 Review
Review

Affiliation:
Departments of Pediatrics, Obstetrics and Gynecology and Pathology, Women & Infants Hospital of Rhode Island, Warren Alpert Medical School of Brown University, Providence, RI 02905, USA
ORCID: https://orcid.org/0000-0003-4911-6234
Affiliation:
Departments of Pediatrics, Obstetrics and Gynecology and Pathology, Women & Infants Hospital of Rhode Island, Warren Alpert Medical School of Brown University, Providence, RI 02905, USA
Affiliation:
Departments of Pediatrics, Obstetrics and Gynecology and Pathology, Women & Infants Hospital of Rhode Island, Warren Alpert Medical School of Brown University, Providence, RI 02905, USA
Email: ssharma@wihri.org
ORCID: https://orcid.org/0000-0002-4331-3467
Explor Immunol. 2022;2:363–382 DOI: https://doi.org/10.37349/ei.2022.00056
Received: November 22, 2021 Accepted: May 09, 2022 Published: June 13, 2022
Academic Editor: Nathalie Lédée, Hôpital Pierre-Rouquès Les Bluets-Drouot, France
The article belongs to the special issue Human Reproduction: Involvement of the Immune System
Among decidual immune cells, regulatory T cells (Tregs) have been unanimously recognized as central contributors to tolerance and maintenance of healthy pregnancy. Numerical and functional downregulation of Tregs or disturbed interaction of Tregs with trophoblasts and other immune cells have been linked to early pregnancy loss such as idiopathic infertility and miscarriage and later-onset adverse pregnancy outcomes including preeclampsia. This review focuses on the mechanisms for regulating the generation, expansion, and function of Tregs, the roles of Tregs in maintaining maternal immune tolerance through crosstalk with trophoblasts and other decidual regulatory immune cells, and how Tregs may play foes to pregnancy and contribute to the programming of pregnancy-related complications. Therapeutic options for implantation failure and adverse pregnancy outcomes are now part of the emerging significance of Tregs in pregnancy tolerance and maintenance.
Pregnancy is a unique physiological process that involves semi-allogeneic fetus growth in the uterus without rejection by the maternal immune system. The mechanisms underlying maternal tolerance toward the fetus have been associated with the decreased antigenic property of the extravillous trophoblasts with no classical major histocompatibility complex (MHC) class I [except human leukocyte antigen (HLA)-C] and class II molecules and the specialized immune cells at the maternal-fetal interface [1]. The pregnant endometrium (decidua) is replete with specialized immune cells, particularly natural killer (NK) cells (about 70%), macrophages (20–30%), regulatory T cells (Tregs; about 10%), and CD4+, CD8+, and dendritic cells (DCs; 5–10%). B cells do not represent a significant number of decidual leukocytes, particularly in humans [2–4]. These immune cells may interact with extravillous trophoblasts for maintaining immune tolerance and for promoting functions such as spiral artery remodeling. Among decidual immune cells, Tregs have been the subject of extensive studies for their roles in implantation and maintenance of pregnancy. In general, it has been described that Tregs play pivotal roles in many immune-associated disorders, including autoimmunity, infections, transplant rejection and cancer, making them attractive therapeutic targets [5–7]. Their clinical importance lies in the observations that reduced numbers and dysregulated function of Tregs are associated with the incidence of autoimmunity [8]. Notably, their elevated numbers have been observed in cancer tissues [9]. Modulation of their numbers and functions has shown therapeutic implications for Tregs in experimental models of autoimmunity and graft-versus-host disease [10].
In this review, we will highlight the characteristics of Tregs at the maternal-fetal interface and the mechanisms by which Tregs contribute to the establishment of maternal immune tolerance. We will further discuss how classical features of Tregs may be compromised in response to inflammation and infection during pregnancy. More importantly, we will explore whether this knowledge can be used to develop preventive and therapeutic approaches.
Inflammation and adaptive immune responses appear to contribute to adverse pregnancy outcomes. In normal pregnancy, embryo implantation is associated with inflammation which is followed by anti-inflammatory period of gestation [11]. Parturition again requires inflammation [11]. Gestation-associated anti-inflammatory mechanisms include the regulatory control by specialized CD4+CD25+ forkhead box protein 3 (FOXP3)+ Tregs [12]. These cells may play a pivotal role in suppressing inflammation and T cell effector activity, enhancing angiogenesis by cross-talking to invading trophoblasts, and interacting with other predominant uterine immune cells such as NK cells [12].
Tregs were rediscovered by Sakaguchi et al. [13] in 1990s. Although immune concept of suppressive T cells was rejected previously [14], reemergence of Tregs revolutionized the research on survival of tissues from cell-mediated effector immune responses and detrimental inflammatory milieu. The field of reproductive immunology has immensely benefitted from Treg’s well-known role in self-tolerance and in maintaining balanced immune responses [15, 16]. Interestingly, Tregs are thought to be essential in the generation of maternal-fetal tolerance in mice and humans [17–19]. Although the CD4+CD25+ phenotype may also be associated with activated CD4+ T cells, Tregs express their signature transcription factor, FOXP3, which activates genes required for suppressor functions and maintains the effector transcriptional program of the Treg lineage cells [20, 21]. These cells produce immunosuppressive cytokines interleukin-10 (IL-10) and transforming growth factor-β (TGF-β), an effector characteristic for exerting immune tolerance at the maternal-fetal interface [22–24]. On the other hand, it is still not known whether uterine Tregs like decidual NK (dNK) cells can turn into pregnancy foes in response to infection, inflammatory triggers and alterations in cytokines and hormones, leading to poor immune tolerance at the maternal-fetal interface [25]. This may explain altered immunological tolerance that has been linked to pregnancy-related complications such as recurrent spontaneous abortion (RSA), pre-term birth, gestational diabetes and preeclampsia (PE) [26].
In early pregnancy, endometrium contains an abundance of Tregs in both humans and mice [15, 19, 27–29]. Robertson et al. [18] showed functional significance of Tregs even at the time of implantation in the context of endometrium exposure to seminal fluid. Their work suggested that seminal fluid which contains cytokines, prostaglandins and growth factors prepared the implantation site to attract and expand Tregs [18]. These newly recruited and induced Tregs (iTregs) are eventually localized to the endometrium (Figure 1). Failure in this process may lead to implantation failure [19, 29–32]. The issue of their significance post implantation is still debated because the experiments in mice show that their post implantation depletion may not severely affect pregnancy outcome [12]. It is noteworthy, however, that transfer of iTregs to an abortion-prone mating or pregnancy challenged by inflammation or stress can be protected by adoptive transfer of Tregs [33, 34].
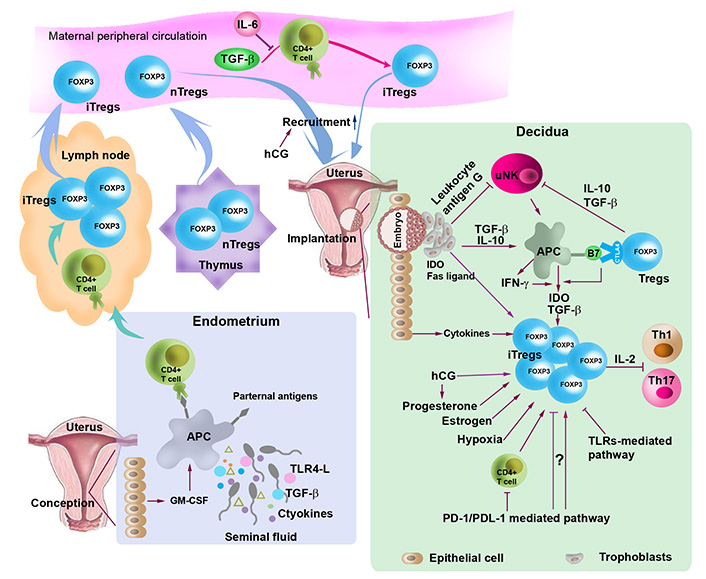
Regulation of the generation, expansion and recruitment to the uterine of Tregs. At conception, TGF-β, toll-like receptor (TLR) 4 ligand (TLR4-L), paternal antigens, and cytokines in seminal fluid and granulocyte-macrophage colony-stimulating factor (GM-CSF) released by uterine epithelial cells stimulate antigen-presenting cells (APCs), which carry paternal antigens, traffic to the lymph node and present the antigens to naive T cells, thereby activating naive T cells. Activated naive T cells proliferate and differentiate into Tregs. During implantation, expanded Tregs from the lymph node and the thymus are recruited into the uterus. Treg generation, expansion and recruitment are regulated by cytokines (e.g., TGF-β and IL-10) hormones [e.g., estrogen, progesterone and human chorionic gonadotropin (hCG)], indoleamine 2,3-doxygenase (IDO), Fas ligand, hypoxic microenvironment, programmed death 1 (PD-1)/PD-ligand 1 (PDL-1) and TLRs-mediated pathways. Tregs inactivate T helper 1 (Th1) and Th17 by sequestering IL-2 and inhibit uterine NK (uNK) cells through TGF-β and IL-10. Treg generation and expansion are also modulated through crosstalk with trophoblasts and uNK cells. nTregs: natural Treg; CTLA4: cytotoxic T-lymphocyte antigen-4
Another issue that has attracted a great deal of attention is thymic or extrathymic origin of uterine Tregs (Figure 1). Although it has been suggested that Tregs are important at the maternal-fetal interface, mice lacking the propagation of extrathymic Tregs due to absence of conserved non-coding sequence 1 (CNS1) enhancer element in the first intron of the FOXP3 gene do not exhibit any gestation-associated abnormalities [35], suggesting that this thymic-extrathymic uterine Tregs issue is still in need of a debate. Furthermore, thymus-derived Helios+ Tregs have been shown to be accumulated in the lymph node draining the uterus during early pregnancy (Figure 1). This may not preclude iTregs from being expanded and accumulated in the periphery during later stages of pregnancy [19].
Characteristically Tregs suppress proliferation of activated autologous CD4+CD25– T cells and this process involves cell-to-cell contact (Figure 1). What is not clear is whether decidual Tregs contribute to the mechanism of known or unknown fetal antigen-specific immune tolerance [17]. Another major caveat that remains unaddressed is whether uterine Tregs maintain their plasticity in response to inflammation and infection during pregnancy. This may also apply to circulating Tregs. Surprisingly, circulating Tregs also follow a similar pattern of pregnancy-dependent increase and decrease. It is possible that increase or decrease in peripheral blood Tregs during pregnancy is dependent on hormonal fluctuations, particularly that of hCG and progesterone [36, 37] (Figure 1). Indeed, our recent observations (Sharma et al., unpublished observations) and those of others suggest that mouse uterine Tregs are responsive to hCG which may promote their recruitment to the pregnant uterus [36]. Tregs significantly decline at term, probably in response to the onset of labor. This agrees with the recent observations of changes in Treg functions in preterm labor deliveries [27, 38, 39].
It has been shown that specialized Tregs that matched well-characterized Tregs markedly increased during pregnancy in mice [33] and humans [40–42]. This led to studies on Tregs in pregnant women who experienced adverse pregnancy outcomes. Decrease in the frequency of Tregs was observed in patients with spontaneous abortion [30] and this also was observed in abortion-prone DBA/2 mated CBA/J mice [43]. Tregs may be of consequence in other pregnancy complications such as PE, preterm birth and gestational diabetes and we discuss this issue elsewhere. It is now generally accepted that Tregs are a specialized subpopulation of T cells that regulate immune tolerance to self-antigens and suppress the immune response of other cells such as effector T cells (Teffs). Decidual Tregs undergo tightly regulated kinetics over gestation. Studies using mouse models demonstrate that Tregs start to rise in the preimplantation phase, reach the peak around gestational day 7.5–13.5 and then decline with advancing gestational age [12, 44]. Likewise, clinical studies show that decidual Tregs in pregnant women exhibit similar regulatory kinetics [44–46].
A recent study has described three types of decidual CD4+ Tregs in healthy human pregnancies: CD25HIFOXP3+, PD-1HIIL-10+, and TIGIT+FOXP3dim Tregs. These three types of Tregs have the ability to suppress the proliferation of CD4+ and CD8+ effector and cytotoxic T cells. Both CD25HIFOXP3+ and PD-1HIIL-10+ Tregs can regulate cytokine production by CD4+ and CD8+ T cells, including interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) [47]. Transcriptional profiling analysis reveals that these three types of Tregs have a distinct pattern of gene expression, which was validated at the protein level using flow cytometry. These differentially expressed genes can increase Treg activation and suppressive effect at the maternal-fetal interface [47].
How decidual Tregs are generated and recruited to the maternal-fetal interface has been the subject of intense investigation. A number of factors are implicated in the generation, expansion, and recruitment of Tregs at the maternal-fetal interface, including seminal fluid, fetal alloantigens, hormones, low oxygen tension, PD-1 pathway, and endometrial mesenchymal stem cells (Figure 1).
A body of evidence has demonstrated a crucial role for seminal fluid priming in eliciting the expansion of the Tregs population in early pregnancy (Figure 1) [46]. In support of this, removal of seminal vesicle gland attenuates Tregs activation and abolishes the expansion of Treg pool in mice [48–50]. In addition, prior seminal fluid contact increases implantation success in in vitro fertilization (IVF) treatment cycles [51]. Seminal fluid contains paternal alloantigens, abundant TGF-β, and TLR4-Ls as well as an array of cytokines [52]. Paternal antigens in seminal fluid in the vaginal lumen stimulate APCs such as DCs and macrophages. Moreover, TGF-β and TLR4-Ls in seminal fluid can provoke an inflammation-like response in the female reproductive tract. These processes can trigger DCs and macrophages to carry paternal alloantigens to the regional lymph nodes and present antigens to naive T cells, leading to a transformation of effector CD4+ cell into iTregs [16, 53] within days of conception and then in the spleen and peripheral blood. Circulating Tregs are then recruited to the implantation site in response to epithelial cell-expressed and -secreted chemokines and their receptors [46]. It is proposed that this process contributes to the establishment and maintenance of immune tolerance to fetal alloantigens during embryo implantation [46].
TGF-β is a pivotal differentiation factor for promoting conversion of peripheral CD4+CD25– naive T cells to CD4+CD25+ Tregs through increasing FOXP3 expression [54]. However, in the presence of IL-6, TGF-β fails to induce the generation of FOXP3 Tregs; instead, IL-6 and TGF-β together increase the differentiation of Th17 cells from naive T cells in mice (Figure 1) [55]. These findings suggest that TGF-β-mediated induction of FOXP3+ Tregs depends on the state of the innate immune system and the production of inflammatory cytokines such as IL-6 [55].
Pregnancy-associated hormones, including estrogen, progesterone, and hCG, are key pregnancy hormones, playing an immune modulatory role (Figure 1). However, whether and how these hormones modulate the expansion of Tregs remains poorly understood. In this regard, Aluvihare et al. (2004) [33] found that the expansion of CD4+CD25+ FOXP3+ Tregs during pregnancy is alloantigen-independent in syngeneically mated female mice, suggesting the role of pregnancy hormones. Additionally, transferring CD4+CD25+ Tregs from syngeneically mated female mice can protect allogeneic fetuses from rejection [33, 56, 57]. These results suggest that Treg expansion may be associated with hormonal modulation. Corroborating studies report that prior to conception and implantation, nTregs expand systemically and accumulate in the uterus in response to increased estradiol at the time of ovulation in mice [44, 58]. In vitro and in vivo experiments have shown that estrogen treatment elicits the expansion of Tregs through binding to estrogen receptor α. In vitro studies demonstrate that estrogen enhances the conversion of CD4+CD25– T cells to Tregs through elevating FOXP3 messenger RNA (mRNA) expression [59, 60]. Estrogen-induced expansion of Tregs occurs during embryo implantation [60]. Likewise, administration of progesterone not only increases the proportion of Tregs in peripheral blood, spleen, and inguinal lymph nodes, but also enhances the suppressive activity of Tregs in ovariectomized mice during midterm pregnancy [61]. Progesterone increases the expansion of Tregs through binding to the nuclear progesterone receptors [61]. On the other hand, progesterone can bind to the glucocorticoid receptor, leading to an increase in the proportion of Tregs and apoptosis of Teffs [62]. A recent study has revealed that progesterone drives the development of nTregs through the osteoclast differentiation receptor, the receptor activator of nuclear factor kappa B (NF-kB) ligand (RANK), and genetic deletion of RANK leads to reduced accumulation of nTregs in the mouse placenta and increases miscarriages [63]. In this regard, knockout of progesterone receptor on CD11c+ DCs in mice decreases the frequency of CD4+FOXP3+ and CD8+CD122+ Tregs, which is associated with intrauterine growth restriction [64]. The progesterone receptor membrane component 1 (PGRMC1) in the endometrial glands is decreased in women with recurrent miscarriage [65]. However, alteration of PGRMC1 expression inversely correlates with the number of infiltrated FOXP3+ Tregs in patients with recurrent miscarriage. This was interpreted by the authors as a compensatory response to the increased uNK endometrial infiltration [65].
hCG is a critical pregnancy hormone that can be detected during the very early days post-implantation, reaches the peak around gestational week 12, and then remains low during gestation until birth [36]. hCG is a trophoblast-produced hormone [66, 67] and has a myriad of functions in pregnancy, including the promotion of progesterone production, implantation and decidualization, angiogenesis, cytotrophoblast differentiation, and immune cell regulation [36]. Low levels of hCG and Treg numbers have been observed in women with spontaneous abortion, raising the question of whether hCG can influence Treg generation and expansion. Indeed, accumulated evidence has shown that hCG functions as a chemoattractant for recruiting Tregs at the maternal-fetal interface, contributing to immune tolerance during pregnancy [37, 68]. Administration of hCG significantly increases the frequencies of Tregs in pregnant but not nonpregnant mice [68]. In vitro studies have shown that hCG can augment the induction of CD4+FOXP3+ Tregs from CD4+FOXP3– T cells, which can be inhibited by blockade of hCG [68]. In abortion-prone mice, administration of hCG during peri-implantation upregulates the accumulation of Tregs in the endometrium, thereby reducing the fetal resorption rate [37].
Interestingly, based on these observations, intrauterine administration of hCG before embryo transfer has been employed as a therapeutic strategy to improve IVF-embryo transplantation outcomes [69–73] and enhance the live birth rate in women with repeated implantation failure [69, 74–76]. Subcutaneous injection of hCG in women prior to IVF can increase the production of anti-inflammatory IL-27 and IL-10 and numbers of Tregs in the peripheral blood while decreasing the abundance of inflammatory IL-17 [77]. Likewise, intramuscular injection of immunoglobulin-combined hCG induces a decrease in the number of Th17 cells and production of IL-17 and IL-6, and an increase in the number of Tregs and abundance of IL-10 and TGF-β1 in patients with unexplained RSA [78]. A recent study reports that intrauterine infusion of hCG enhances recruitment of Tregs to the endometrium in patients with repeated implantation failure, which may be mediated by inducing the production of chemokine C-C motif chemokine ligand 2 (CCL2) [79]. Taken together, hCG plays pivotal roles in attracting Tregs to the endometrium and elevating the numbers and suppressive capacity of Tregs at the maternal-fetal interface.
PD-1 receptor and PDLs, PDL-1 and PDL-2, are expressed on hematopoietic cells, including conventional CD4+ and CD8+ T cells, nonhematopoietic cells, and cancer cells. PDL is a type-1 transmembrane protein. The binding of PDL to the inhibitory checkpoint molecule PD-1 transmits inhibitory signals that limit the immune responses of effector and cytotoxic T cells, regulate the tolerance and exhaustion of T cells, and resolve inflammation. Thus, the PD-PDL pathway plays an inhibitory role in regulating the magnitude of adaptive immune response and tolerance. Notably, blockade of PD-1 and PDL-1 has been utilized for efficacious treatment of over 20 types of cancers [80]. The role of PD-1-PDL-1 negative costimulatory pathway in the maintenance of tolerance has been described in multiple transplant models, a diabetes model, and at the maternal-fetal interface of an alloantigen-specific CD4+ TCR transgenic mouse model [81, 82].
Studies from Guleria et al. [81] provided for the first time that blockade of PDL-1 using anti-PDL-1 monoclonal antibody leads to an increase in the rate of spontaneous fetal resorption in allogeneic pregnancy mouse model, and the expression of PDL-1 on the surface of Tregs is essential for their suppressive function and maternal immune tolerance [83]. PDL-1 has the ability to enhance and sustain FOXP3 expression and promote iTreg development and suppressive function through downregulation of phosphor-Akt, mechanistic target of rapamycin (mTOR), and extracellular signal-regulated kinase 2 (ERK2) and concomitant with an upregulation of phosphatase and tensin homolog (PTEN) [84]. PDL-1–/–PDL-2–/–Rag–/– naive CD4+ T cells display a marked reduction in iTreg conversion and rapid onset of a fatal inflammation [84]. A deficiency of PDL-1 induces apoptosis of Tregs, decreases the ratio of Tregs to Teffs, and promotes Th1 and Th17 cell development and expansion, leading to fetal rejection and reduction in fetal survival [82, 83]. Furthermore, neutralization of IL-17 or adoptive CD25+FOXP3+ Treg transfer restores Treg numbers, thereby rescuing the effects of PDL-1 deficiency on fetal survival rate [82]. In addition, interaction of PDL-1 with PD-1 has been shown to induce the conversion of human Th1 cells into Tregs [85].
In contrast, a most recent study demonstrates an inhibitory effect of PD-1 on Tregs in mice in which deletion of PD-1 can be selectively induced in Tregs by tamoxifen administration. Selective deletion of PD-1 in Tregs in tissues such as the thymus and spleen of mice leads to an increase in their immunosuppressive capacity. Interestingly, PD-1-deficiency in Tregs ameliorates experimental autoimmune encephalomyelitis and protects nonobese diabetic mice, the mouse model of spontaneous autoimmunity, from developing type 1 diabetes. Mechanistically, the phosphatidylinositol 3-kinase (PI3K)-AKT pathway is responsible for the increased suppressive activity of PD-1 deficient Tregs [86].
TLRs are a large family of pattern recognition receptors that are expressed both on trophoblasts and immune cells [87, 88]. TLRs expressed on CD4+CD25+ Tregs can regulate the expansion and suppression of Tregs [89, 90]. Activation of TLR9 by oligodeoxynucleotide (ODN)1826 decreases the proportion of Tregs in the decidua of abortion-prone mice [91]. In vitro experiments show that TLR9 activation inhibits the proliferation of Tregs and induces apoptosis of Tregs through caspase pathway. These TRL9-mediated effects can be abrogated by antagonist ODN [91]. Thus, TLR9-mediated signaling plays an inhibitory role in the generation of Tregs. In contrast, TLR4 knockout mice exhibit reduced pregnancy rate, increased mid-gestation fetal loss and fetal growth restriction, failed expansion of peripheral and thymic CD25+FOXP3+ Tregs, and fewer Tregs expressing Ki67 proliferation marker and suppressive function marker CTLA4. This suggests that TLR4 is a positive mediator for the generation of Tregs [92]. Thus, TLR members may play differential roles in the regulation of Tregs.
Hypoxic microenvironment in tumors has been shown to promote recruitment of Tregs, which in turn enhances immune tolerance and angiogenesis in tumors [93, 94]. Interestingly, the physiological hypoxic microenvironment also occurs at the maternal-fetal interface during early pregnancy. Emerging evidence has shown a role for hypoxia in the recruitment and expansion of Tregs during early gestation. It has been proposed that hypoxia-inducible factor-1α (HIF-1α) deficiency can reduce the recruitment of dNK cells into the maternal decidua [95], suggesting that hypoxia promotes the infiltration of dNK cells into the maternal-fetal interface. It is unclear whether a hypoxic environment directly induces recruitment of Tregs to the decidua during early gestation. However, recruited dNK cells can enhance the induction and expansion of Tregs at the maternal-fetal interface [96].
The role of mesenchymal stem cells from different origins such as bone marrow, adipose tissue, and endometrium in induction of Tregs has recently emerged. A recent study by Aleahmad et al. [97] shows that pretreatment of menstrual mesenchymal stem/stromal cells (MnSCs) with IFN-γ and IL-1β increases the ability of Tregs to inhibit the proliferation of anti-CD3/CD28-stimulated peripheral blood mononuclear cells (PBMCs), while non-treated MnSCs decreases the frequency of Tregs. This effect is regulated through IL-6, IL-10, TGF-β, and IDO [97]. These results suggest a pivotal role of endometrial stem cells in local induction of Tregs in the endometrium.
The semi-allogenic fetus is capable of inducing maternal immune response during pregnancy; however, the fetus is not rejected, rather tolerated by the maternal immune system. The mechanisms underlying the immune balance between tolerating fetal growth and simultaneously defending a pathogen infection involve a complicated interplay of maternal immune cells and trophoblasts. Studies based on human and animal experiments have shown that Tregs regulate immune tolerance through cell-cell contact dependent and independent pathways.
Contact-dependent immune-suppressive activities of Tregs are mediated by IL-2 receptor α (IL-2Rα; CD25) and CTLA4, both of which are expressed on Tregs. Neutralization of circulating IL-2 with anti-IL-2 monoclonal antibodies inhibits Treg proliferation, leading to autoimmune diseases [98–101]. IL-2 and IL-2Rα-deficient mice die from severe lymphoproliferation and autoimmune disease in early life. IL-2 and IL-2Rα-mediated signaling are essential for the development and maturation of Tregs both in the thymus and in the periphery [102]. IL-2 has higher affinity to IL-2R on Tregs than Teffs, thereby preferentially promoting the expansion of iTregs. Consumption of IL-2 by Tregs suppresses the expansion and activity of Th17 cells [103]. On the other hand, CTLA4 on Tregs has a higher affinity to CD80/CD86 on APCs than CD28 on Teffs. The binding of CTLA4 to CD28 induces trans-endocytosis, thereby removing CD28 from APCs. CD28 is a costimulatory molecule that can increase the activation of Teffs. Thus, CTLA serves as a suppressive regulator of Tregs for inhibiting Teffs through competition with CD28.
In addition to the cell-cell contact dependent pathway, in vivo studies have shown that Tregs also mediate immune tolerance through secreting IL-10 and TGF-β, both of which play crucial roles in regulating the expansion and suppressive activity of Tregs at decidual microenvironment [104]. IL-10 expressed in Tregs exerts an auxiliary role in enhancing their expansion and immunosuppressive activities. IL-10 deficient mice or mice treated with IL-10R blockers suffer from chronic enterocolitis and intestinal inflammation. Mice with IL-10R-deficient Tregs have high levels of IL-17 and are susceptible to developing severe colitis. Mice with selective disruption of IL-10 expression in Tregs exhibit spontaneous colitis [104]. Although the role of TGF-β in regulating the function of Tregs is controversial, many studies have shown that TGF-β secreted by Tregs contributes to maintenance immunosuppressive function through binding to TGF-β receptor. This conclusion is supported by a body of in vitro and in vivo evidence. TGF-β treatment increases the conversion of naive T cells into Tregs [105]. Selective depletion of TGF-β expression in Tregs or administration of anti-TGF-β antibodies inhibits the suppressor function of Tregs. Tregs restrain immune responses through a mechanism that is dependent on the expression of FOXP3 [104]. Peripheral blood Tregs isolated from TGF-β knockout mice exhibited a dramatically reduced FOXP3 expression; however, the addition of TGF-β rescues FOXP3 abundance. Moreover, TGF-β enhances the conversion of peripheral CD4+CD25– naive T cells to CD4+CD25+ Tregs through induction of FOXP3 expression [54]. Mice with impaired TGF-β signaling have significantly reduced numbers of peripheral CD4+CD25+ Tregs [106, 107] and readily develop colitis. These pathological alterations in mice can be prevented by transferring wild-type Tregs. Thus, TGF-β signaling is required for the in vivo expansion and suppressive capacity of Tregs [106]. These data indicate an essential role for TGF-β in maintaining peripheral Tregs in vivo [107].
Interestingly, after delivery, alloantigen specific Tregs have the capability to maintain an immune memory for tolerance to pre-existing fetal antigens. These Tregs can rapidly proliferate during subsequent pregnancy [108]. In line with these results, the incident rate of adverse pregnancy complications such as PE and miscarriage is reduced in the secondary pregnancy with a short interval from the first pregnancy [109].
Tregs-mediated immune tolerance also involves crosstalk between Tregs and other immune cells as well as trophoblasts at the maternal-fetal interface. Crosstalk among these cells plays an essential role in regulating trophoblast invasion and in promoting spiral artery remodeling [12, 110]. dNK cells are the major population of immune cells (about 70%) in human and rodent decidua during early pregnancy and possess unique phenotypic and functional properties [96, 110]. dNK cells play a pivotal role in regulating Treg induction and their immunosuppressive activity [96]. dNK cells reside in close contact with CD14+ macrophages in decidual tissues. Vacca et al. [96] showed that dNK cells when co-cultured with CD14+ macrophages exhibit an increase in the production and secretion of IFN-γ, which in turn enhances the expression and release of IDO and TGF-β by CD14+ macrophages. IDO, an enzyme that catabolizes tryptophan into L-kynurenine, can prevent T cell activation and promote Treg induction through tryptophan deprivation. L-kynurenine inhibits the secretion of IFN-γ by peripheral blood NK cells, but not dNK cells. Thus, dNK cells when interacting with CD14+ macrophages can promote Treg proliferation and their immunosuppression through IFN-γ-induced IDO and TGF [96]. This interaction-mediated induction of Tregs is dependent on CTLA4 but not PD-1 on Tregs [91]. Interestingly, co-culture involving either NK cells or CD14+ cells isolated from peripheral blood failed to alter the induction and immunosuppression of Tregs. Thus, cell-cell interactions in the decidua may be important in maintaining Treg pool. Since dNK cells and Tregs decrease after second trimester, it is possible that they require each other for their self-maintenance.
Trophoblasts can regulate the induction of Tregs at the maternal-fetal interface through multiple pathways. For instance, trophoblasts express IDO and Fas ligand, which in turn mediate the induction of Tregs [111, 112] (Figure 1). Fas ligand can induce apoptosis of activated maternal T cells that express Fas receptors [112]. Trophoblast also express HLA-G, a non-classical MHC class I molecule, which in turn inhibits NK cell cytotoxicity, mediates T cell activation through the induction of tolerogenic DCs, and elicits the expansion of Tregs. In addition, trophoblasts can secrete inhibitory cytokines such as IL-10 and TGF-β [113, 114] and express PDL-1 [115], all of which can regulate the proliferation and expansion of Tregs (Figure 1).
As discussed above, generation, recruitment, and expansion of Tregs are regulated by a myriad of factors such as seminal fluid priming, hormones, hypoxia, cytokines, and interaction with other immune cells and trophoblasts. Therefore, it is possible that dysregulation of these factors may alter the microenvironment that negatively influences the quantity and function of Tregs, leading to pregnancy-related complications (Figure 2). Pathogenic conversion of FOXP3+ Tregs into Th17 cells has been reported in autoimmune arthritis [116]. Although it is unclear whether Tregs can directly convert into inflammatory Th17 cells in response to pathological conditions at the maternal-fetal interface [117], dysfunction and reduced numbers of Tregs have been associated with adverse pregnancy complications such as RSA, implantation failure, and PE [118]. Emerging evidence has shown that the accumulation of immune-suppressive Tregs in pregnant mice may increase infection susceptibility to intracellular pathogens [119], and that Tregs may transform to toxic Th cells in the inflammatory milieu (Figure 2) [120].
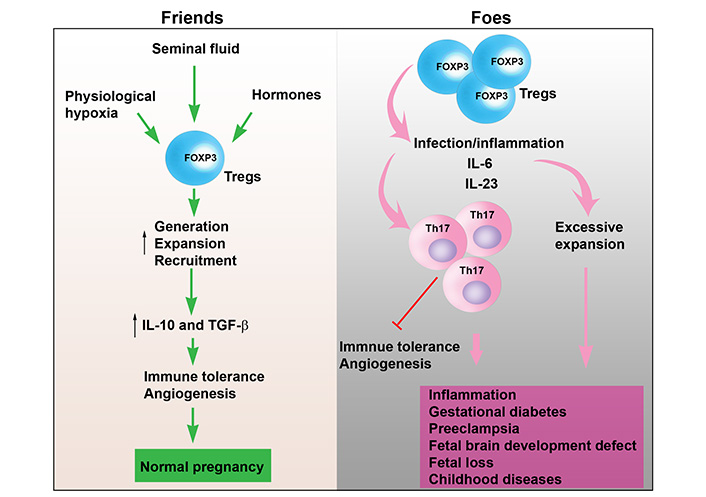
Uterine Tregs as “Friends” or “Foes”. In normal pregnancy, various factors such as seminal fluid, physiological hypoxia, and hormones elevate the generation, recruitment and local expansion of Tregs in the uterine microenvironment. This leads to elevated production and release of IL-10 and TGF-β from uterine Tregs. As a result, immune tolerance is maintained, and angiogenesis is induced. Thus, uterine Tregs function as “Friends” promoting normal pregnancy. However, under pathological conditions, Tregs can be converted into Th17 cells in response to infection and inflammatory cytokines, such as, TNF-α, IL-6 and IL-17, leading to inhibition of immune tolerance and angiogenesis. These cytokines released from inflammatory Tregs can contribute to inflammation, gestational diabetes, PE, fetal brain development defects, fetal loss, and childhood diseases. In addition, excessive expansion of Tregs may increase susceptibility to intracellular bacteria. Therefore, uterine Tregs may act as “Foes” contributing to adverse pregnancy outcomes, cytokine storm, and fetal brain development
RSA is characterized by three or more consecutive pregnancies. Although the etiology of this pregnancy adversity is still unclear, accumulating evidence has shown impaired immune tolerance as a contributing factor to RSA. In the mouse model of abortion, diminished activity of Tregs and subsequent accumulation of paternal alloantigen specific Th1 cells were observed in the decidua of abortion mice [43]. Notably, adoptive transfer of Tregs from normal pregnant mice protects against fetal rejection [43, 121]. Likewise, a significantly reduced quantity and function of Tregs was observed at the maternal-fetal interface of abortion-prone mice or mice with abortion induced by progesterone antagonist [61]. Furthermore, administration of TLR4 antagonist E5564 to abortion-prone mice increases the proportion of Tregs and expression of FOXP3 mRNA and proteins and decreases inflammatory response, thereby reducing embryo absorption rate [122].
Clinical studies have demonstrated a markedly reduced proportion and function of Tregs and an imbalance of the Th1, Th2, Th17 and Tregs in the decidua and peripheral blood from patients with spontaneous abortions or recurrent implantation failure compared to controls [30, 118, 122–126]. The expression of FOXP3, a determinant for the function of Tregs, was found to be significantly lower in both the decidua and peripheral blood from women with unexplained RSA [122, 125]. In support of this, Tregs from women with RSA exhibit reduced ability to expand during the pre-implantatory phase relative to those from fertile women [118]. In addition, increased ratios of Th1/Th2 and Th17/Tregs have been detected in peripheral blood from patients with RSA [126–128]. In vitro studies have revealed that Tregs from patients with RSA display inhibited ability to suppress the proliferation of Th17 cells and their secretion of IL-17 via cell-cell contact and mediation by IL-10 and TGF-β [118]. Additionally, higher levels of TLR1, TLR3, TLR4, TRL8, and TLR9 gene expression were observed in the decidua of patients with RSA [89]. Given that TLRs activation can inhibit the proliferation of Tregs [91], increased levels of TLRs may contribute to inhibition of Tregs expansion and suppressive activity in women with RSA. Taken together, decreased proportion and function of Tregs or their conversion to Th17 cells in response to inflammation [117] may disturb maternal immune tolerance against fetal rejection and subsequently increase inflammation, thereby leading to implantation failure and abortion.
PE is a multifactorial, pregnancy-specific syndrome characterized by new onset of hypertension at or after 20 weeks of gestation. It is a leading cause of maternal morbidity and mortality and affects 5–8% of all pregnancies. While the etiology of PE is still unclear, accumulating evidence suggests that impaired maternal immune tolerance contributes to the pathophysiology of PE. Inadequate paternal antigen-specific immune tolerance has been associated with an increased risk of PE. An epidemiological study unraveled that women inseminated with donor sperm have an increased risk of PE compared to those with their partner’s sperm [129]. Primiparous pregnancy, long interval between pregnancies, first pregnancy after changing partner, and reduced time length of exposure to seminal fluid have been linked to a substantially elevated risk of developing PE [130–132]. These clinical observations point to an important role of paternal antigen exposure-induced immune tolerance and immunological memory in the mechanisms for maintaining normal pregnancy and protecting against the development of PE during subsequent pregnancies [132].
As discussed earlier, the expansion of FOXP3+ Tregs is essential for maternal-fetal immune tolerance in normal pregnancy. Experimental data indicate that abnormalities of Tregs in the decidua and peripheral blood contribute to the etiology of PE [133–137]. For instance, reduction in FOXP3 expression and elevation in Th17-specific transcription factor retinoic acid related orphan receptor (ROR)ɤt and IL-17 presence have been observed in peripheral blood from patients with PE. An imbalance of Th17 cells/Tregs and anti-/pro-inflammatory cytokines illustrates a disturbed immune tolerance in PE patients [138]. In addition, many studies demonstrate that the expansion and suppressive activity of Tregs declines in PE patients [26, 136]. In vitro experiments show that a subset of CD14+ APCs, CD14+DC-specific intercellular adhesion molecule 1-grabbing nonintegrin (SIGN)+ APCs, from healthy pregnant women significantly increase the induction of iTregs; however, these cells from PE patients have a decreased ability to induce iTregs. Thus, these results suggest that reduced expansion of iTregs in the decidua of PE patients may be caused by decreased proportion of CD14+DC-SIGN+ APCs in the decidua [26].
FOXP3+ Tregs play an essential role in maintaining the balance between immune suppression and immune activation for host defense against infection at the maternal-fetal interface [12, 119]. It has been reported that influenza A virus infection in pregnant women has serious consequences as evidenced by markedly higher rates of hospitalization, morbidity and mortality [139]. It has been shown that expanded FOXP3 Tregs markedly impair host defense and increase susceptibility to intracellular bacterium Listeria monocytogenes in pregnant mice relative to nonpregnant controls at mid-gestation, and ablation of maternal Tregs beginning mid-gestation reverses the pregnancy-associated susceptibility to bacterial infection [119]. These results may explain why this bacterium has a striking predilection for disseminating infection in pregnant women [140]. Furthermore, mice containing IL-10-deficient Tregs exhibit stronger resistance to Listeria monocytogenes infection compared to controls in pregnant [119] or nonpregnant mice [141], and cell intrinsic IL-10 is required for Treg-mediated infection susceptibility [119]. Additionally, Adetunji et al. [142] reported that inactivation of Tregs using TNF receptor II (TNFR2) antagonistic antibody remarkably reduces Brucella abortus bacterial burden systemically and locally in reproductive tissues, suggesting that highly suppressive CD4+FOXP3+TNFR2+ Tregs are responsible for the persistent of Brucella abortus infection.
Importantly, Tregs may switch from suppressive to inflammatory phenotype in the aberrant environment. Emerging evidence has shown that nTregs can convert into Th1, Th17 and T follicular helper cells in an inflammatory milieu rich with inflammatory cytokines, such as IL-1, IL-6, and IL-23 [120]. Mechanistically, toxic conversion of nTregs may be caused by downregulation of FOXP3 expression. nTregs-converted Th17 cells can produce IL-17 and chemokine receptor CCR6 [120]. In contrast, iTregs are stable in the inflammatory milieu because they are resistant to conversion into Th1, Th2, and Th17 cells [120]. These observations suggest that nTregs and even iTregs may play distinct roles in controlling the adaptive immune response [120].
Tregs may also be subjected to transdifferentiation and become hybrid Treg-Th17 cells in response to temporal viral infections. These cells may acquire inflammatory phenotype by virtue of producing IL-17 and cause fetal brain development abnormalities. Their depletion or conversion to normal Tregs in mid-pregnancy may reduce the toxic effects caused by temporal viral infections (Jash and Sharma, unpublished results) [117].
Tregs account for critical immune cell population proportion in the decidua during early pregnancy. Numerous studies have demonstrated an essential role for Tregs in successful pregnancy through maintaining immune tolerance at the maternal-fetal interface. Treg generation, expansion and recruitment to the endometrium are initiated and regulated by multiple stimuli including exposure to seminal fluid, paternal antigens, the cytokines and chemokines, hormones, and interaction with trophoblasts and other immune cells such as dNK cells (Figure 1). Reduced numbers and activity of FOXP3+ Tregs in the peripheral blood and decidual tissue have been linked to the pathophysiology of infertility, recurrent spontaneous miscarriage, and PE. Emerging clinical studies have shown that targeting Tregs is a promising therapeutic strategy for improving pregnancy outcomes in IVF patients and treating pregnancy complications, including spontaneous abortion and PE.
APCs: antigen-presenting cells
CTLA4: cytotoxic T-lymphocyte antigen-4
DCs: dendritic cells
dNK: decidual natural killer
FOXP3: forkhead box protein 3
hCG: human chorionic gonadotropin
IDO: indoleamine 2,3-doxygenase
IFN-γ: interferon-γ
IL-10: interleukin-10
IL-2Rα: interleukin-2 receptor α
iTregs: induced regulatory T cells
IVF: in vitro fertilization
NK: natural killer
nTregs: natural regulatory T cells
PD-1: programmed death 1
PDL-1: programmed death-ligand 1
PE: preeclampsia
RSA: recurrent spontaneous abortion
Teffs: effector T cells
TGF-β: transforming growth factor-β
Th1: T helper 1
TLR: toll-like receptor
TLR4-L: toll-like receptor 4 ligand
Tregs: regulatory T cells
uNK: uterine natural killer
SS supervised the work. All authors contributed to writing the manuscript and approved the submitted version.
The authors declare that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported in part by the NIH P20 GM121298, 3P20GM121298-04W1 and P30 GM114750 grants and Brown University Seed Award. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Gustaaf Albert Dekker, Pierre Yves Robillard
Kushaan Khambata ... Satish K. Gupta
Gursaran P. Talwar ... Krishna M. Ella
Julia Szekeres-Bartho
Shigeru Saito ... Sayaka Tsuda
Noémie Abisror ... Arsene Mekinian
Ruchi Sachdeva, Rahul Pal
Betcy Susan Johnson, Malini Laloraya
Alison McCallion ... Chandrakant Tayade
Pier Luigi Meroni ... Francesco Tedesco
Marijke M. Faas
Alaa Kazhalawi ... Nathalie Lédée
Chiara Agostinis ... Roberta Bulla
Mickey V. Patel ... Charles R. Wira
Thanh Luu ... Joanne Kwak-Kim
