
Abstract
Human γδ T cells are unconventional lymphocytes that function in innate and adaptive immune responses and immunosurveillance. These cells show potent cytotoxicity against tumor cells in a major histocompatibility complex unrestricted manner and have recently gained considerable attention as a sparkling star for clinical immunotherapy. Clinical immunotherapy trials with activated γδ T cells are tolerated well. However, clinical benefits are still unsatisfactory. Therefore, anti-tumor effects need to further increase the cytotoxicity of γδ T cells via several mechanisms, including the novel nitrogen-containing bisphosphonate products, adjuvant use with a bispecific antibody and chimeric antigen receptor, co-immunotherapy with γδ T cells plus immune checkpoint inhibitors, and adoptive immunotherapy with Vδ1 T cells and T cells engineered to express a defined γδ T cell receptor. Here, this article describes the crucial role of γδ T cells in anti-tumor immunity, concludes transduction strategies and summarizes the different development of novel approaches for clinical applications and cancer immunotherapy, which may be effective in overcoming current therapeutic limitations.
Keywords
γδT cell, immunotherapy, adoptive cell therapyIntroduction
Human γδ T cells are the unique T cell subset by expression of γδ heterodimeric T cell receptor (TCR) on the cell surface. These cells only account for 1–5% of T cells in peripheral blood (PB), lymphatic circulation, and mucosal tissues but undergo rapid expansion in response to pathogens, inflammation and tumor. γδ T cells share innate and adaptive immune cells [1]. Unlike conventional αβ T cells, γδ T cells are non-major histocompatibility complex (MHC)-restricted in different antigen recognition mechanisms. First, γδ T cells undergo somatic recombination and express rearranged TCR genes when maturing in the thymus as conventional αβ T cells. However, γδ T cells are activated independence of antigen processing and presentation by MHC molecules given that γδ TCR and other functional receptors expressed on γδ T cells can directly recognize ligands and trigger their activity.
Except γδ TCR, γδ T cells express other functional receptors to distinguish between tumor cells and normal tissue. The natural killer (NK) group 2 member D (NKG2D) receptor expressed on γδ T cells recognizes a broad and structurally diverse range of the NKG2D ligands (NKG2DL) including MHC I chain-related molecules A/B (MICA/B) and cytomegalovirus unique long 16 (UL16)-binding proteins 1–6 (ULBP1–6), which are generally absent on normal cells but upregulated on tumor cells [2]. DNAX accessory molecule 1 (DNAM1) also contributes to the activation of γδ T cells, which recognizes nectin-2 and polyoma virus receptor (PVR). Furthermore, other natural cytotoxicity receptors (NCR), such as NKp30 and NKp44, help γδ T cells in recognizing leukemia cells [3–6].
Human γδ T cells are divided into two main populations in accordance with their TCR usage of the Vδ1 or Vδ2 chain. The majority of γδ T cells in PB is Vδ2 subset, which is paired with the Vγ9 chain. Vδ1 subset resides in mucosal epithelial tissues and has adaptive features. Vγ9Vδ2 T cells are known to identify phosphoantigens (PAgs) such as (E)-4-hydroxy-3-methylbut-2-enyl pyrophosphate (HMBPP) synthesized in the bacterial isoprenoid biosynthesis pathway, and isopentenyl pyrophosphate (IPP), which are intermediates of the cholesterol synthesis pathway in eukaryotic cells. Besides, butyrophilin subfamily 3 member A1 (BTN3A1) and BTN2A1 molecules are considered to play an important role in Vγ9Vδ2 T cell activation by PAgs [7–10]. PAgs bind to the intracellular B30.2 domains, which recruits the cytoskeletal adaptor protein periplakin, the Rho family member B (RhoB), and the enzyme that cleaves guanosine triphosphate (GTP; GTPase) [11, 12], which rearranges cytoskeleton and reduces BTN3A1 membrane mobility. Subsequently, BTN3A1 undergoes conformational change and associates with the extracellular domain of BTN2A1, which can directly bind to the germline-encoded regions of Vγ9 [8, 9]. The metabolic reprogramming of tumor cells increases the activity of mevalonate pathway and bacterial infection, leading to PAg upregulation. Nitrogen-containing bisphosphonates (NBPs), such as zoledronate acid (ZOL) and alendronate acid (ALD), inhibit farnesyl diphosphate synthase (FDPS), which are the rate-determining enzyme in the mevalonate pathway, resulting in the intracellular accumulation of IPP [13]. Besides, F1-ATPase-related structure has been detected in tumor cells, and the apolipoprotein A-I (apo A-I) and DNA mismatch repair protein human MutS homolog 2 (hMSH2) expressed on the surface of target cells are also considered to be actively recognized by Vγ9Vδ2 TCR [14]. Moreover, the subset expressing the Vδ1 chain paired with any Vγ chain is mostly distributed in the mucosa and frequently co-expresses functional receptors of innate immune cells. Vδ1+ γδ T cells can recognize lipids and glycolipids presented by CD1 molecules. In addition, Vγ9Vδ2 and Vδ1+ γδ T cells are activated by heat shock proteins (HSP).
After activation, γδ T cells exert cytotoxic effects against cancer cells in several pathways similar to conventional cytotoxic T cells. First, γδ T cells secrete molecules, such as pore-forming molecule perforin and pro-apoptotic protease granzyme B, to kill cancer cells. Except the perforin-granzyme axis, γδ T cells induce cancer cell apoptosis through Fas ligand (FASL) and tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) binding to receptors on cancer cell, including Fas and TRAIL receptor (TRAILR). Besides, CD16 known as Fcγ receptor III binds to antibodies, resulting in induced antibody-dependent cellular cytotoxicity (ADCC) by γδ T cells. γδ T cells also exert anti-tumor function through indirect effects by influencing other immune responses. Activated γδ T cells secrete cytokines, such as TNF-α, interferon-gamma (IFN-γ), and interleukin-2 (IL-2). In addition, γδ T cells work as antigen presentation cells (APC) to activate peptide-specific T cells and stimulate NK cell cytotoxicity through 4-1BB [15].
Based on their ability to produce cytokines, the functional phenotype of γδ T cells is highly plastic and can be divided into several subtypes. γδ T cells polarize to T-helper 1 (Th1)-like pattern under Th1-priming conditions, including PAg activation and cytokines IL-2, IL-12, and IL-15. Th1-like γδ T cell is the main functional population that exerts cytotoxicity effects against tumor cells [16, 17]. Under Th2-priming condition (IL-4 and anti-IL-12 antibody), γδ T cells are driven to the Th2-like phenotype that produces IL-4 [16]. IL-21 is a potent immunomodulatory cytokine which enhances the effector functions of NK cells and cytotoxic T cells. IL-21 also contributes to acquiring the follicular Th phenotype by human Vγ9Vδ2 T cells, whose surface expresses C-X-C chemokine receptor type 5 (CXCR5). This phenotype of γδ T cells secretes C-X-C chemokine ligand type 13 (CXCL13) to attract the B cell to germinal centers [18, 19]. Except for effector cells, γδ regulatory T cells (Tregs) or inhibitory γδ T cells can regulate immune balance and maintain immune tolerance [20–22]. γδ T cells control the immune responses in vivo models of multiple organs from inflammation, allergy, and cancer, such as kidney, lung, heart, and brain [23, 24]. The forkhead box P3 (Foxp3)-positive Tregs-like phenotype can be induced in the presence of IL-15 and transforming growth factor-β (TGF-β) and exert immunosuppressive through multiple mechanisms [25, 26]. γδ Tregs can secrete TGF-β and IL-10, which inhibit the activity of T cells and induce Tregs [27–33]. Through combined stimulation with cytokines, like IL-1β, IL-6, IL-23, TGF-β, γδ T cells can be driven to the IL-17-producing phenotype (γδ T17) [34]. γδ T17 is considered to be a pro-tumor phenotype attracting attention, which has been proven in various rodent models. In the fibrosarcoma-bearing mouse model, tumor-filtrating γδ T cells are the primary source of IL-17, which promotes tumor metastasis by inducing the angiogenesis via transcription of vascular endothelial growth factor (VEGF) and angiopoietin-2 (Ang-2) [34]. γδ T17 is induced in the tumor microenvironment and recruits tumor-promoting macrophages and neutrophils, thus inhibiting anti-tumor immune response [14, 35]. The CD39+ γδ T cells are a new type of γδ Tregs found in human colorectal cancer [31]. CD39 and CD73 expression on γδ T cells can hydrolyze and dephosphorylate adenosine triphosphate (ATP) to adenosine, which binds to adenosine receptors and subsequently exerts strong suppressive capacity [31, 36]. γδ T cells also suppress the proliferation of CD4 T cells through checkpoint molecules such as programmed cell death (PD) ligand 1 (PD-L1) [37, 38]. The galectin-1 secreted by γδ T cells is thought to be a potential regulatory molecule induced by myeloid-derived suppressor cells activated by the toll-like receptor 5 (TLR5)-dependent signal [39].
In conclusion, the role of γδ T cells is very complicated and context-dependent. Immunotherapy with activated γδ T cells is tolerated well. However, the clinical benefits are unsatisfactory. Therefore, the cytotoxicity of γδ T cells via various mechanisms needs to be further increased.
New methods to enhance γδ T cell cytotoxicity
Novel nitrogen-containing bisphosphonate products
Boosting Th1-phenotype γδ T cells in patients with cancer for their independence of MHC molecules is attractive, and the loss of MHC I molecules is one of the common immune evasion mechanisms. Aminobisphosphonates were once thought to be the ideal drug because they can induce effector Vγ9Vδ2 T cell activation and expansion. They are commercially available and used in many clinical trials worldwide, such as in osteoporosis and bone metastasis in cancer treatment [13]. However, most clinical trials boosting Vγ9Vδ2 T cells in vivo show disappointing responses (Table 1) [40]. Findings may be related to the energy and exhaustion of γδ T cells in patients, which prevent γδ T cells from responding to NBPs [41]. Moreover, the efficacy of NBPs is challenged by their pharmacokinetic properties in vivo [42]. Less than 1% of NBPs are absorbed when given orally, and NBPs have a short half-life in PB because they are rapidly excreted absorbed by bone tissue. PAgs, such as bromohydrin pyrophosphate (BrHPP), have a short half-life period in the plasma [43]. NBPs are hydrophilic and enter cells through fluid-phase endocytosis. These reasons result in poor tissue accumulation except for bone.
Associated clinical trials of γδ T cells immunotherapy
| Clinical trial identifier | Phase | Start date | Status | Cancer type | Interventions | Outcome measures |
|---|---|---|---|---|---|---|
| NCT01404702 | I | August, 2011 | Terminated | Neuroblastoma | ZOL, IL-2 | Safety, tumor response, etc. |
| NCT00582790 | II | August, 2003 | Terminated | Kidney cancer | ZOL, IL-2 | OS, PFS, safety, etc. |
| NCT02781805 | I | August, 2016 | Terminated | Breast neoplasms | ALD | Percentage change of γδ T cells |
| JPRN-UMIN000008097 | I | June, 2017 | Not recruiting | Esophageal cancer | Autologous γδ T cells | Safety, PFS, OS, etc. |
| JPRN-UMIN000007878 | II | May, 2012 | Not recruiting | MM | Autologous γδ T cells | TTP, PFS, safety, etc. |
| JPRN-C000000336 | I, II | March, 2006 | Not recruiting | NSCLC | Autologous γδ T cells | Safety, PFS, QOL, etc. |
| JPRN-UMIN000006128 | II | August, 2011 | Not recruiting | NSCLC | ZOL-expanded autologous γδ T cells | PFS, QOL, safety, etc. |
| NCT03790072 | I | November, 2018 | Completed | AML | Allogeneic γδ T cells | Safety, DLT, CR, etc. |
| NCT04008381 | I | September, 2019 | Recruiting | AML | Allogeneic γδ T cells | RR, safety, OS, etc. |
| NCT04518774 | I | August, 2020 | Recruiting | Hepatocellular carcinoma | Allogeneic γδ T cells | Safety, DLT, QOL, etc. |
| NCT04735471 | I | March, 2021 | Recruiting | Lymphoma | Allogeneic γδ T cells | DLT, OS, PFS, etc. |
| NCT04165941 | I | February, 2020 | Recruiting | Brain tumor adult | DRI modified γδ T cells | DLT, safety, TTP, etc. |
| NCT03533816 | I | January, 2020 | Recruiting | Leukemia, MDS | Allogeneic γδ T cells | DLT, safety, RFS, etc. |
| NCT04911478 | - | August, 2021 | Enrolling by invitation | Lymphoma | Allogeneic γδ T cells | Safety, ORR, PFS, etc. |
| NCT05015426 | I | August, 2021 | Recruiting | AML | aAPC-expanded γδ T cells | MTD, RFS, OS, etc. |
| NCT05001451 | I | August, 2021 | Recruiting | AML | Allogeneic Vδ1+ γδ T cells | Safety, DLT, etc. |
| NCT00562666 | I | February, 2008 | Terminated | Hepatocellular carcinoma | γδ T cells | Safety, tumor response |
| JPRN-UMIN000028370 | II | July, 2017 | Recruiting | AL, lymphoma, solid tumor | Autologous γδ T cells, nucleoside (acid) analogues | DFS |
| NCT04107142 | I | December, 2019 | Unknown status | Colorectal cancer, TNBC, Sarcoma, nasopharyngeal Carcinoma, PC, GC | NKG2DL-targetting CAR-grafted γδ T cells | DLT, safety, PFS, etc. |
| NCT05302037 | I | April, 2022 | Not yet recruiting | Cancer | NKG2DL-targetting CAR-grafted γδ T cells | DLT, safety, PFS, etc. |
| NTR6541 | I | January, 2017 | Recruiting | AML, MDS, MM | TEG001 | DLT |
| NCT04688853 | I | May, 2021 | Recruiting | MM | TEG002 | Safety, DLT, OS, etc. |
aAPC: artificial APC; AL: acute leukemia; AML: acute myeloid leukemia; CAR: chimeric antigen receptor; CR: complete remission; DFS: disease free survival; DLT: dose-limiting toxicities; DRI: drug resistant immunotherapy; GC: gastric cancer; MDS: myelodysplastic syndromes; MM: multiple myeloma; MTD: maximum tolerated dose; NSCLC: non-small cell lung cancer; ORR: objective response rate; OS: overall survival; PC: prostate cancer; PFS: progression-free survival; QOL: quality of life; RFS: relapse-free survival; RR: relative risk; TEG: T cell engineered to express a defined γδ TCR; TNBC: triple-negative breast cancer; TTP: time to progression
The bisphosphonate prodrugs were designed to overcome the problem of pharmacodynamic restriction. The highly hydrophobic bisphosphonate prodrug, tetrakis-pivaloyloxymethyl 2-(thiazole-2-ylamino) ethylidene-1,1-bisphosphonate (PTA) can efficiently permeate cell membranes and be hydrolyzed to active acid form, 2-(thiazole-2-ylamino)ethylidene-1,1-bisphosphonate (TA), by intracellular esterase to block FDPS [44, 45]. PTA induces the intracellular accumulation of IPP more efficiently than ZOL [46]. Subsequently, PTA is 1000-fold more potent than ZOL in stimulating Vγ9Vδ2 T cells in PB mononuclear cell (PBMC) ex vivo. Furthermore, Vγ9Vδ2 T cells expanded by PTA reach a higher purity (> 98%), and the number is 20% higher on average while preserving their cytotoxic activity compared with ZOL [44]. After adoptive transfer in nonobese diabetic (NOD)/Shi-scid/IL-2R gamma(null) mice (NOG mice), highly enriched Vγ9Vδ2 T cells (> 98%) result in high circulating Vγ9Vδ2 T cells. NBP prodrugs can be optimized by introducing a fluorine atom which is helpful in the stabilization of NBPs [47]. Therefore, PTA can be a better stimulator of Vγ9Vδ2 T cells than NBPs to prepare large numbers used in adoptive immunotherapy for cancer.
In addition, using nano-technology to deliver NBPs and synthetic nucleotide pyrophosphate is attractive [42, 43]. NBPs, such as ALD and ZOL, encapsulated within liposome [liposomal ALD (L-ALD) and liposomal ZOL (L-ZOL)] and L-ALD do not exert an improved ability to sensitize tumor cells to destruction by Vγ9Vδ2 T cells, and the use of L-ZOL in vivo is prohibited by profound toxicity and sudden mouse death [48–51]. L-ALD can help NBPs go through the cell membrane and has a superior ability to sensitize epithelial ovarian cancer (EOC) and melanoma xenograft effectively to Vγ9Vδ2 T cell adoptive immunotherapy [48, 49]. The positron emission tomography tracking of 89Zr-label shows higher Vγ9Vδ2 T cell number in L-ALD-treated tumors and less in bone than ALD-treated [52]. Targeted liposomes have been formulated to increase the efficacy of Vγ9Vδ2 T cells and NBP combination immunotherapy. NBPs encapsulated within folate-targeted (FT) liposomes (FT-L-ZOL and FT-L-ALD) sensitize folate receptor-α+ EOC cell lines in a 10-fold lower concentration than liposomal-NBP (L-NBP) and free NBP [48]. The αvβ6 integrin-targeted L-ALD can enter the αvβ6-positive cells line via receptor-mediated endocytosis and improve the sensitization of the αvβ6 positive cell line to Vγ9Vδ2 T cells [53]. However, no additional advantage is observed in the metastatic lung mouse model compared with L-ALD.
γδ T cells as antibody adjuvant
Based on the capacity of ADCC, γδ T cell adoptive immunotherapy can also benefit from the treatment of monoclonal antibody (mAb). The cytotoxicity of Vγ9Vδ2 T cells against malignant B cells and breast cancer increases in the presence of antibodies targeting CD20 and human epidermal growth factor receptor 2 (HER2) via CD16-mediated ADCC [54–56]. Furthermore, this result is confirmed and expanded in a non-human macaque model in vivo. The research also found that PAg promotes Vγ9Vδ2 T cell binding to mAb target cells and formation of immunologic synapses [55]. Vγ9Vδ2 T cells exhibit more significant cytotoxicity against Ewing’s sarcoma and neuroblastoma cell lines through opsonization by ganglioside antigen (GD2) antibodies [57–59]. The combination therapy of γδ T cells and GD2 antibody significantly prolongs the survival of humanized neuroblastoma model.
Bispecific T cell engagers (BiTEs) targeting T cells and tumor antigens through the coupling of single-chain variable fragments (scFv) can help γδ T cells recognize and kill tumor cells in the absence of PAgs. BiTEs are interesting candidates to overcome this hurdle as they bind immune effector and tumor cells and recruit effector cells to the tumor lesion. The bispecific antibody targeting PD-L1 and CD3 improves the anti-tumor effect of Vγ9Vδ2 T cells against NSCLC [60]. The (Her2 × CD3) bispecific scFv specifically binds to γδ T cells and Her2-positive cell lines and enhances γδ T cell-mediated lysis against pancreatic ductal adenocarcinoma (PDAC). Besides, the [(Her2)2 × Vγ9] bispecific tribody, which consists of 2 scFv specifically binding to HER2 and 1 Fab directed to γδ TCR subunit Vγ9, has a superior activity compared with (Her2 × CD3), which may be due to the bivalent targeting of tumor cells and different qualitative signaling via Vγ9 TCR [61]. Subsequently, compared with PAgs, [(Her2)2 × Vγ9] induces minimal cell death of Vγ9Vδ2 T cells [62]. Recombinant immunoligands consisting of human CD20 scFv and MICA or ULBP2 enhance the γδ T cell-mediated lysis of lymphoma and leukemia cells [63]. BiTE with scFv binding TCR gamma V region 9 (TRGV9) and scFv binding CD123 induces the activation of Vγ9Vδ2 T cells and cytolytic activity of CD123-positive AML cell lines by Vγ9Vδ2 T cells without eliciting cytokine storm, which occurs to patients accepting CD3-direction immunotherapy [64].
Nanobodies are variable antigen-binding regions from heavy chain-only antibodies, naturally occurring in camelids. The application of nanobodies is advantageous for bispecific antibodies because of their simple structure. Nanobodies contain only heavy chains and lack the Fc-region, resulting in low immunogenicity and the ability to reach the antigen easily [65]. de Bruin et al. [66, 67] devised a series of nanobodies specifically binding to Vγ9 and Vδ2 TCR, which can be powerful tools for cytometry, immunocytochemistry, Vγ9Vδ2 T cell isolation through magnetic-activated cell sorting (MACS), and blocking the activation signal of Vγ9Vδ2 TCR mediated by NBPs. Later, de Bruin et al. developed a novel bispecific Vγ9Vδ2 T cell engager, and the anti-epidermal growth factor receptor (EGFR) nanobody joins the anti-Vγ9Vδ2 TCR nanobody. This (EGFR × Vγ9Vδ2 TCR) nanobody induces the γδ T cell-mediated lysis of EGFR positive tumor cells and enhances the γδ T cell-mediated inhibition of colon adenocarcinoma cell line growth in vivo [65]. Besides, the exertion of cytolytic activities against normal keratinocytes by γδ T cells is barely observed even in the presence of a high concentration of these bispecific nanobodies. Another bispecific Vγ9Vδ2 T cell engager consisting of Vδ2 TCR-specific nanobody and CD1d-specific nanobody enhances the Vγ9Vδ2 T cell-mediated cytotoxicity against chronic lymphocytic leukemia (CLL) cell line [68]. The overview of γδ T cell adoptive immunotherapy with antibody adjuvant is given in Table 2.
Research of γδ T cells with antibody adjuvant
| Name | Target | Tumor type | Reference |
|---|---|---|---|
| mAb | |||
| hu14.18 | GD2 | Neuroblastoma/Ewing’s sarcoma | [57–59] |
| Rituximab & GA101 | CD20 | B cell lymphoma/CLL | [54–56] |
| Trastuzumab | HER2 | Breast cancer | [54, 55] |
| Alemtuzumab | CD52 | Lymphoma | [55] |
| BiTE | |||
| Her2-CD3 & (Her2)2 × Vγ9 | HER2 & CD3/Vγ9 | PDAC | [61, 62] |
| Vγ9/CD123 bispecific antibody | CD123 & Vγ9 | AML | [64] |
| Y111 | PD-L1 & CD3 | NSCLC | [60] |
| Bispecific nanobody | |||
| 7D12-5GS-6H4 | EGFR & Vδ2 | Colon adenocarcinoma | [65] |
| anti-CD1d-Vδ2 | CD1d & Vδ2 | CLL | [68] |
γδ T cells plus immune checkpoint therapy
Immune checkpoint receptors (ICRs) are expressed on the surface of most leukocytes, including γδ T cells, and prevent the immune response from overt activation and damage to tissue. The majority of ICRs inhibit cell activation through a mechanism that intracellular immunoreceptor tyrosine inhibitory motifs (ITIM) or intracellular immunoreceptor tyrosine switch motifs (ITSM) recruit and phosphorylate Src-homology-2 phosphatases 1 (SHP-1) and SHP-2, which dephosphorylate substrates and inhibit cell activation. Otherwise, ICRs counteract the activating signals by competing with activating receptors for ligands. Cytotoxic T lymphocyte antigen-4 (CTLA-4) binds to B7 ligands with higher affinity than CD28, and T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT) limits CD226-mediated cell activation in the same way.
Vγ9Vδ2 T cells transiently upregulate PD-1 after PAg-mediated TCR activation [69, 70]. But Vγ9Vδ2 T cells from cord blood express PD-1 for 28 days after PAg stimulation [71]. The activation and effector function of γδ T cells is controlled by PD-1. Bone marrow (BM) Vγ9Vδ2 T cells from MM are largely PD-1-positive and anergic to PAg stimulation [72]. After binding to PD-L1, cytotoxicity against tumor cells and cytokine production of PD-1+ Vγ9Vδ2 T cells, such as IFN-γ, TNF-α, and IL-17A, is impaired [69, 71, 73]. Blockade of PD-1 signal can restore the inhibition. Vγ9Vδ2 T cells proliferation and degranulation in response to PAg are partially recovered in the presence of anti-PD-1 mAb [72]. Furthermore, anti-PD-1 mAb potentiates CD16-mediated ADCC of follicular lymphoma cells by Vγ9Vδ2 T cells [74]. B- and T-lymphocyte attenuator (BTLA) is strongly expressed on resting Vγ9Vδ2 T cells and inversely correlated with differentiation. BTLA negatively regulates Vγ9Vδ2 TCR-mediated activation and attenuates proliferative capacities following herpes virus entry mediator (HVEM) ligation [75, 76]. Antagonistic anti-HVEM or BTLA mAb increases Vγ9Vδ2 T cell proliferation in co-culture with HVEM+ tumor cells and prevents terminal differentiation [75, 76]. Vγ9Vδ2 T cells upregulate T cell immunoglobulin and mucin domain-containing 3 (Tim-3) at the early stage of TCR stimulation and keep a high level for more than 18 days [77]. Upon Galectin-9 ligation, Tim-3 promotes Vγ9Vδ2 T cells apoptosis and inhibits the cytotoxicity of colon cancer cells [77, 78]. Blocking Tim-3 can significantly increase proliferation [79], inhibit apoptosis of Vγ9Vδ2 T cells, and enhance their cytotoxicity [77, 78].
γδ T cells in adoptive immunotherapy
On the basis of the advantages of γδ T cells for immunotherapy, clinical trials have been conducted in the last decades [35, 80]. Associated clinical trials are listed in Table 1. In 2003, the first γδ T cell-mediated immunotherapy clinical trial was carried out in non-Hodgkin lymphoma (NHL) and MM patients with the injection of NBP [41]. γδ T cell-mediated immunotherapy is proven to be feasible and tolerated well. However, the clinical outcome is unsatisfactory. Studies on other types of cancers, such as refractory PC, renal cell carcinoma, melanoma, AML, breast cancer, and neuroblastoma also reach the same conclusion [19, 81–83]. Adoptive transfer of γδ T cells expanded ex vivo is explored in metastatic renal cell carcinoma, NSCLC, melanoma, AML, pancreatic cancer, colorectal cancer, GC and MM [84–94]. The precise reason for the unsatisfying clinical outcome remains unclear, but a number of assumptions have been raised. One of the reasons is that γδ T cells possess regulatory functions on immune responses. And whether the Foxp3, the marker of Tregs, is necessary for γδ Tregs, which means the subsets of γδ T cells can’t be sorted out through a distinctive marker. Surveys have explained that γδ T cells exert immunosuppressive function after γδ TCR stimulation [25, 37]. Moreover, the systematic intravenous injection of γδ T cells can not achieve enough effector-target (E-T) ratio to eliminate cancer cells at the tumor site. The adoptively transferred γδ T cells trafficked predominantly to the lungs at first and migrated to the liver and spleen [88].
Vδ1 T cell adoptive immunotherapy
In the past decades, non-Vδ2 T cells, including Vδ1 T cells, do not get much attention in immunotherapy as Vγ9Vδ2 T cells due to their low abundance among PB γδ T cells and lack of protocols for expansion. The Vδ1 T cell has its advantages as a candidate for adoptive immunotherapy. Vδ1 T cells also recognize the target cell independence of MHC molecules and are minimally susceptible to activation-induced cell death [3, 4]. Vδ1 T cells recognize lipid antigens presented by CD1 and NKG2DL through γδ TCR and NKG2D and target B7-H6-positive cancer cells through NCR, such as NKp30 and NKp44 [5, 6]. The “two-step” Vδ1 T cell expansion protocol can achieve more than 1000-fold expansion of Vδ1 T cells. MACS sorted-γδ T cells are cultured with several cytokines and anti-CD3 mAb for three weeks, and Vδ1 T cells can be acquired. Th adoptive Vδ1 T cells transfer inhibits tumor growth and dissemination in vivo [4, 6].
CAR-γδ T cells
CARs comprise an scFv ectodomain targeting a cell surface molecule specific for tumor-associated antigens (TAAs) and endodomain that transduces the co-stimulation signal [95]. CAR-T therapy has shown remarkable success in some cases but still suffers from some limitations. The main side effects of CAR-T therapy are on-target/off-tissue and off-target toxicities induced by attacking non-malignant host cells. Several mechanisms cause the phenomenon. First, CAR recognizes antigens on the surface of normal host cells expressing TAAs, or CAR-T targets normal tissue through cross recognition. Second, anergized self-reactive T cells are re-activated by signaling through CARs [96]. Besides, the cytokine release syndrome is another side effect of T cells expansion in vivo and the release of toxic cytokine levels [97]. CAR-T cells must be individually fabricated for each patient to avoid the risk of graft-versus-host disease (GVHD). Therefore, a universal cellular treatment needs to be devised. Researchers have developed several approaches by using T-cell precursors, NK cells, or γδ T cells as carrier cells [98].
CAR-γδ T cells, which are generated from Vγ9Vδ2 T cells engineered with CAR, show anti-tumor efficacy. Studies showed that Vγ9Vδ2 T cells with CD19-specific CAR exert an anti-tumor effect against CD19+ leukemia and γδ T cells with GD2-specific CAR efficiently and specifically lyse Ewing’s sarcoma and neuroblastoma cells [99, 100]. Interestingly, CAR-γδ T cells retain the ability to take up tumor antigens, and the cross presents the processed peptide to conventional T cells [95]. CARs provide γδ T cells with the ability to recognize and activate by TAAs. CAR-γδ T cells can sense metabolic dysregulation like normal Vγ9Vδ2 T cells, thus reducing the risk of off-target effect. CD19-specific CAR-γδ T cells show CD19 dependent activity against tumor cells and cytotoxicity against CD19-negative target cells via γδ TCR recognizing PAg. For these reasons, CAR-γδ T cells provide an idea for overcoming the barrier of CAR-T therapy [101].
Investigators also searched for mechanisms that enhance the safe application of CAR-γδ T cell therapy. Second-generation CAR endodomain contains CD3ζ providing TCR signals (signal 1) and co-stimulatory domains (signal 2) such as CD28, 4-1BB, CD27, or ox40 in the presence of target antigen to bypass the requirement for MHC-restricted antigen presentation. Once CAR binds to TAAs, TCR and co-stimulatory signals are stimulated, and T cells are activated. At the same time, off-tumor toxicity remains a concern [95]. Therefore, Fisher et al. [102] devised a new kind of GD2-DAP10 CAR-γδ T cell, in which separated receptors transduce the two signals for γδ T cell activation. Signal 1 is provided through sensing the metabolic dysregulation of target cells by native γδ TCR. In contrast, the co-stimulation signal is transduced by the endodomain of CAR recognizing TAAs, such as GD2. CAR-γδ T cells are fully activated after these two separate receptors recognize their distinct ligands. In the current study, GD2-DAP10 CAR-γδ T cells without CD3ζ do not show toxicity against GD2+ normal cells that do not activate the γδ TCR signal. Furthermore, Fleischer et al. [103] designed the novel non-signaling CARs (NSCARs), which lack signaling/activation domains. CD5- or CD19-NSCAR-modified γδ T cells exhibit enhanced antigen-directed cytotoxicity against T cell-acute lymphocytic leukemia (T-ALL) or B cell-acute lymphocytic leukemia (B-ALL) cell lines in contrast to αβ T cells. NSCARs are hypothesized to enhance γδ T cell-mediated anti-leukemia effect by improving the interaction between γδ T cells and leukemia cell lines expressing TAAs. Upon co-culture with target cells, NSCAR-modified γδ T cells showed greater degranulation. Harrer et al. [96] engineered γδ T cells through RNA electroporation. Melanoma-associated chondroitin sulfate proteoglycan-specific CAR-γδ T cells specifically exhibit lysis against melanoma cells without a background cytokine secretion. Clinical studies are currently underway to evaluate the safety and efficacy of anti-CD19 and anti-CD20 CAR-γδ T cells in hematologic malignancies and anti-CD7 CAR-γδ T cells in CD7-positive T cell-derived malignant tumors (NCT02656147, NCT04702841, NCT04735471). The overview of CAR-γδ T cell adoptive immunotherapy is listed in Table 3.
CAR-γδ T cell-based immunotherapy research
| Target | Endodomain | Tumor type | Reference |
|---|---|---|---|
| GD2 | TCRξ | Neuroblastoma | [100] |
| GD2 | CD28-CD3ξ | Neuroblastoma/Ewing’s sarcoma | [95] |
| GD2 | DAP10/CD28-CD3ξ | Neuroblastoma/Ewing’s sarcoma | [102] |
| CD19 | Non | T-ALL | [103] |
| CD19 | CD28-CD3ξ | B cell lymphoma/CLL | [99, 101] |
| CD19 | TCRξ | B cell lymphoma/CLL | [100] |
| MCSP | CD28-CD3ξ | Melanoma | [96] |
| CD5 | Non | B cell lymphoma/CLL | [103] |
MCSP: melanoma-associated-chondroitin-sulfate-proteoglycan
TEGs
The major barrier to adoptive immunotherapy is the generation of auto-reactive αβ T cells. Therefore, CAR-T and αβ T cells engineered to express tumor-specific αβ TCR are developed. However, reprogramming αβ T cells with defined γδ TCRs is hampered by MHC restriction [104]. Although Vγ9Vδ2 T cells exert a strong anti-tumor reactivity against various solid and hematologic malignancies, most clinical trials reported limited tumor control by transferred Vγ9Vδ2 T cells. One of the reasons may be that the functional phenotype of γδ T cells is highly plastic. Even when from the same donor, the anti-tumor reactivity of individual Vγ9Vδ2 T cell clones is variable due to the different amino acid sequences of Vγ9Vδ2 TCR complementary determining region three domains [105, 106]. Therefore, TEGs are introduced to immunotherapy. Their ability to recognize antigens in an MHC-unrestricted manner and sense metabolic dysregulation is based on the Vγ9Vδ2 TCR. αβ T cells can also possess these properties through the transfection of Vγ9Vδ2 TCR. By picking the Vγ9Vδ2 TCR that binds to tumor cells efficiently, the problem of functional diversity of Vγ9Vδ2 T cells can be solved.
The first Vγ9Vδ2 TCR chain used for redirecting αβ T cells is clone G115, which has been reported to recognize PAgs and bind to ApoAI and F1-ATPase [107, 108]. CD8+ αβ T cells engineered to express Vγ9Vδ2 TCR clone G115 exert cytotoxicity functions against a broad panel of tumor cells, ignore normal tissue and the anti-tumor effect involved F1-ATPase. CD4+ TEGs can induce the maturation of dendrite cells as conventional Vγ9Vδ2 T cells. Moreover, IL-12p70 secretion by dendrite cells can be detected after incubation with CD4+ TEGs compared to conventional γδ T cells [107].
Methods to purify engineered T cells are also introduced to TEGs. γδ TCR is a strong αβ TCR competitor for the components of the CD3 complex and reduces the expression of αβ TCR. So Straetemans et al. [109, 110] utilize Good Manufacturing Practice (GMP)-grade anti-αβ TCR beads to reduce the fraction of non-transduced and poorly transduced αβ T cells. After purification, TEGs with αβ T cells depletion significantly increase the anti-tumor activity against malignant cells and reduce allo-reactivity towards PBMC from healthy donors. The GMP-grade manufacturing of TEGs is sufficient to produce the drug product TEG001, which has been proven in a clinical trial (NTR6541). TEG001 showed anti-leukemia effects while remaining not harmful to human normal cells in a mouse model [111]. TEG002 shares the same Vγ9Vδ2 TCR as TEG001 and exerts a cytotoxicity effect against neuroblastoma organoids. Straetemans et al. [112] also proved that the insertion of Vγ9Vδ2 TCR does not increase the risk of TEG001 cells for malignant transformation. A Phase I clinical trial has also been initiated to investigate the safety of TEG002 (NCT04688853).
Besides Vγ9Vδ2 TCR, other subtypes of γδ TCR are transferred to αβ T cells. Based on the paradoxical phenomenon that human cytomegalovirus (CMV) reactivation after allogeneic stem cell transplantation (allo-SCT) reduces the risk of leukemia relapse, researchers found that CMV-induced Vδ1 T cells can show cytotoxicity against leukemia cells [113]. αβ T cells engineered to express this type of Vδ1 TCRs are capable of recognizing leukemia cells. Otherwise, Vγ5Vδ1 T cell, the subgroup of γδ T cells derived from tumor tissue, recognizes colorectal adenocarcinoma and Epstein-Barr virus (EBV)-transformed lymphoblastoid cells in the presence of human leukocyte antigen (HLA)-A*24:02. Interestingly, HLA-A*24:02-mediated recognition does not rely on the presentation of antigen peptides. TEG011 expressing Vγ5Vδ1 TCR functions the same as Vγ5Vδ1 T cell [114]. Furthermore, TEG011 does not exhibit off-target toxicity in nontumor tissues in humanized HLA-A*24:02 transgenic mice [115]. The single-cell analysis of TNBC revealed that Vγ4aVδ5 T cells are abundant in tumor tissues. TEG-C132 expressing Vγ4aVδ5 TCR is reactive to endothelial protein C receptor. Thus, the anti-tumor activity of TEG-C132 is observed [116]. However, given that the specific mechanism of how γδ TCR recognizes antigens remains unclear, we still can not distinguish which types of patients with tumors will benefit from TEG therapy. Overview researches of TEGs-based immunotherapy are given in Table 4.
TEGs-based immunotherapy research
| TCR type | TEG name | Tumor type | Reference |
|---|---|---|---|
| Vγ9Vδ2 G115 | - | Leukemia | [107] |
| γG11/δ5 | - | Hematological malignancy | [109] |
| Vδ1 | - | Leukemia | [104] |
| Vγ9Vδ2 G115 | - | MM | [108] |
| Vγ9Vδ2 clone 5 | TEG001 | AML | [110, 111] |
| Vγ5Vδ1 | TEG011 | Colorectal adenocarcinoma & EBV transformed lymphoblastoid cells | [114, 115] |
| Vγ4aVδ5 | TEG-C132 | TNBC | [116] |
| Vγ9Vδ2 clone 5 | TEG002 | Neuroblastoma | [117] |
“-” indicates no TEG names are available
Conclusions
Although γδ T cells are a small population of T cells in PB, they attract remarkable attention for their ability to exert potent cytotoxicity against broad types of tumor cells in an MHC-unrestricted manner. In the current review, we discussed several novel strategies to improve the anti-tumor effects of γδ T cell-based immunotherapy (Figure 1). Nonetheless, clinical trials show limited benefit from γδ T cell-based immunotherapy. The precise reason remains unclear, but several assumptions have been raised. One of the hypotheses is that the functions of activated γδ T cells are plastic, and several subsets of γδ T cells exert immunoregulatory functions. In addition, boosting the function of γδ T cells in vivo by NBPs and IL-2 was limited by the pharmacokinetic properties of NBPs. Therefore, NBP prodrugs and encapsulating within liposomes are designed. NBP prodrugs have an excellent potential application for its high effect on activating γδ T cells. Adoptive transfer can also benefit from this effect because γδ T cells stimulated by NBP prodrugs achieve high purity (> 98%). Functional and subset diversity may be another reason. By selecting the γδ TCR that has high affinity or tumor cell and transfers to αβ T cells, TEGs may avoid the functional diversity of γδ T cells. Moreover, the systematic intravenous injection of γδ T cells cannot achieve enough E-T ratio to eliminate cancer cells at the tumor site. Consequently, strategies to improve cytotoxicity and recruit γδ T cells need further research. CAR-redirected γδ T cells have a good way of recognizing specific tumor cells. The anti-tumor effect of γδ T cell-based immunotherapy can be enhanced via the CD16-mediated ADCC. BiTEs recruit γδ T cells to tumor sites through binding to γδ T cells and tumor cells. In summary, given that γδ T cells are heterogeneous, more clinical trials need to be thoroughly delineated and utilized to maximize the efficacy of immunotherapy by using γδ T cells.
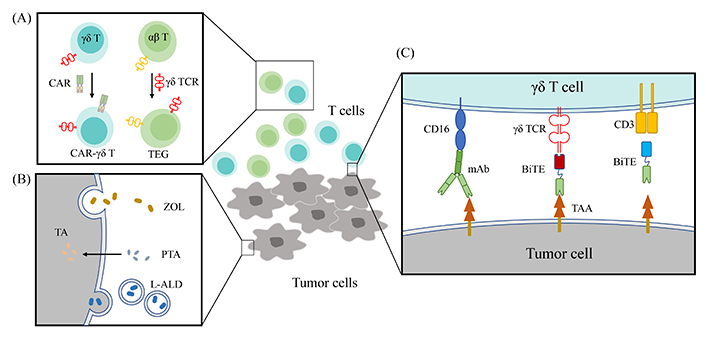
Strategies to improve the anti-tumor effect of γδ T cells. (A) CAR redirected γδ T cells have a better way of recognizing specific tumor cells. αβ T can possess the advantages of recognizing antigens in an MHC-unrestricted manner and a sense of metabolic dysregulation through transfection Vγ9Vδ2 TCR; (B) NBPs are hydrophilic and enter cells through fluid-phase endocytosis. The highly hydrophobic bisphosphonate prodrug, PTA can efficiently permeates cell membranes and be hydrolyzed to active acid form, TA by intracellular esterase to block FDPS. NBPs are delivered using nano-technology; (C) the cytotoxicity of Vγ9Vδ2 T cells against malignant cells increases in the presence of antibodies targeting TAAs. BiTEs targeting γδ T cells and tumor antigens recruit γδ T cells to the tumor site
Abbreviations
| ADCC: | antibody-dependent cellular cytotoxicity |
| ALD: | alendronate acid |
| AML: | acute myeloid leukemia |
| ATP: | adenosine triphosphate |
| BiTEs: | bispecific T cell engagers |
| BTLA: | B- and T-lymphocyte attenuator |
| BTN3A1: | butyrophilin subfamily 3 member A1 |
| CAR: | chimeric antigen receptor |
| CDR3: | complementary determining region 3 |
| CLL: | chronic lymphocytic leukemia |
| DLT: | dose-limiting toxicities |
| EGFR: | epidermal growth factor receptor |
| FDPS: | farnesyl diphosphate synthase |
| GC: | gastric cancer |
| GD2: | ganglioside antigen |
| HER2: | human epidermal growth factor receptor 2 |
| HLA: | human leukocyte antigen |
| HVEM: | herpes virus entry mediator |
| ICR: | immune checkpoint receptor |
| IL-2: | interleukin-2 |
| IPP: | isopentenyl pyrophosphate |
| L-ALD: | liposomal alendronate acid |
| L-ZOL: | liposomal zoledronate acid |
| mAb: | monoclonal antibody |
| MDS: | myelodysplastic syndromes |
| MHC: | major histocompatibility complex |
| MM: | multiple myeloma |
| NBP: | nitrogen-containing bisphosphonate |
| NK: | natural killer |
| NKG2D: | natural killer group 2 member D |
| NKG2DL: | natural killer group 2 member D ligands |
| NSCAR: | non-signaling chimeric antigen receptor |
| NSCLC: | non-small cell lung cancer |
| OS: | overall survival |
| PAgs: | phosphoantigens |
| PB: | peripheral blood |
| PC: | prostate cancer |
| PD: | programmed cell death |
| PD-L1: | programmed cell death ligand 1 |
| PFS: | progression-free survival |
| PTA: | tetrakis-pivaloyloxymethyl 2-(thiazole-2-ylamino)ethylidene-1,1-bisphosphonate |
| QOL: | quality of life |
| RFS: | relapse-free survival |
| scFv: | single-chain variable fragments |
| TA: | 2-(thiazole-2-ylamino)ethylidene-1,1-bisphosphonate |
| TAAs: | tumor-associated antigens |
| TCR: | T cell receptor |
| TEG: | T cell engineered to express a defined γδ |
| TGF-β: | transforming growth factor-β |
| Th1: | T-helper 1 |
| Tim-3: | T cell immunoglobulin and mucin domain-containing 3 |
| TNBC: | triple-negative breast cancer |
| TNF: | tumor necrosis factor |
| Tregs: | regulatory T cells |
| TTP: | time to progression |
| ZOL: | zoledronate acid |
Declarations
Author contributions
JZ and XJ wrote the initial draft of the manuscript; JZ prepared the figure; HZ and WW organized literature for manuscript; XW and ZJ revised the final draft. All authors read and approved the submitted version.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This study was supported in part by grants from the National Natural Science Foundation of China (Nos. 81800143, 81770150, 81200388, and 82170220), Natural Science Foundation of Guangdong Province (No. 2020A1515010817 and 2022A1515010313), the Science and Technology Program of Guangzhou City (No. 202201010164), National Innovation and Entrepreneurship Training Program for Undergraduate (No. 202010559081), Guangdong’s Innovation and Entrepreneurship Training Program for Undergraduate (Nos. 202010559081 and 202210559083), “Challenge Cup” National Undergraduate curricular academic science and technology works by race (No. 21112027) and Guangdong College Students’ Scientific and Technological Innovation (Nos. CX22438, CX22446, and CX21285). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2022.