 Review
Review

Affiliation:
1SH/Scientific-Hemostasis, Scientific Director and Consultant in Hemostasis and Thrombosis Diagnostics, 95130 Franconville, France
Email: jean.amiral@scientific-hemostasis.com
ORCID: https://orcid.org/0000-0002-0680-9172
Affiliation:
2HYPHEN BioMed (HBM), 95000 Neuville sur Oise, France
Affiliation:
2HYPHEN BioMed (HBM), 95000 Neuville sur Oise, France
Affiliation:
2HYPHEN BioMed (HBM), 95000 Neuville sur Oise, France
Affiliation:
2HYPHEN BioMed (HBM), 95000 Neuville sur Oise, France
Explor Immunol. 2022;2:604–621 DOI: https://doi.org/10.37349/ei.2022.00071
Received: January 13, 2022 Accepted: June 13, 2022 Published: August 31, 2022
Academic Editor: Narinder K. Mehra, Indian Council of Medical Research, India
In ultrarare cases, patients vaccinated with DNA adenovirus vector vaccine against severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), develop a vaccine-induced immune thrombotic thrombocytopenia (VITT), with a high incidence of fatal cases. The causative agent is the development of platelet factor 4 (PF4)-dependent antibodies that resemble heparin-induced thrombocytopenia (HIT) complication, although many differences can be noticed in clinical presentation, antibody reactivity, involved epitopes on the PF4 protein, and pathological mechanisms. From the literature review, and the experience of HIT and testing a few plasmas from patients with VITT, this review analyzes the possible mechanisms, which show the strong immunoglobulin G (IgG) antibody reactivity to PF4 alone, in the absence of heparin, and to a lesser extend to stoichiometric complexes of PF4 and heparin (H-PF4). In addition, much lower heparin concentrations are required for inhibiting antibody binding to PF4. These concentrations are much lower than those required for disrupting the stoichiometric H-PF4 complexes. This confirms that IgG antibodies responsible for HIT bind preferentially to PF4, to epitopes that are readily masked by low concentrations of heparin. These antibodies are at a much higher concentration than the current ones observed for HIT, keeping a strong reactivity even for plasma dilutions as high as 1/500 to 1/5,000, whilst the current dilution for testing heparin-dependent antibodies in HIT is 1/100. Although VITT anti-PF4 antibodies can be detected with the current anti-H-PF4 enzyme-linked immunosorbent assays (ELISAs) designed for HIT, some assays have low sensitivity or are unreactive, like lateral immunofiltration methods or chemiluminescent automated assays. The preferred method should concern the use of capture assays using PF4 coated solid surfaces. This report proposes that the immune response is only targeted to the binding domain of PF4 with the hexons present on the adenovirus vector, through an epitope spreading mechanism, without any exposure of neo-epitopes on PF4 protein.
The clinical severity of the coronavirus disease 2019 (COVID-19) pandemic can be better controlled with the introduction of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) vaccines. Two major vaccination technologies are being used in western countries. They are based on the injection of a viral genetic code, which induces the synthesis of the viral spike protein by the vaccinated host, and therefore the development of specific antibodies capable of blocking the viral infection [1–3]. This concerns either messenger RNA (mRNA) vaccines (Pfizer-NG-Biotech; Moderna) [4]; or the use of an adenovirus vector for introducing DNA [5], which codes for the spike protein, then synthesized by immunized cells (ChAdOx1 from Astra Zeneca, Johnson and Johnson Ad26.COV2.S from Janssen, Sputnik V from Russia). More traditional approaches are proposed in other countries like China (inactivated viral particles from Sinovac), and other vaccines with new or existing technologies are under current development (recombinant spike protein from Sanofi, inactivated viruses from Valneva, purified non-infectious viral fragments from Novavax). Although the ratio of benefit/risk is highly positive for all vaccines, some differences concerning efficacy and side effects have been described. More especially, rare but severe and life-threatening complications have been reported for the vaccines using the adenovirus vector, more particularly for the Astra Zeneca and the Janssen ones, used in western and some developing countries [6–8]. The most severe side effect concerns the development of thrombotic thrombocytopenia, with the occurrence of multiple thromboses, sometimes developing at unusual sites, and associated with massive platelet destruction and blood activation [2, 9–14]. Disseminated intravascular coagulation (DIC) can be present in some patients. Blood activation markers are usually very elevated, with especially a huge concentration of D-dimer [6, 7]. These complications have now been named: vaccine-induced thrombotic thrombocytopenia or vaccine-induced immune thrombotic thrombocytopenia (VITT) [5–7, 15]. When these vaccine side effects occur, a high incidence of fatal cases has been reported. Fortunately, these complications concern only very rare cases, in the range of 0.0001–0.002% of vaccinated persons [13, 16]. A tendency to note these complications more frequently in females than in males was reported, but this is not totally confirmed by more extensive statistics [11, 14]. Other very rare side effects have also been described for RNA vaccines, like myocarditis occurring more often with Moderna vaccine, and in young persons [4], but will not be discussed in this article. Mechanisms responsible for the COVID-19 vaccine side effects are not always well-understood [17–19]. However, for the adenovirus vector vaccines, the complication is obviously of an immune origin, as the concerned patients develop a high positivity of antibodies to heparin-platelet factor 4 (H-PF4) complexes when tested with the anti-H-PF4 enzyme-linked immunosorbent assays (ELISAs), that are proposed for the diagnosis of heparin-induced thrombocytopenia (HIT). Very likely it is allo-immune because the autoimmune complication is induced by a non-self-component forming a complex with a self-component, which is the autoantibody target. Surprisingly, not all anti-H-PF4 HIT assays show the same positive reactivity when plasma or serum from affected patients are tested [7, 20]. The Acustar™ HIT-IgG (PF4-H) chemiluminescence-based assay and the rapid individual immuno-filtration tests like the STIC Expert HIT™ remain frequently negative. In this review we only focus on the immune complications associated with the adenovirus vector-based vaccines, reviewing the possible inducing causes, their pathogenicity, and the laboratory detection of the immunoglobulin G (IgG) developed IgGs to platelet factor 4 (PF4). We also review how these immune reactions can be characterized.
What causes the immune thrombotic thrombocytopenia, in the rare, vaccinated patients developing this complication, is the generation of PF4-dependent antibodies, which induce a massive and life-threatening platelet activation with thromboembolism, and thrombosis occurring at unusual sites and possibly evolving to DIC [5–7]. This complication has been described as a HIT-like syndrome, resulting from an immune response to PF4 complexed with polyanions, not yet identified, as heparin or heparin-like material is absent [18, 21, 22]. This statement is supported by the strong reactivity of IgG antibodies to H-PF4 complexes, evidenced in most, if not all, patients with VITT. However, a major pending question concerns the nature of polyanions, which can react with the positively charged PF4 through electrostatic interactions, like heparin polysaccharides. The vaccines themselves do not contain any anionic polysaccharide which could react with PF4, and, as the injection is usually performed into the shoulder muscle, no significant concentrations of PF4 are expected to be present for reacting with vaccine components. Special attention is required on the adenovirus vector itself, which is composed of an envelope formed by hexons, and a few pentons and it exposes some fiber filaments which help to bind and enter the cells which are infected [23, 24], as shown on Figure 1. Former studies did not report any binding of adenoviral hexons or pentons with PF4, but rather with coagulation factor X (FX). The adenovirus reactivity with PF4 was recently investigated by Baker et al. [24]. Concerning cell entry mechanisms, adenovirus fibers are known to react with a cell receptor, coxsackie and adenovirus receptor (CAR), which favors cell viral infection. Anyhow, PF4 and adenovirus interactions are not likely to occur, because PF4 concentrations are not expected to be significant at the injection site. Another consideration concerns DNA, an electronegative sequence, which could bind to the highly positive PF4 protein. Recently the mechanisms were better elucidated, and the binding of PF4 to the electronegative adenovirus surface was described in the comprehensive study by Baker et al. [24]. PF4 bound onto the adenovirus surface could become immunogenic and induce the generation of autoantibodies.
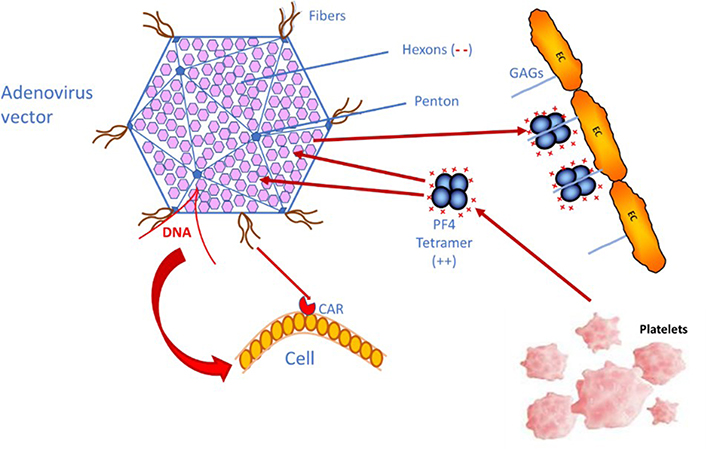
Simplified schema showing how the adenovirus vector used for DNA vaccines can enter human cells through the binding to CAR, and how PF4 could react with the hexon motives, through electrostatic interactions, if both come into accidental contact. PF4 is either released from platelet α-granules upon activation or can be released from the endothelial pool, where PF4 binds to endothelial cells (ECs) glycosaminoglycans (GAGs). (--): electronegative charges on GAGs; (++): electropositive charges on PF4
For the rare VITT complication cases, a wrong injection route has been suspected, and the vaccine could then enter a blood vessel. Vaccine components, and especially adenoviruses, can then disseminate into the blood circulation and bind to some blood or vascular cells for which they have some tropism, like ECs and platelets, and this induces rapid activation of the innate immune system [25–28]. In blood circulation, adenoviruses can induce thrombocytopenia, and generate a cytokine storm syndrome, and thrombosis. If the vaccine enters blood circulation, contact of adenoviruses with PF4 is then a high probability, especially if it induces an inflammatory and immune response, which can release PF4. This contact is faster when significant concentrations of PF4 are already present in pathological states, where it is bound onto ECs’ GAGs. This hypothesis needs however to be demonstrated, but this could be the mechanism, which induces the PF4-dependent auto-immunization. Yet, the recent study by Baker et al. [24] confirms the binding of adenovirus vectors, used for the various DNA vaccines, to human cells by adhering through the fiber structures to CAR, a transmembrane-bound protein receptor exposed on many cells, like epithelial and endothelial, but also in heart and brain. Baker’s study [24] also shows that PF4 binds to adenovirus envelope proteins, particularly to electronegative hexons (at the interface of 3 hexons), and this could explain the pathogenic generation of IgG autoantibodies to PF4.
Although the VITT syndrome is reported to be like HIT, because the patients develop reactive antibodies to H-PF4, mechanisms and pathogenesis could be significantly different [22]. Firstly, these patients did not receive heparin or any polysaccharidic polyanion which can bind strongly to PF4 and induce the immune response. Secondly, the PF4-dependent antibodies developed in VIIT, are targeted to the same PF4 sequence, which binds to heparin-like-polysaccharides and is located on the ring of positive charges surrounding the PF4 tetramer [29]. This electropositive PF4 tetramer ring binds strongly to the heparin sulfate groups supporting electronegative charge [30]. VITT antibodies behave then like heparin for binding to PF4, with the exception that they are not a long polysaccharide chain tightening one or more PF4 tetramers and inducing the exposure of neo-epitopes.
When tested with the HIT PF4-dependent ELISAs, VITT antibodies are reactive with multimolecular H-PF4 complexes [5–7, 20]. In HIT, when heparin is withdrawn, the harmful effect of antibodies is stopped. Conversely, in VITT the pathogenic effects are lasting, and can only be controlled in surviving patients with non-heparin anticoagulant treatments, or Ig infusions, corticoids, and/or anti-aggregates [15, 31, 32]. The reported fatal incidence remains however high to very high [13, 16]. Thirdly, we have the experience of very rare thrombotic thrombocytopenic complications occurring in patients with oro-pharyngeal infections, associated with the development of cerebral thrombosis, which is not controlled by heparin-like anticoagulant therapy, but unexpectedly it worsens with this treatment [33]. In 2010, we described one of these cases, which concerned a young girl, who was followed over time. That patient developed cerebral thrombosis, and when treated with heparin, she rapidly had thrombocytopenia. She was tested for H-PF4 antibodies and was found highly positive, already a few hours after the onset of heparin treatment [33]. This anticoagulant was immediately withdrawn. That patient did not receive any heparin before the occurrence of thrombosis, and nothing explained the so rapid development of thrombocytopenia, with positivity for antibodies to H-PF4. A complementary investigation showed that this patient had antibodies to PF4 alone at a significantly higher concentration than to H-PF4. These autoantibodies progressively decreased but were still detectable 6 months after their first detection. The development of these antibodies was probably induced by the viral infection, and that mechanism, although less critical, looks like that occurring in VITT rather than that involved in HIT. This complication remains fortunately extremely rare, and not well-understood yet, but some questions arise: i) could the viral agent enter blood vessels to generate a PF4-dependent immune response? ii) was there a bleeding lesion at the infectious sites, which could facilitate the contact with blood cells, then their activation provoked by the inflammatory and the immune response? If yes, this could induce platelets’ attraction, their activation, and PF4 release, which becomes then available for reacting with viruses. We had the opportunity to test various other cases highly positive for antibodies to PF4 alone, which were detected because the patient’s developed thrombosis, and thrombocytopenia occurred immediately following heparin treatment. These cases resemble the spontaneous HIT-like cases reported by Warkentin and Greinacher [17].
The development of PF4-dependent antibodies, in the absence of heparin or any polysaccharidic polyanion, remains a highly ununderstood phenomenon. Recent studies show that the VITT complication has a very low incidence, and can affect healthy persons, with no predisposing risk [13, 16]. This complication was first reported to affect more young females than other individuals, but this is not confirmed by the most recent statistics, which show a close incidence in males. However, VITT could occur more frequently in younger people, probably due to their more reactive immune system. If concerned, a misdirected injection stimulus could produce a higher autoimmune response and pathology than in older persons. The factors favoring VITT occurrence remain however unknown, and the accidental wrong injection route remains only a hypothetic plausible cause yet confirmed. There are too few reported cases to analyze reliable statistics. No significant associations with antecedents of autoimmunity, former HIT, SARS-CoV-2 infection, or any identified risk factor have been demonstrated until now in patients with VITT [34–37].
Various studies have investigated patients vaccinated with adenovirus vector vaccines for the development of blood hyper-coagulability or anti-H-PF4 antibodies. No significant elevation of blood activation biomarkers, like D-dimer, or occurrence of any thrombocytopenia was detected. When vaccinated people were compared with a matched group of non-vaccinated persons, no significant differences were observed [38–40]. Only, a very slight increase of anti-H-PF4 antibody reactivity was reported, but without clinical significance [39]. In another study, a group of vaccinated individuals was monitored for the development of PF4-dependent antibodies. While most of them remained negative, antibodies to H-PF4 complexes were observed in about 1% of cases, but very few with significant reactivity [38]. The presence of these antibodies in a few cases, with very low or borderline concentrations compared to the very high ones observed in VITT, is questionable about their specificity or significance, and they must be considered with great caution [38–40]. This lack of significant PF4-dependent immune response in vaccinated individuals could be an argument for the involvement of yet non-identified factors in VITT, or of an accidental occurrence. If present, non-identified risk factors could contribute to the PF4-dependent immune response in very rare, vaccinated persons. Their eventual presence could explain the possible lower statistical incidence following the 2nd shot.
Recent articles have reported that VITT is induced by the development of anti-PF4-polyanion antibodies, like those responsible for HIT, in affected patients [13, 16]. This development occurs about 5 days to > 15 days following the first injection of DNA-adenovirus vector vaccines, sometimes later, and the incidence is thought lower following the 2nd shot, suggesting, if confirmed, the hypothesis of a patient-specific favoring context. This risk factor could be additive to the accidental wrong injection route hypothesis. A possible inducing cause has been suggested to be the interaction of the vaccine DNA spike-coded protein with the adenoviral vector [37]. This is however not fully in line with the induction of very high concentrations of anti-H-PF4 IgG antibodies, and the ultra-rare occurrence of VITT, mainly after the first shot. The few studies, which have investigated the occurrence of anti-H-PF4 antibodies following the adenovirus vector vaccine injection, show their possible development in a few percentages of vaccinated patients, which is far above the very rare cases reported for VITT, which incidence is only 0.001% to 0.0001% [13, 16, 38, 39]. Moreover, the reported anti-H-PF4 reactivities remain very low, and often in the grey zone. In these individuals, the PF4-dependent immunization mechanism, should it be present, is then borderline, and without any comparison to what occurs in VITT.
The key question remains to understand the causes that can induce the development of PF4-dependent antibodies in the rare patients concerned by that complication. A pre-existing sensitization to PF4 is possible, but it remains unlikely as it is expected to concern a higher proportion of patients, as reported by Greinacher et al. [41]. The expected incidence should be like that of HIT, in the range of 0.2–3% [30], or higher as many heparin-treated patients develop asymptomatic and transitory antibodies [42]. On the other hand, some infectious diseases could induce the development of anti-PF4-polyanion antibodies, including polyphosphates, with a persistent T-cell memory which renders the immune response anamnestic and faster following a new PF4-dependent stimulation. This context is thought to occur for some HIT complications. Yet, PF4 has an opsonin activity by binding to infectious agents’ polysaccharides or polyphosphates, and this favors the initiation of the innate immunity, then that of the PF4-dependent immune response [43]. Usually, the body’s immune system remains capable to distinguish between self and non-self-components. Although infections are frequent, and the primary development of anti-H-PF4 antibodies is a rather frequent phenomenon in heparin-treated patients [42], only a few subsets of them develop HIT pathogenic antibodies, and this is highly favored by the presence of clinical contexts like inflammation, the existence of pathological thrombogenic sites, and platelet activation, and by the antibody concentration [30]. The causes provoking HIT could rely on individual factors, not yet understood, or the random presence of heparin and PF4 concentrations in treated patients, which meet the conditions to generate a significant amount of macromolecular H-PF4 complexes, which stimulate the immune system. The persistence of H-PF4 complexes in blood circulation, and onto platelets and ECs, provokes antibody binding to H-PF4 complexes present on cell surfaces and platelet Fcγ-RIIA receptors. This induces platelet activation, aggregation, thrombocytopenia, and sometimes thrombosis [44].
For VITT, the following arguments suggest that the mechanism is obviously different: i) heparin or heparin-like polysaccharide are not present; ii) no electronegative polyanion chain is available for binding PF4; iii) the PF4 targeted epitopes by VITT antibodies are on the tight structure involved in binding to heparin or polyanions; iv) only PF4, without any PF4 binding protein, is required for capturing antibodies; v) inhibition of antibody binding to PF4 occurs for very low heparin concentrations. Concerning the adenovirus vector interactions, it can bind to CAR on human cells, and PF4 can react with adenovirus at the interface of 3 hexons. This binding remains very different from that of PF4 to polyanions. These features are in line with the recent study by Singh et al. [22], and the behavior of adenoviruses when introduced into blood circulation: they can bind to platelets, erythrocytes, and leukocytes [25–28].
It is necessary to identify what can induce the development of pathogenic amounts of high concentration and affinity PF4-dependent antibodies in the ultrarare VITT cases. Some hypotheses have been forwarded for understanding the development of these antibodies. A possible accidental shot into a blood vessel could allow the contact of the adenovirus vector with platelets, initiating a harmful pathological loop. However, should a wrong injection occur, the immunogen stimulus will remain at a low concentration, as the virus does not replicate, and VITT can develop following a single shot. Baker et al. [24] have reported the binding of PF4 to the virus surface, through its interaction with hexons. The immune system could be misdirected and fooled by the viral complex, therefore developing antibodies not only to the virus itself but also to the intimately bound human proteins, PF4 in this case. This mechanism has been already described for some allo-immune complications occurring with infectious diseases and inducing thrombosis subsequently to antibody binding to hemostasis or platelet proteins for example [33, 45–50]. It is schematized in Figure 2. In VITT, the immune response is only directed to the tight electropositive PF4 amino acid sequence involved in its binding to hexons [24, 29]. Other possibilities could be linked to individual contexts or risk factors, with a propensity to develop an autoimmune reaction in parallel to the vaccine response. Present evidence suggests that the VITT causative agent is more the adenovirus itself than the vaccine adjuvants (which are not the same for the various adenovirus vector vaccines), or DNA (although DNA is an electronegative chain structure). The possible role of an isolated strong inflammatory reaction at the injection site, provoking an increased vessels’ permeability, and blood leakage from surrounding vessels is an alternative hypothesis to the wrong injection route. It can favor the release of PF4 and its binding to the adenovirus vector, then leading to the generation of PF4-dependent antibodies in some individuals.
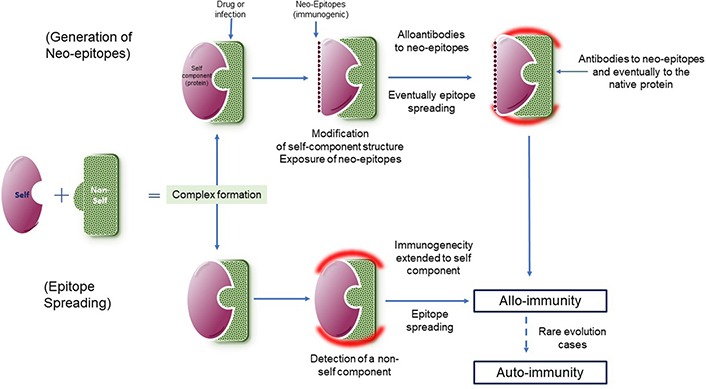
Complex formation between a self-component (purple) and a non-self-one (green) and possible mechanisms involved in the generation of antibodies to complexes, extended to the body’s proteins (red halo), which can produce autoimmune complications. On top, the complex formation alters the self-component structure and exposes neo-epitopes, that can induce the generation of antibodies. At the bottom, antibodies are generated to the non-self-component (green) and extended to the self-component (green) through epitope spreading. The reaction is alloimmune as targeted simultaneously to the non-self-component, which is required for the immunization process. In rare cases, the immune response to the self-component remains, although that to the non-self-component has disappeared, and becomes then autoimmune, and chronic
PF4-dependent antibodies responsible for VITT have been identified by testing patients’ plasma or serum with anti-H-PF4 ELISAs, proposed for the diagnosis of HIT. Interestingly, in VITT, not all immuno-assays for H-PF4 can catch the PF4-dependent IgG antibodies [7, 20, 51]. Lateral immuno-filtration assays and chemiluminescent-based assays frequently fail to detect those antibodies, although they correctly detect the H-PF4 antibodies developed in HIT. These differences result from the targeted epitopes on PF4, which are not the same in VITT or HIT. In addition, conversely, to HIT, antibodies developed in VITT are more reactive to PF4 alone than to H-PF4 [6, 22], and this is confirmed by our experience from testing a few patients’ plasmas with this complication. When present, PF4-dependent antibodies are often able to activate platelets in HIT but can also be asymptomatic [30, 42, 44]. In VITT, no observation of asymptomatic patients with the presence of high concentrations of PF4-dependent antibodies has been reported.
The present recommendations for the diagnosis of VITT are now to test plasma or sera from suspected patients with a conventional H-PF4 ELISA, proposed for the diagnosis of HIT, excluding the chemiluminescent based methods or immuno-filtration techniques [15, 31, 51]. In addition, functional HIT assays with PF4 supplementation can also be used [6]. The different behavior of these assays for capturing PF4-dependent antibodies is not understood, but there is evidence that the PF4 epitopes are not exposed in VITT in the same way as in HIT. In addition, it has recently been shown, that various groups of PF4 epitopes can react with heparin-dependent antibodies in HIT, whilst only the ring of positive charges, which spreads around the PF4 tetramer and binds heparin, is reactive with VITT antibodies [24, 29]. The amino-acid sequence involved in these epitopes has been identified and concerns the same PF4 electropositive structure which binds to heparin or electronegative polyanions. Furthermore, heparin or polysaccharide polyanions totally inhibit the binding of VITT IgG antibodies to these PF4 epitopes, even at a low concentration, which is far below that required for disrupting multimolecular H-PF4 complexes by the excess of heparin, therefore inhibiting the binding of HIT antibodies. Mechanisms involved in HIT or VITT highly differ: in VITT PF4 reactive epitopes are masked by binding to heparin, whilst in HIT, the multimolecular complexes are dissociated by an excess of heparin.
The reactivity of PF4-dependent IgG antibodies with the various anti-H-PF4 ELISAs is discussed in the article by Platton et al. [20]. The Zymutest™ HIA assay, that we designed for the HIT diagnosis, is highly performing for capturing the anti-PF4 antibodies responsible for VITT, as reported in various studies [6, 20, 32, 51]. What could explain the different reactivities and sensitivities between assays is the presentation of PF4, the ratio of PF4 to heparin, and the formation of heparin and PF4 complexes, then their coating onto the solid phase (micro-ELISA plate, magnetic latex beads, or nitro-cellulose membrane, through adsorption or covalent coupling). The target epitopes could be masked by heparin or by the binding to a solid surface. The Zymutest™ HIA assay has the particularity to offer a dynamic binding of PF4-dependent immune complexes to heparin immobilized onto the solid surface [52], therefore capturing antibodies developed in VITT.
We tested various samples with ELISA, using PF4 or H-PF4 coated plates, and a tagged antibody targeted to human IgGs and coupled to horse radish peroxidase. Color development was obtained with tetra-methyl-benzidine containing hydrogen peroxide (TMB/H2O2). Inhibition assays are performed by incubating the assayed samples with a heparin or PF4 concentration range, then testing for the residual antibody binding to coated plates. The KKO monoclonal antibody [43, 53] was used as a control for the reactivity of HIT antibodies to H-PF4 and PF4.
From our experience of testing plasmas from patients with VITT, the use of PF4 alone, coated onto a solid surface, without heparin or any polyanion, offers the best capture antigen for antibodies. The most striking observations we did are: i) a much higher antibody reactivity to H-PF4 in VITT than that observed in HIT (for example, VITT patient samples need to be diluted up to 1/500 or even 1/5,000 to yield similar reactivities than those obtained usually for HIT at the 1/100 dilution), suggesting an antibody concentration far above that of HIT; ii) reactivity is stronger or much stronger to PF4 alone than to H-PF4 stoichiometric complexes; iii) inhibition of antibody binding to PF4 or H-PF4 coated plates is observed for a much lower heparin concentration than in HIT, as shown on Figure 3.
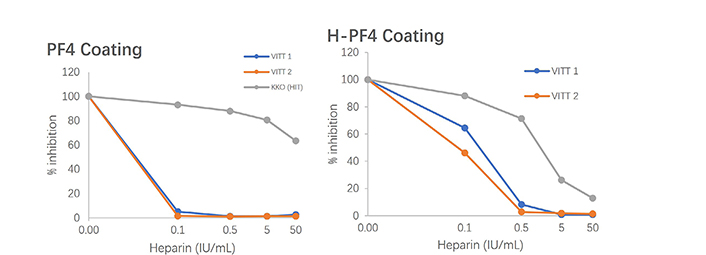
Inhibition of VITT antibodies binding to PF4 or H-PF4 coated plates tested by ELISA, comparatively to the HIT mimicking antibody (KKO); much lower concentrations of heparin totally inhibit binding to PF4, and a lesser extent to H-PF4 in VITT than in HIT
It shows an example of the inhibition of VITT or HIT antibody binding to H-PF4 or PF4 by increasing heparin concentrations (Figure 3). The HIT antibody behavior is illustrated by the monoclonal antibody mimicking HIT, KKO [43, 53]. We propose that the best and most sensitive assay for detecting VITT pathogenic IgGs is to use a capture solid phase coated with only PF4. We do not know if the development of IgGs is preceded by the development of IgM isotopes, and if IgA isotypes can be present as in HIT, as too few observations have been reported until now.
Understanding the mechanisms of the immune thrombocytopenia and thrombosis induced by the adenovirus vaccines was illustrated by the discovery by various groups that plasma from affected patients contained highly reactive IgGs targeted to H-PF4 complexes when tested with the PF4-based ELISAs used for the diagnosis of HIT [5–7]. The rationale to test anti-H-PF4 antibodies in these patients relied on the similarities between VITT and HIT: i) occurrence of rare cases with thrombocytopenia and thrombosis at unusual sites (cerebral sinus thrombosis, splanchnic vein, etc.); ii) time of onset of thrombocytopenia and thrombotic complications following the vaccine injection; iii) positive anti-H-PF4 immunoassays, and platelet aggregation assays, enhanced by PF4 supplementation, positive at low but not high heparin concentration. However, VITT patients received no heparin, and the hypothesis that another polyanion, of unknown origin, could be involved in binding to PF4 and producing immunogenicity was proposed [17]. The target antigen for HIT is obtained usually by preparing stoichiometric complexes of PF4 and heparin, at a proportion of about 27 international units (IU) of heparin per mg of PF4 [30]. This ratio is in the range of 150–200 μg of low molecular weight (MW) heparin per mg of PF4, depending on its MW and specific activity. The heparin regular disaccharide sequences and the regular pentasaccharide antithrombin (AT) binding sequence, all expose highly electronegative residues (mainly on sulfate groups) and are involved in the binding to PF4. Therefore, the heparin ponderal concentration matters more than its anticoagulant activity for forming the stoichiometric reactive complexes with PF4, able to bind HIT antibodies [52]. A heparin length of at least 12 to 14 saccharides is required for spanning around the PF4 tetramer structure and exposing the immunogenic epitopes [30], as shown in Figure 4.
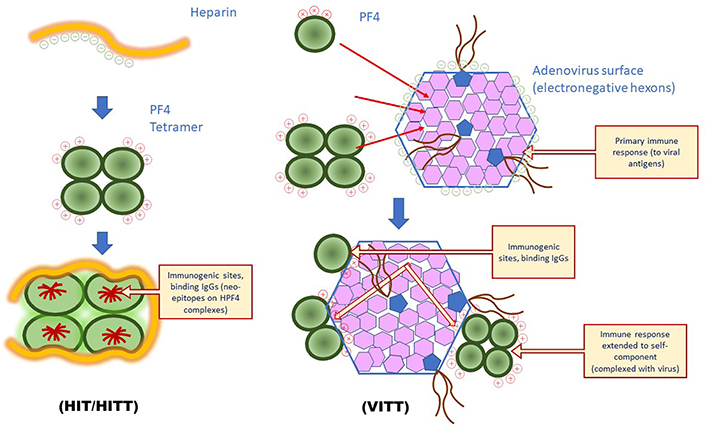
Differences in PF4-dependent immune response involved in HIT or VITT. In HIT binding of PF4 to electronegative polyanions induces the exposure of neo-epitopes, target for antibodies, which can extend to other PF4 epitopes through epitope spreading; in VITT, the ultrarare binding of PF4 to adenovirus vector is the probable stimulus for inducing the generation of antibodies to a restricted structure located at the PF4 positive amino acid sequence, involved in the binding to electronegative hexons: antibodies first target these hexons before the reaction is extended to the associated PF4 sequence through epitope spreading. HITT: HIT with thrombosis
In HIT, antibody reactivity is first targeted to the neo-epitopes exposed on the stoichiometric H-PF4 complexes, but a subset of patients has an extended reactivity to the whole PF4 molecule and can react with PF4 in the absence of heparin or any electronegative polyanion. The accepted pathological HIT mechanism is that these antibodies bind to H-PF4 complexes, when present at the right stoichiometric concentration in blood circulation; these complexes can then interact with platelet FγRII-A receptors, which induces platelet activation, aggregation, thrombocytopenia, and sometimes thrombosis [44]. However, we have hypothesized that antibodies not only bind to FcγRII-A, but also to PF4, which can be present on the platelet surface, and on that of endothelial and other blood cells, directly or through heparin [52–54]. This double binding enhances antibody binding to platelets, their activation, aggregation, and destruction, which amplifies the pathological process. This direct PF4 binding onto platelet surface was confirmed in 2000 by Newman and Chong [54]. Moreover, we have some evidence that anti-PF4 antibodies can develop at a pathogenic concentration and induce thrombosis in some clinical situations, in the absence of heparin. In very rare cases of oro-pharyngeal infections, we noted that patients can develop cerebral sinus thrombosis, which is not cured when heparin therapy is started, but thrombotic complications rapidly worsen with the occurrence of thrombocytopenia [33]. This requires an immediate withdrawal of heparin and the use of another anticoagulant. We had the possibility to investigate other and similar rare cases, with always a prominent reactivity to PF4 alone, although antibody binding to H-PF4 complexes was always present with high reactivity, at a lesser extent than to PF4 (unpublished data). Warkentin and Greinacher [17] also reported spontaneous HIT-like symptoms in the absence of heparin.
For VITT, PF4-dependent antibodies, bind to PF4 alone with a higher reactivity than to H-PF4 complexes as reported by Greinacher et al. [6]. These antibodies activate platelets in a HIT-like mechanism, i.e. in presence of low heparin or low-molecular-weight heparin (LMWH) concentrations but not a high one, and activation of platelets is enhanced by PF4 supplementation. This questions if a polyanion could be involved in this reactivity. In their recent study, Baker et al. [24] very elegantly demonstrated that PF4 can bind with high affinity to the electronegative surface of the viral hexons present on the adenovirus vector envelope, whilst adenoviral fibers bind preferentially to a CAR receptor present on many cells, and favor virus entry. Interestingly, the PF4 epitopes reactive with VITT antibodies, concern a much more restricted sequence than that involved in HIT. This PF4 electropositive domain contains many lysine and arginine residues and is masked by heparin-binding. Our experience confirms the heparin inhibition of VITT antibody binding, at very low heparin concentrations, as shown in Figure 3. Reactivity of PF4-dependent antibodies developed in VITT is then totally different from those present in HIT and is in line with the report of Singh et al. [22]. For capturing those antibodies, only PF4 is required, although H-PF4 complexes remain very reactive, provided that electropositive PF4 epitopes remain free and available. This availability can highly differ between immunoassays, which explains their different behavior for capturing VITT antibodies. PF4 tetrameric presentation or its binding to heparin or polyanions is not required for capturing VITT antibodies. In addition, other characteristics differentiate VITT from HIT: anti-PF4 antibodies are at a much higher concentration; platelet count nadir is usually much lower; D-dimer concentrations are much higher (up to 80–100 μg/mL). In our experience, blood activation markers, like procoagulant extra-cellular vesicles (EVs) or EVs exposing fissue tactor (TF), are elevated to very elevated and parallel D-dimer variations, which is expected from the extensive blood activation.
Pathogenicity of antibodies to PF4 responsible for VITT results from their direct binding onto PF4 exposed on platelets, ECs, or leukocytes, through direct binding or by the intermediary of cell-surface aminoglycans, or polyphosphates [43, 53]. We know that circulating activated platelets can bind more heparin, and therefore PF4, than platelets at rest [55], and this can contribute to an enhancing loop effect, by releasing more PF4 by newly activated platelets, and to increased binding of antibodies. As was observed in patients with oro-pharyngeal infections when PF4 is present, thrombocytopenia and thrombosis can occur in the absence of heparin, and the disease worsens if heparin therapy is initiated. Pre-existing thrombosis and rapid onset of thrombocytopenia following anticoagulation with heparin is strong evidence that antibodies bind to PF4 alone, already present on the platelet surface, but possibly also onto ECs and leukocytes. A similar but amplified mechanism could occur in VITT, as illustrated in Figure 4.
Thiele et al. [35] reported that the concentration of anti-PF4 antibodies remains high in patients with VITT when followed over time. This does not match with our experience, and with the antibody kinetics course in most allo-immune complications, which is usually transitory, and not evolving into auto-immune diseases: antibody concentration progressively decreases over time, at a rather low rate, and its pathogenicity disappears within a few months (3 to 6 in most cases). The impression that antibodies remain constant with time comes probably from the use of the standard sample dilution in H-PF4 ELISAs, i.e. 1:100 dilution currently. To follow the quantitative evolution of antibody concentration, it should be necessary to test much higher dilutions, and in our experience plasma samples from patients with VITT must be diluted much more than the usual 1:100 dilution, up to 1:500 or 1:5,000, eventually more. We speculate that the higher the IgG antibody concentration in patients with VITT, and more severe the complications. Obviously, these latter are also depending on the patient global clinical status and are highly influenced by the presence of possible risk factors, which can render antibodies more harmful.
The present therapeutic protocol for treating patients with VITT is to not use heparin, from the experience of HIT, and because oro-pharyngeal patients with anti-PF4 antibodies have their condition worsened with heparin, which confirms the complete counter-indication of that anticoagulant. Alternative and non-polysaccharidic anticoagulants must be considered, along with antiaggregant, or Ig infusions used to treat the auto-immune complication, and eventually anti-inflammatory drugs. This is not the goal of that review, which just focuses on mechanisms, but we remind the published recommendations [15, 31, 32].
VITT complication resembles very rare immune complications occurring during infectious diseases, or during treatments like heparin. We already discussed the immune thrombotic complications developed in oro-pharyngeal disease cases, probably induced by adenovirus infection, and which were associated with the development of anti-PF4 antibodies. These immune complications should be characterized as allo-antibodies rather than auto-antibodies, as they disappear progressively when the disease heals, and the non-self-component at the origin of the immune response is no more present. By contrast, the autoimmune response characterizes cases where the response remains and becomes chronic, in the absence of the non-self-component. The mechanisms and the causes involved in these rare complications suggest that the infectious agent binds strongly to a self-component (a protein generally), which misdirects the immune response. It then does not discriminate between the self and the non-self, which are intimately associated in the same complex [56]. The immune system is then unable to identify the self-body protein from the aggressive pathogenic antigen, especially if this contact is prolonged. Fortunately, the usual immune response is appropriate, because the antigen formation between the self and the non-self can remain rare and the immune system keeps its selectivity. Alternatively, patients with auto-immune complications could have an accidental pre-immunization, and therefore the T-cell memory promotes a rapid immune response. Lastly, the inducing cause could be associated with rare individual risk contexts or genetic factors.
The lessons that we learned from the generation of autoantibodies to thrombin in patients treated with fibrin glue [45], but also from the understanding of HIT, illustrate the development of these auto-immune complications. The complex formation between external components and self-proteins, or the introduction of an exogenous protein sharing enough epitopes with the self-one, can induce an immune response with epitope spreading [57]. The latter is first targeted to the non-self-epitopes, but it can be extended to the self-epitopes present on the same molecular structure or complex. The autoantibodies to thrombin in fibrin glue illustrate this mechanism. This surgery product, used to accelerate healing, contained human fibrinogen and bovine thrombin. Many patients receiving this fibrin glue for stopping bleeding or favoring wound healing, developed antibodies, first targeted to bovine thrombin only, and without any complication, but antibody reactivity rapidly extended to the human thrombin itself, then producing microvascular thrombosis and DIC. The selective immune response to bovine thrombin (i.e. to its distinct epitopes differing from human thrombin), was misdirected to the specific human epitopes through the epitope spreading process [57]. This is shown in Figure 2. Other examples are available like those responsible for thrombotic events in rare chicken pox/varicella infections, with anti-coagulation protein S (S) antibodies, or adenovirus infections inducing antibodies to prothrombin, rarely to FX, and associated with a lupus anticoagulant (LA) like syndromes [45, 49]. In both cases, high concentrations of antibodies to protein S or prothrombin were identified. We recently had another example, by studying rare, acquired coagulation factor V deficiencies, associated with a second-line antibiotic therapy in patients resistant to the first line. This deficiency was induced by the development of antibodies to coagulation factor V (FV) [58]. This list is not limitative, and many other examples could be cited, like the anti-platelet antibodies which can generate transitory or chronic thrombocytopenia subsequently to induction of autoantibodies to platelet surface proteins during viral infections like with Epstein Bar, or cytomegalovirus [47, 48]. The immune response is then extended to the generation of autoantibodies to platelet surface glycoproteins (GPs), like GP Ib-IX, or GP IIb-IIIa. In some patients, this autoimmune disease, developing during viral infection, can become chronic with the persistence of platelet autoantibodies, and leads to idiopathic thrombocytopenic purpura. Some of the immune responses, which can generate autoantibodies to the body’s proteins, are summarized in Table 1.
Examples of autoantibody generation in situations where an infectious component or a drug bind to a blood protein
| Disease | Antibody target | Clinical symptoms | Clinical severity | Antibody clearance (time) | Chronic autoimmune disease evolution |
|---|---|---|---|---|---|
| Oropharyngeal infection | PF4 | Cerebral and other thrombosis | + to +++ | 3–6 months | Unknown |
| Chicken pox | Protein S | Phlebitis, periheral thrombosis, renal failure, limb extremity necrosis | ++ to +++ | 3–6 months | Possible |
| Oropharyngeal infection | Prothrombin | Bruise, prolonged clotting time, bleeding | +/– to +++ | 3–6 months | Possible |
| EBV or CMV infection | GP IIb-IIIa | Thrombocytopenia | + to +++ | > 6 months | Frequent |
| Influenza infection, others | ADAMTS-13 | iTTP | ++ to +++ | Possible | |
| Healing with fibrin glue (bovine thrombin) | Thrombin | Thrombosis, DIC | ++ to +++ | > 3 months | Not reported |
| Various infections | B2-GP1, prothrombin | LA, prolonged clotting time, thrombosis, bruise | +/– to +++ | 3 to > 6 months | Possible |
| Autoimmune complications, lymphoproliferative disorders, others | Factor XIII | LA-like, delayed hemorrhagia | ++ to +++ | Under investigation | Possible |
| Antibiotherapy for antibioresistance | Factor V | Thrombosis, DIC | ++ to +++ | Unknown | Unknown |
EBV: Epstein-Bar virus; CMV: cyto megalo virus; iTTP: idiopathic thrombotic thrombocytopenic purpura; +: weak ; ++: moderate; +++: strong ; +/–: borderline
From our experience of HIT laboratory study and of antibody pathogenicity mechanisms, and from the literature analysis of the immune thrombotic thrombocytopenic syndrome developing very rarely following the vaccination to SARS-CoV-2 with DNA adenovirus vector vaccines, in this article we review the characteristics of the VITT PF4 dependent antibodies. We illustrate it with data from our laboratory investigation and we propose our understanding of the mechanisms involved. There is no evidence that these antibodies are targeted to a well-defined structure located on the electropositive domain of PF4, as very nicely demonstrated by Huynh et al. [29] and discussed by Klok et al. [16]. Singh et al. [22] also demonstrated that antibodies are oligoclonal, comprising 3 different monoclonals. Obviously, VITT antibodies markedly differ from those targeted to H-PF4 and developed in HIT.
The first identification of these PF4-dependent antibodies came from the observation of VITT clinical similarities with HIT [5–7]. Severe thrombocytopenia occurs, developing 5 to > 15 days from vaccination, along with thrombotic events, frequently at unusual sites like cerebral sinus or splanchnic vein, and can evolve to DIC. The association of thrombosis with thrombocytopenia suggested a HIT-like mechanism [13, 16, 18]. Laboratory testing confirmed this association, and when evaluated with the anti-H-PF4 ELISAs, used for the diagnosis of HIT, plasmas from VITT patients contain high concentrations of reactive IgGs, although no heparin was present. In addition, these antibodies can activate platelets in functional assays, and supplementation with PF4 increases this reactivity [6]. Many authors suggested then that an unknown polyanion could be involved in the generation of PF4-dependent immunogenicity in VITT, but this polyanion has not been identified yet [20]. Indeed, polyphosphates can bind to PF4 and induce platelet activation if complexed with antibodies [43, 53], but antibody-PF4 complexes can activate ECs and release tissue factors [59] or induce platelet activation and death [60]. Michalik et al. [61] have reported the presence of electronegative contaminants like cell debris and proteasomes in the vaccine preparations, and this could increase the risk to develop anti-PF4 antibodies in addition to ethylenediamine tetra-acetic acid (EDTA) induced capillary leakage, leading to an increased risk to develop VITT [61]. Recently, Greinacher et al. [62] demonstrated the presence of vaccine components forming complexes with PF4 and VITT patients’ antibodies on the platelet surface. The authors suspected the complex formation of PF4 with DNA or polyphosphates, generating motives capable to generate and bind antibodies, then inducing thrombocytopenia and thrombosis [62]. The capillary leakage favored by EDTA associated with the inflammatory response could stimulate that process.
However, if some similarities with HIT suggest a close mechanism for VITT, many differences are nevertheless present. Thrombocytopenia uses to be much more severe in VITT remains than in HIT; thrombotic complications are worse with huge levels of D-dimer and sometimes a decrease of fibrinogen concentration; lethality is higher; conversely, to HIT antibodies, reactive with all assays, antibodies are detected mainly with anti-H-PF4 ELISAs, whilst most of them are missed with chemiluminescent or lateral flow migration assays; supplementation with PF4 contributes to increasing the functional assays’ reactivity. In addition, Huynh et al. [29] have shown that the target epitope for VITT antibodies on PF4 is located at the heparin electronegative polyanions’ binding site, i.e. they bind to the same PF4 site as heparin. By contrast, HIT antibodies are reactive with neo-epitopes generated through the stoichiometric binding of PF4 tetramers with heparin or electronegative polyanions, and this reactivity can be extended in some patients to other PF4 epitopes. Antibodies are then polyclonal [22, 30]. Our limited experience of testing a few plasmas from patients with VITT suggests that the reactivity to PF4 is very different in VITT compared to HIT. VITT antibodies are much more reactive to PF4 alone than to H-PF4 complexes, and PF4 reactivity is inhibited by very low concentrations of heparin, unable to disrupt H-PF4 complexes. Antibody concentration, which is associated with disease severity, is 5 to 50-fold higher in VITT than the current concentrations measured in HIT (from our experience; data not yet published). This high antibody concentration suggests massive immunogenicity stimulated in the ultrarare patients developing VITT. The follow-up of vaccinated patients over time did not show significant differences when compared to a matched non-vaccinated group for the presence of anti-H-PF4 antibodies, although borderline or low anti-H-PF4 antibody concentration was detected in a few percent and is unable to activate platelets [35, 38]. Furthermore, blood activation biomarkers like D-dimer remained unchanged [40]. The pathogenic antibodies, although with a higher tendency to develop in younger patients, are fortunately only present in 0.002–0.001% of vaccinated patients, suggesting an accidental factor or an unknown risk factor predisposing to this complication.
The study by Baker et al. [24] has demonstrated the capability of PF4 to bind to the adenovirus vector surface, at the interface of 3 hexons, through electrostatic interactions, even though weaker than the interaction of PF4 with heparin, as it can be displaced with intermediary salt concentrations. This binding is not expected to alter the PF4 structure and to expose neo-epitopes, conversely to its binding with stoichiometric concentrations of heparin. This suggests an immune response to the adenovirus vector, which is extended to the PF4 binding domain bound to viral hexons through an epitope spreading process. A similar immune response to PF4 has been observed in very rare cases of viral infections as previously discussed [33]. This could only occur if enough concentrations of PF4 are available for binding to the viral envelope hexons. This is unlikely to occur when the vaccine is correctly injected into the shoulder muscle. We could however speculate that a massive inflammatory response could attract leukocytes and platelets and induce some blood leakage with the release of PF4, but should it be the case, the occurrence rate should be expected to a higher incidence in vaccinated patients, who use to develop an inflammatory response. The ultrarare VITT complications could then result from an accidental shot into a blood vessel. Yet, when introduced into blood circulation adenovirus vector could bind to platelets, induce inflammation, and release PF4, which binds back to the adenovirus surface, as shown in mice [25–28]. With adenovirus vector vaccines, the viral dose is too low for inducing thrombocytopenia, but PF4 can be released at contact sites with platelets, bind to adenovirus, and form immunogenic adenovirus-PF4 complexes. It has been shown that there is no relationship between antibodies to SARS-CoV-2 and the development of PF4-dependent antibodies [36]. It is then unlikely that the spike protein is involved in that complication. As shown in Figure 4, our understanding is that the strong immune response is targeted to the adenovirus and in rare cases could be extended to the PF4 bound onto the viral envelope, and results in the development of acute PF4 autoimmunity. Generated antibodies are then targeted to the well-defined PF4 electropositive site involved in the binding to hexons. This site is the same as that interacting with heparin [29]. PF4 antibodies can activate platelets, by directly binding to PF4 present on their surface or by forming immune complexes captured by the FcγRIIA receptors, producing then platelet aggregation and thrombosis. The binding of antibodies to PF4 is abrogated in vitro by low concentrations of heparin. Interestingly, this observation is in line with the recent report from Singh et al. [22] showing that therapeutic concentrations of heparin neutralize the PF4 antibody binding in patients with VITT.
Interactions of adenovirus vectors with blood proteins were reported for coagulation FX, which can bind to the viral envelope, onto hexons [23]. One could fear FX to also be a target for the immune response associated with adenovirus vector vaccines, and that it could be decreased, especially in patients with VITT. To our knowledge, no FX-induced deficiency has been reported following vaccine injections or in patients with VITT. Probably, the binding of PF4 is much stronger than that of FX, and this chemokine can become associated more intimately with adenoviral hexons [24].
The limitations of this study concern the few cases of VITT which were analyzed in our laboratory, but a deep analysis of literature has been performed to understand the possible mechanisms. They are discussed respectively to our laboratory observations and experience from HIT. Complementary studies are needed to investigate the incidence of the other PF4 binding polysaccharides, heparin-like material or polyanions, in VITT antibody reactivity. Conversely to heparin, fondaparinux or ultra-short low MW heparins (with less than 12 or 10 saccharide units), can bind to PF4 through electrostatic interactions, but they are unable to alter the PF4 structure because they are not long enough to wrap around the PF4 tetramer. We anticipate that these materials can also inhibit VITT antibodies binding to PF4. We tested DNA, which is an electronegative chain, but it has a low inhibitory effect on VITT antibodies binding to PF4, much weaker than that of UFH or enoxaparin. The last study must be done by testing combinations of the adenovirus viral vector with PF4 to analyze the impact on capturing VITT antibodies, and to document the co-existence of antibodies to the viral envelope with those to PF4, or to PF4 complexed with hexons.
Therefore, the ultrarare antibodies developed in VITT and responsible for thrombocytopenia and thrombosis are targeted to PF4. They present a stronger reactivity to PF4 alone than to H-PF4 complexes, and the binding is inhibited by very low concentrations of heparin. This is a significantly different reactivity from that of anti-H-PF4 antibodies developed in HIT, and antibody concentrations are much higher in VITT than in HIT. Detecting these antibodies should be better performed using a solid phase coated with PF4 alone, rather than with H-PF4 complexes.
CAR: coxsackie and adenovirus receptor
DIC: disseminated intravascular coagulation
ECs: endothelial cells
ELISAs: enzyme-linked immunosorbent assays
FX: factor X
GAGs: glycosaminoglycans
GP: glycoprotein
HIT: heparin-induced thrombocytopenia
H-PF4: heparin-platelet factor 4
IgG: immunoglobulin G
LA: lupus anticoagulant
MW: molecular weight
PF4: platelet factor 4
S: coagulation protein S
SARS-CoV-2: severe acute respiratory syndrome coronavirus-2
VITT: vaccine-induced immune thrombotic thrombocytopenia
JA designed the studies and analyzed the results; EL, MV, DR prepared the technical protocols, did the figures and tables, contributed to the literature survey and realized the experimentations; GR redacted analytical protocols, reviewed data, and participated to literature review and synthesis. All authors contributed to manuscript revision, read and approved the submitted version.
JA is consultant for HYPHEN BioMed SAS, EL, MV, DR and GR are employees of HYPHEN BioMed, company which commercializes the Zymutest HIA IgG assay.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

