 and CD69 was observed, and importantly, the mice exhibited increased sensitivity to dextran sulfate induce colitis [51]. At a functional level, the GLP- 1R, which plays a critical role in the regulation of body weight, co-purified with TCR γδ and TCR αβ in IEL preparations. A GLP-1 agonist increased cyclic adenosine monophosphate (cAMP) in γδ T cells. Moreover, GLP-receptor (GLP-R) deficiency enhanced sensitivity to dextran sulfate, and dysregulated genes and microbiota [52]. Supporting these findings, it was shown that integrin β7 (a component of homing α4β7 and αEβ7molecules), deficient mice lack IEL. These mice are resistant to an HFD due to the lack of absorption of the incretin GLP-1 by the absence of GLP-1R expressing γδ and αβ T cells, which serve normally act as a “sink” for GLP-1, thus indirectly contributing to the generation of obesity in conditions of high obesity inducing nutrients [35]. Interestingly, γδ T cells themselves appear to control the absorption of one such class of nutrients. Indeed, the machinery for digestion and absorption of carbohydrates is controlled by γδ T cells via suppression of IL-22 by ILC and nutrient availability altered the distribution and transcriptome of γδ T cells [52].
NRF2–/– mice, which served to produce a model for human AMD, were fed HFD. Fifty-eight percent of the mice on the HFD developed advanced retinal lesions, and IL-17 detected in the eye was produced by γδ T cells [53].
One of the corollaries of obesity is hypercholesterolemia. A high cholesterol diet-induced an oxidant stress-dependent increased miR101Cm which decreased Tet methylcytosine dioxygenase 1 (TET1) in hematopoietic stem cells (HSC). This in turn reduced the number and function of terminally differentiated NKT and γδ T cells in the thymus, the colon submucosa, and during early tumorigenesis [54]. Furthermore, ablation or inhibition of cytochrome P450 family 27 subfamily A member 1 (CYP27A1), the enzyme responsible for the rate-limiting step in 27-hydroxycholesterol biosynthesis, significantly reduces metastasis in relevant animal models of cancer. Enhanced metastasis by 27-hydroxycholesterol requires myeloid immune cell function, via an increase of the number of polymorphonuclear neutrophils and γδ T cells at distal metastatic sites. The pro-metastatic actions of 27-hydroxycholesterol require both polymorphonuclear neutrophils and γδ T cells and result in a decreased number of cytotoxic CD8+ T lymphocytes. Therefore, 27-hydroxycholesterol functions as a biochemical mediator of the metastatic effects of hypercholesterolemia through its actions on γδ T cells and polymorphonuclear neutrophils [55]. Diet-induced obesity increased IL-6 producing macrophages resulting in increased C-C Motif chemokine ligand 20 (CCL20) that attracted CCR6+ pro-tumoral γδ T cells to chemically induced colon adenocarcinoma. Moreover, inhibition of γδ T cells blocked tumor progression [56]. Finally, it has been shown that IFNγ producing γδ T cells are dependent upon glycolysis for their metabolism and function, whereas IL-17 producing γδ T cells are dependent upon oxidative phosphorylation and have increased mitochondrial mass. Tγδ17 cells exhibit high lipid uptake and storage, are increased in obese mice, and are pro-tumoral in tumors of obese mice. By contrast, γδ T cells producing IFNγ were more active in the presence of glucose and were anti-tumoral [57].
As summarized in graphical form in Figure 1, AT and obesity appear to have an important impact upon the function and numbers of γδ T cells in a variety of ways, that, for the most part, imply a shift toward a tumor supportive phenotype, while mitigating anti-tumoral effects. A priori γδ T cells in healthy ATs, even in the absence of obesity, appear to be skewed toward the production of IL-17, which is involved in physiologic control of thermogenesis and sympathetic innervation. Adipocytes appear to produce factors that drive DC to a more immunosuppressive state in response to γδ T cell stimulation. As obesity develops e.g., under the influence of an HFD, a more pro-inflammatory phenotype with a shift from IFNγ and toward IL-17 producing cells takes place in a variety of conditions including psoriasis, asthma, and cancer. IL-17 as discussed in the introduction is considered pro-tumoral by effects on VEGF and pro-tumorigenic neutrophils. Thus, γδ T cells in peripheral tissues of obese animals, are substantially altered: in the skin, they are reduced and lose their ability to maintain epithelial integrity. Likewise, γδ T cells are reduced among gut IEL, rendering the gut more susceptible to signals inducing chronic inflammation, thus exposing it to a pro-tumoral environment, while simultaneously driving the appearance of an enhanced γδ T17 pro-tumoral phenotype. When γδ T cells are exposed to proinflammatory conditions such as ozone in the lung, or imiquimod in the skin, obesity serves as an additional driver toward polarization of these tissue-resident γδ T cells to an IL-17 producing phenotype, predisposing these patients to harbor pro-tumoral γδ T cell populations in the skin and lung. All in all, along with the tendency of obesity, and the associated hypercholesterolemia to drive a reduction of IFNγ, these analyses suggest that a major result of obesity, is a total body reduction of the ratio of anti-tumoral IFNγ to pro-tumoral IL-17 γδ T cells as well as the induction of immunosuppressive γδ T cell phenotypes. Thus, as summarized in Figure 2, we conclude that in conjunction with numerous other immune mechanisms some of which were described above, the effects of obesity and its related conditions on γδ T cells play a relevant ancillary role in the increased incidence of cancer in obese patients. Further studies of the effects of obesity and ancillary conditions on γδ T cells will be required to completely understand how effects of obesity on these largely pro-tumoral effects could be mitigated.
ACM: adipocyte-conditioned media
AT: adipose tissue
CCR6: C-C motif chemokine receptor 6
CD4: cluster of differentiation 4
CRC: colorectal carcinoma
DC: dendritic cells
GLP-1: glucagon-like peptide-1
GLP-1R: glucagon like peptide-1 receptor
HFD: high fat diet
IEL: intraepithelial lymphocytes
IFNγ: interferon γ
IL-17: interleukin-17
IL-17RC: interleukin-17 receptor C
ILC-1: type 1 innate lymphoid cells
IR: insulin resistance
KD: ketogenic diet
MHC: major histocompatibility complex
MICA: major histocompatibility complex class I polypeptide-related sequence A
NK: natural killer
NKG2D: natural killer receptor group 2 member D
PB: peripheral blood
PD1: programmed death 1
PPAR-β: peroxisome proliferator-activated receptor β
ST2: suppression of tumorigenicity 2
TCR: T cell receptor
Tg: transgenic
TL1A: tumor necrosis factor-like cytokine 1A
TNFα: tumor necrosis factor α
Treg: regulatory T
V: variable
VAT: visceral adipose tissue
WAT: white adipose tissue
The author contributed solely to the work.
The author declares that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Xiangjin Zhang ... Wei He
Huidi Wang ... Jia Yin
Chang Liu ... Wei He
Joseph M. McGraw, Deborah A. Witherden
Yue Wang ... Wei He
Rupert Handgretinger ... Manon Queudeville
Gabrielle M. Ferry, John Anderson
Juan-Pablo Cerapio ... Jean-Jacques Fournie
Alessandro Poggi, Maria Raffaella Zocchi
Navdeep Jhita, Sunil S. Raikar
Jessica Da Gama Duarte ... Andreas Behren
