
Abstract
Complement component 1q (C1q) is the recognition molecule of the classical pathway of the complement system that can bind to an array of closely spaced antigen-bound immunoglobulin G (IgG) and IgM antibodies. In addition to its involvement in defence against a range of pathogens and clearance of apoptotic and necrotic cells, C1q has also been implicated in immune and non-immune homeostasis. C1q is locally produced by immune cells such as monocytes, macrophages, and dendritic cells. C1q is also synthesized by decidual endothelial cells, thus acting as a link between decidual cells and trophoblasts, as well as contributing to the remodelling of spiral arteries. Furthermore, C1q is produced by the extravillous trophoblasts (EVTs) invading the decidua. As a pro-angiogenic molecule, C1q is also important for normal placentation processes as it favors the active angiogenesis in the developing decidua. These observations have been validated by C1q gene knock-out mice which showed pre-eclampsia (PE)-like symptoms, characterized by hypertension, proteinuria, glomerular endotheliosis, and increased soluble fms-like tyrosine kinase-1 (sFlt-1)/placental growth factor (PlGF) ratio, and increased oxidative stress. The role of C1q in normal and adverse human pregnancy is being studied extensively due to its absence or low level as a likely precipitating factor for the development of PE.
Keywords
C1q, complement system, pregnancy, pre-eclampsiaIntroduction
Complement component 1q (C1q) is the first recognition subcomponent of the complement (C) classical pathway [1]. Its reported functions in a range of biological processes including tissue remodelling, aging, neuronal synapse homeostasis, and immunomodulation, in addition to the classical pathway activation, are simply remarkable [2, 3]. By virtue of the local production of C1q by immune and non-immune cells, its role in pregnancy and its complications has also been examined recently [4, 5]. C1q expression can have both positive and negative effects on pregnancy outcomes. As regards its positive contribution to pregnancy, C1q has been shown to be an essential player in the correct placentation process. It is synthesized by extravillous trophoblasts (EVTs) to help their connection with the matrix of the maternal decidua, thus easing their invasive capacity towards the inner layer of myometrium and migration next to the spiral arteries [6]. Furthermore, the endothelial cells of spiral arteries synthesize C1q exploiting it as a bridge between them and EVTs, which place themselves in the walls of spiral arteries and cause the remodelling of the vessel [7]. C1q is also a pro-angiogenic factor that promotes neoangiogenesis and wound healing [8]. In a knock-out mouse model, C1q deficiency caused the development of a dysfunctional placenta and pre-eclampsia (PE)-like symptoms, suggesting that lack of C1q could be one of the causes of defective placentation and PE in human pregnancies [5].
With regard to the pathological consequences, C1q deposition is associated with the activation of the classical pathway of the C. Excessive C activation can cause angiogenetic factor imbalance and an impaired invasion of the decidua by trophoblasts, thus leading to incorrect placentation [5, 9]. Activation of the C has been observed in the pathogenesis of PE; pre-eclamptic women have decreased serum levels of C components due to consumption and increased deposition of activated C components in the placenta [10, 11]. With regard to C1q, its levels in pre-eclamptic women sera have been shown to be reduced as compared to controls; this may be linked to consumption of C1q consequent to C activation, or to the binding of C1q to apoptotic bodies and debris found at higher levels in pre-eclamptic patient circulation, because of the hypoxic conditions within the placenta [12].
In this review, we aim to first examine the contributions of C in the pathophysiology of pregnancy. Subsequently, we closely review the important role that C1q plays in placentation and angiogenesis.
The C system
The C system is the most potent effector system of innate immunity, being the most ancient defense mechanism against pathogens. The C system includes three different activation pathways, classical, lectin, and alternative [13, 14].
The first component of the classical pathway, the C1 complex, is formed by C1q and the two serine proteases C1r and C1s [15]. The C activation is primarily triggered by the binding of C1q to either immunoglobulin G (IgG) or IgM bound to an antigen [16]. Moreover, the classical pathway can be activated following the recognition by C1q of several endogenous ligands, including apoptotic and necrotic cells [17, 18], extracellular matrix (ECM) proteins [19], the fluid phase pattern recognition molecule pentraxin 3 (PTX3) [20] or the acute-phase C-reactive protein (CRP) [21]. Following ligand binding, C1q conformational change leads to the auto-activation of the associated C1r, which in turn cleaves and activates C1s [22]. The proteolytic activity of C1s induces the cleavage of C4, releasing the anaphylatoxin C4a and the C4b fragment, and C2, producing C2a and C2b fragments, which takes part in the formation of the classical pathway C3-convertase (C4b2a), in addition to releasing C2b. This convertase is responsible for the initiation of the common terminal pathway [16] (Figure 1). The lectin pathway starts upon the binding of mannose-binding lectin (MBL), ficolins (M, L, and H), or collectin-11 to mannose-rich carbohydrate surfaces. These proteins form a C1-like complex with MBL-associated serine proteases 1, 2 (MASP1, 2) and MASP2 like C1r cleaves the C components C4 and C2 resulting in the formation of the C3-convertase [23] (Figure 1). The alternative pathway is activated by spontaneous low-level hydrolysis of the central component of the C system, C3, to form C3(H2O), in the so-called process of “tick-over” C3(H2O) is able to bind to factor B, which is cleaved by factor D to form the fluid phase C3-convertase, C3(H2O)Bb (Figure 1).
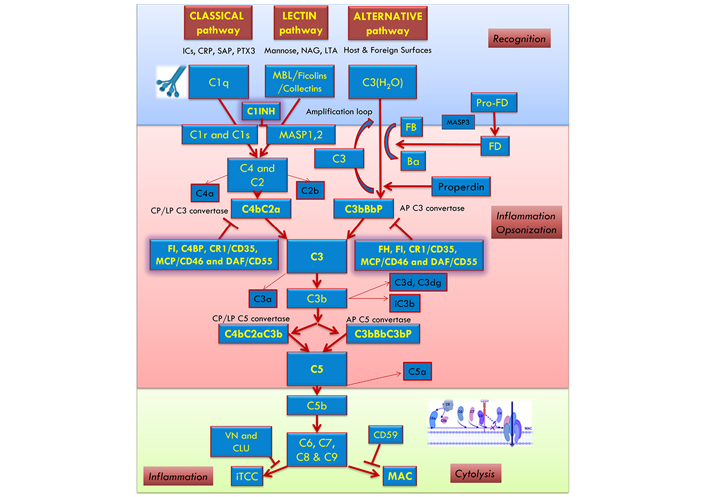
The C activation occurs through three pathways: classical, alternative, and lectin. The recognition and activation phases in each pathway converge on the formation of C3 convertase. The amplification phase starts when the C3 convertase cleaves C3, and C3b attachment to C3 convertases changes their substrate specificity, allowing them to become C5 convertase. The C5 convertase cleaves C5, starting the last phase (Terminal-Attack phase), in which C5b forms the membrane attack complex (MAC) with C6, C7, C8, and C9, resulting in cell perturbation. AP: alternative pathway; C1INH: C1-inhibitor; C4BP: C4b-binding protein; CLU: clusterin; CP: classical pathway; CR1: C receptor 1; DAF: decay-accelerating factor; FB: factor B; FD: factor D; FH: factor H; FI: factor I; ICs: immunocomplexes; iTCC: inactive terminal C complex; LP: lectin pathway; LTA: lipoteichoic acid; MCP: membrane cofactor protein; NAG: N-acetylglucosamine; Pro-FD: pro-factor D; SAP: serum amyloid P component; VN: vitronectin
All three C activation pathways converge on the common C3 molecule, which is cleaved by the C3-convertase (C4bC2a for lectin and classical pathways, C3bBbP for alternative pathway) into the anaphylatoxin C3a and the opsonin C3b [13, 14, 24].
In order to prevent excessive or aberrant effects due to its activation, C is subjected to the tight control of several regulators. For instance, factor I is responsible for the cleavage of C3b to its inactive form, inactivated C3b (iC3b) [25]. However, some C3b can bind to C-activating surfaces and associate with factor B, further cleaved by Factor D, leading to the formation of the predominant alternative pathway C3-convertase (C3bBb) involved in a rapid amplification loop. The binding of properdin enhances the stability of this highly labile convertase [26]. The binding of another C3b-fragment to the C3-convertase generates the C5 convertase, which consists of C3bBbC3b in the alternative pathway and C4b2a3b in the classical or lectin pathway. C5 convertase can cleave the C5 into C5a, another potent anaphylatoxin, and C5b, which in turn is able to sequentially interact with C6, C7, C8, and C9 to form the C5b-9 complex, also known as terminal C complex (TCC) [27]. If TCC is completely inserted into a cell membrane, it is called MAC. The C5b-9 complex promotes the lysis of cells and microorganisms when in sufficient quantities [13, 14, 28].
Physiology of the C system in the placenta
Locally in the placenta, the semi-allogeneic fetal antigens on the trophoblasts constituting the villi are continuously exposed to the maternal blood stream, and hence, C components. The contact between the maternal immune system and fetal antigens is also established during trophoblast invasion. Moreover, the mother commonly develops alloantibodies during pregnancy that could start the C cascade. Therefore, it is vital that the trophoblasts possess protective mechanisms against an inappropriate activation of the C system, in order to efficiently act in the placentation process. Indeed, studies have demonstrated that trophoblasts express both messenger RNA (mRNA) and protein level regulatory proteins of the C system, such as decay-accelerating factor (DAF or CD55), and MCP (or CD46), and CD59. The maternal-fetal interface is rich in C inhibitors. The murine equivalent for C inhibition is the protein “C receptor 1 related protein y (Crry)”; studies in mice showed that homozygous Crry deficiency resulted in the death of the embryo 10 days post-conception, suggesting an adverse outcome for an embryo that is unprotected from the maternal C attack. The additional proof of the involvement of the C system in fetus loss was the observation of a positive outcome in pregnancy of mice deficient for Crry and C3. In humans, polymorphisms of CD46 are associated with increased spontaneous abortions. The same mutation in women affected with systemic lupus erythematosus (SLE) was associated with PE. Furthermore, genetic mutations in C components and regulatory proteins lead to an increased risk of PE [11]. For instance, genetic variants of C receptors CR3 and CR4 were associated with PE onset [29]; moreover, the presence of mutations in C regulatory proteins in patients with PE, SLE, and/or antiphospholipid antibodies, resulted in a further confirmation of the association between C activation and disease pathogenesis [30].
In the maternal circulation during pregnancy, the C system has increased activity. Richani et al. [31] demonstrated the presence of increased concentrations of C3a, C4a, and C5a in the sera of 134 healthy pregnant women compared to non-pregnant controls (P < 0.001). The concentrations were stable during the whole gestation, from 20 weeks to the term. The upregulation of the C system is thought to ensure the protection of the fetus from possible pathogens circulating in the mother’s blood [31]. Circulating levels of C components were found significantly higher in pregnancy also based on a case-control study by Derzsy et al. [32].
The relevance of C activation in pathological pregnancy outcomes was recently highlighted in other systemic rheumatologic diseases, such as rheumatoid arthritis, psoriatic arthritis, and Sjögren syndrome [33].
The C system and PE
PE is a heterogeneous disorder that affects multiple organs; the prevalent feature of the syndrome is endothelial damage. The International Society for the Study of Hypertension in Pregnancy in 2014 defined PE as the new onset after 20 weeks of gestation of de novo hypertension with either proteinuria or end-organ dysfunction. The placenta is the key organ in the pathogenesis of PE since the removal of the placenta has been demonstrated to resolve the PE-associated symptoms [34]. The common feature is a defective deep invasion of the trophoblast that causes an impaired remodelling of the spiral arteries of the uterus [35]. The latter remains with a shallow diameter and high resistance, leading to inadequate placental perfusion. Indeed, histologic examination of placentas from pre-eclamptic patients revealed atherosclerosis in the arteries and arterioles, fibrin deposition, and infarcts [36]. Placental ischemia causes the release of soluble placental factors, such as soluble fms-like tyrosine kinase-1 (sFLt-1) and soluble endoglin (sEng), into the circulation of the mother leads to a widespread maternal endothelial dysfunction [34]. The hypothesis that placenta hypoxia is implicated in the development of PE was first proposed in 1989 by Roberts et al. [37]. Subsequently, Makris et al. [38] observed that surgically induced placental ischemia in rats and baboons induced hypertension and proteinuria, the main features of PE.
Excessive C activation can lead to placental dysfunction and is also associated with PE [10]. The C system can be activated via a range of mechanisms such as maternal antibodies [antiphospholipid and angiotensin II type-1 receptor (AT1) autoantibodies]. Ye et al. [39] reported a statistically significant correlation between the circulating levels of C3a and C5a anaphylatoxins in PE and the AT1 autoantibodies in severe PE, suggesting that the C system is the pathogenic effector of the autoantibodies. Furthermore, inadequate tolerance towards the exposed fetal antigens (which are semi-allogeneic) seems to be responsible for fetal injury and C activation can occur through the recognition of apoptotic and necrotic cells. Inflammation due to hypoxic conditions, caused by impaired angiogenesis, can be a possible cause of C activation in the placenta. Mechanically induced ischemia in the placenta has been found to be associated with increased C activation in a rat model [40]. It has also been recently demonstrated that sFlt-1 levels are directly linked to C activation in PE [41]. sFlt-1 is a scavenger receptor for placental growth factor (PlGF) and vascular endothelial growth factor (VEGF), preventing angiogenic function. It is expressed in the developing placenta, being a fundamental factor for the angiogenic homeostasis during pregnancy, as it modulates local PlGF and VEGF availability [42]. In the maternal circulation of PE women, the anti-angiogenic factor sFlt-1 is increased whilst the free pro-angiogenic factor PlGF is reciprocally decreased since it binds to the soluble receptor sFlt-1; therefore, the ratio sFlt-1/PlGF is higher. It is unclear yet whether the elevation of sFlt-1 is a primary or secondary event in PE pathogenesis. Placental C activation marker C4d, MAC, and sFlt-1 expression were quantified using immunofluorescence microscopy by Yonekura Collier and co-workers [41]. C4d and MAC were strongly correlated with sFlt-1 level in the placenta; increased placental C activation and damage were observed in PE, in correlation with sFlt-1 expression [41].
The lectin pathway seems to play an important role in C-mediated damage during pregnancy. Mating between CBA/J female and DBA/2 male mice yields an established model of immune-mediated early pregnancy loss; this is a model of PE as well since C activation in these pregnant mice seems to be implicated in the high rate of pregnancy loss observed in CBA/J mice [43]. MBL-A was present at the implantation sites of the abortion-prone mating combination CBA/J × DBA/2 that was undetectable in BALB/c-mated CBA/J mice. C4 and C3, but not C1r, could also be localized at the implantation sites of CBA/J × DBA/2, confirming the specific activation of the lectin pathway. In this model, pregnancy failure was prevented by the administration of neutralizing antibodies against C5, as well as by Polyman2, a mannose polymer, which binds to MBL-A with high affinity, and inhibits the lectin pathway activation. Higher levels of ficolins have also been found in pre-eclamptic placentae, suggesting that the lectin pathway is also activated likely in order to remove apoptotic cells [44, 45]. A recent study by Belmonte et al. [46] showed intense staining of PE placentae for ficolin-3, MASP1, and MASP2, supporting the hypothesis that the activation of C system is due to the lectin pathway that proceeds with the cleavage of C4 and C3 and the assembly of the TCC.
Overall, C cleaved products, in particular C4d, were detected at a significant level in pre-eclamptic placentae, demonstrating an increased activation of the classical pathway. Furthermore, an increased mRNA expression of C regulatory proteins (CD55, CD59) was observed in pre-eclamptic placentae, suggesting a compensatory mechanism to counterbalance an excessive C activation. The degree of C4d deposition and CD55 and CD59 upregulation were associated with the severity of the PE disease [47]. Activation of the alternative pathway also occurs in the placenta: Tedesco et al. [48] found consistent C3 deposition in the placentae of pre-eclamptic women. The components of the terminal pathway, in particular C9 neoantigen, were detected at a higher level in the pre-eclamptic placenta; the TCC may therefore be involved in the pathogenesis of PE by lysing the cells with consequent release of inflammatory products [49]. Another pathogenic role of the C was demonstrated by Girardi et al. [50]: C component C5a shifts the trophoblasts to an anti-angiogenic profile, with the low secretion of PlGF and an increase in sFlt-1 in C1q−/− mice models.
The C system in pre-eclamptic maternal circulation
In the sera of pre-eclamptic patients, even higher activation of the C system takes place compared to healthy pregnancies [51, 52]. Regarding the activation of the classical C pathway, Agostinis et al. [53] and Jia et al. [54] demonstrated that pre-eclamptic patients present lower blood levels of C1q and C4; the consumption of the C factors suggests an activation of the classical pathway or the binding of C1q to apoptotic bodies and debris [53]. However, in a recent study by Dijkstra and colleagues [55], the circulating levels of C1q were analyzed using an in-house enzyme-linked immunosorbent assay (ELISA) in three large cohorts in Europe and no changes were observed. Another study demonstrated higher levels of the activation product C4d in PE [32]. Increased levels of MBL were also found in PE compared to controls; being MBL an acute phase protein, this could reflect on the intravascular inflammation seen in PE [45, 56]. On the contrary, ficolins (H, L) were found at lower levels in the plasma but at high concentrations in the PE placenta [45, 46]. As a marker for the activation of the alternative pathway, fragment Bb, was significantly increased in pre-eclamptic patients compared to controls [57]. In general, anaphylatoxins C3a and C5a and terminal component MAC occur at higher plasma levels in the circulation of pre-eclamptic patients [58].
The contribution of C to PE is supported by the epidemiological observation that women affected by autoimmune diseases with consequent excessive C activation (such as SLE) show an increased likelihood to develop PE [59]. Common clinical features between atypical hemolytic uremic syndrome (aHUS), a disease characterized by excessive activation of the alternative C pathway, and hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome, a severe variant of PE, along with the findings of several studies [60–62], have confirmed the role of C activation in PE pathogenesis.
It is noteworthy that a limited but significant minority of patients present with symptoms resembling those of HELLP syndrome, aHUS, thrombotic thrombocytopenic purpura, and thrombotic microangiopathy (TMA) diseases exhibit endothelial dysfunction and aberrant activation of C and coagulation cascades. Lokki and Heikkinen-Eloranta [63] proposed that pregnancy-induced TMA in severe PE, in a subtype of PE patients present with a C dysregulation-mediated TMA-like disease (C-mediated TMA in PE) results from C-mediated thrombo-inflammation.
In humans, aspirin has been shown to reduce the risk of preterm PE, if started before 16 weeks of gestation [64]. It has been suggested that C3 and factor B downregulation, induced by aspirin, may improve the invasion of trophoblasts in the spiral arteries of the uterus [65].
The C components as predictive markers of PE
A few studies have investigated the expression of C components in early pregnancy stages. In the first study suggesting the C component as a possible predictive marker for PE before 20 weeks of gestation, a higher concentration of fragment Bb above the 90th percentile was found to be associated with a 3-fold increased risk of developing PE [57]. A recent prospective study, that analyzed the activation of the C system throughout the entire course of pregnancy, reported an alteration of the classical and alternative pathways occurred as early as the first trimester [54]. More specifically, the levels of factor B, factor H, C1q, C3c, and C4 were considerably higher in the first trimester in pre-eclamptic women compared to controls. A statistically significant correlation between C3c and PE was found, suggesting it could be used as an early predictive biomarker. The levels of the C components became similar between normal and PE groups by the advanced stages of pregnancy. This finding is contrary to the elevation of the C components C3a, C5a, and MAC after the onset of PE [10]. This biphasic correlation study revealed a period of initial C activation during the first trimester that can affect placentation and subsequently lead to PE; a second spike occurs after the clinical onset of PE, when C activation, due to the ischemic state of the placenta, contributes to the progression of the disease, causing trophoblastic damage and release of vasoactive substances [51].
C1q, the recognition subcomponent of the C classical pathway
C1q is the recognition molecule of the classical pathway that binds a range of immune and non-immune target ligands, including antigen-bound IgG and IgM [66]. The structure of C1q is classically described as a bouquet of tulip, with six subunits formed by the C-terminal heterotrimeric globular head C1q (gC1q) domain and N-terminal triple-helical collagen-like region holding the subunits together as a bouquet. Each subunit is a heterotrimer formed by three chains, C1qA, C1qB, and C1qC [67, 68]. The three polypeptides are synthesized by three distinct genes on chromosome 1p: C1qA, C1qB, and C1qC. The major source of C1q is the monocyte/macrophage lineage [69]. C1q can bind to a diverse range of self, nonself, and altered self-ligands, mostly via its ligand-recognizing gC1q domain while the collagen-like C1q (cC1q) domain can bind a number of putative receptors [2]. The gC1q domain recognizes several molecular patterns including modified-self and non-self-antigens, representing the key to C1q versatility [70]; on the other side, the cC1q is implicated in the C1q-mediated chemotaxis and removal of apoptotic cells by macrophages [71].
C1q is produced by the endothelial cells of the decidual spiral arteries
The fact that the C interacts with endothelial cells, bringing about changes in their morphology and function [72, 73], suggested that similar mechanisms may be involved in the endothelial cell remodelling which occurs in the decidua during pregnancy. Bulla et al. [7] revealed by immunohistochemical analysis that C1q was abundantly deposited on the endothelium of spiral arteries in the placenta; this pattern represented a pregnancy-associated condition since it was not observed in the case of any other vascular epithelial cells, such as skin, kidney, and brain, nor in the endometrium of the non-gravid uterus. It was also found that C1q was synthesized locally by and bound to the decidual endothelial cells (DECs). Furthermore, C1q binding was detected in clusters when the endothelial cells were in contact with migrating trophoblasts, likely acting as a molecular bridge for adhesion between the two cell populations; the cell-to-cell adhesion could be mitigated by blocking anti-C1q antibodies. In conclusion, the study proved that only during pregnancy the endothelial cells of spiral arteries gain the ability to synthesize C1q and to bind it on the cell surface. DECs-bound C1q is recognized by globular C1q receptor (gC1qR) on trophoblasts migrating into the endothelium, contributing to the pseudo-vascularisation of the spiral arteries and acting as a bridge between decidual endothelium and endovascular trophoblasts [7].
C1q is produced by the EVTs invading the decidua
In order to reach the perivascular area of spiral arteries, cytotrophoblasts invading the decidua bind to the ECM proteins such as fibronectin, laminins, and glycoproteins [74, 75]; C1q plays an important role in this process. The most abundant protein in the ECM of the decidua, especially in the column of invading cytotrophoblasts, is fibronectin, followed by laminin. These proteins bind to integrins, the adhesion molecules on the surface of cytotrophoblasts that have differentiated into EVT [76]. Elastin microfibril interfacer 1 (EMILIN1) was earlier identified as a connecting molecule between ECM and trophoblasts [77]. EMILIN1 belongs to the C1q family by virtue of harboring a gC1q domain [2]. Thus, Agostinis et al. [6] performed immunohistochemical analysis for C1q in first trimester placentas. C1q was abundantly localized in the stroma of the decidua, especially in the columns of invading trophoblasts and next to the spiral arteries. C3 and C4 were not present at the same level, excluding the defensive function of C1q in this context. Interestingly, the staining for C1q was positive inside the cytoplasm of the EVTs. Reverse transcription polymerase chain reaction (RT-PCR) of the mRNA extracted from the placenta was performed to determine whether cytotrophoblasts were able to synthesize and secrete C1q. Peripheral blood-derived macrophages were used as a positive control. Invading cytotrophoblasts expressed all three chains of C1q, while the stromal cells of the decidua only the C1qA chain [6]. Therefore, C1q is produced by invading trophoblasts, as well as by the endothelial cells of the spiral arteries. Furthermore, the ability to synthesize C1q was acquired only during the transformation to the invasive phenotype of trophoblasts; only the trophoblasts expressing the invasive phenotype marker, human leukocyte antigen G (HLA-G), were positive for C1q. Thus, invading trophoblasts are able to synthesize and secrete C1q for their adhesion to the ECM proteins found in the stroma of the decidua and for their migration towards the perivascular area of spiral arteries [6].
Using blocking antibodies for inhibiting the process revealed that C1q bound to both gC1qR and α4β1 receptors on trophoblasts which activated extracellular signal-regulated kinase 1 (ERK1)/mitogen-activated protein kinase (MAPK) signalling, involved in the adhesive and migratory functions [6]. It is worthwhile to note that C1q genetically deficient mice have a higher frequency of fetal death and low-size babies at birth. The implantation site had impaired labyrinth development, impaired vascular remodelling, and edema in the stroma. The columns of invading trophoblasts were reduced as well as the clusters of trophoblasts near the spiral arteries. In conclusion, the absence of C1q may impair placentation and can have a role in the pathogenesis of PE [6].
C1q has a proangiogenic effect
As described above, C1q is present on the endothelial cells of newly formed decidua during pregnancy. Thus, C1q could potentially also be expressed at other sites where vessels were newly formed, such as wound healing areas. Bossi et al. [8] investigated wound healing sites with immunohistochemical analysis: C1q was found to be present on the endothelium of the vessels, as well as in the stroma and in scattered inflammatory cells; C1r, C1s, C3, and C4 were absent. RT-PCR revealed a higher level of C1q mRNA expression in wound tissues compared to its woundless counterpart, confirming that endothelial cells produce C1q in conditions requiring neo-angiogenesis [8]. Having established the C1q function in angiogenesis, its ability to promote cell permeability, proliferation, and migration was investigated. The permeability-promoting capacity of C1q was assessed by adding fluorescein-5-isothiocyanate (FITC)-labelled albumin to a monolayer of endothelial cells in the presence of C1q; albumin leaked from the layer, as it did in the case of bradykinin (positive control). As regards to C1q pro-proliferative ability, endothelial cells were incubated with C1q and then analyzed for the expression of the proliferation marker ki-67, which doubled as compared to untreated control. Migration was demonstrated by scratch assays and transwell model systems, using VEGF as a positive control: the number of migrated cells was counted, highlighting a positive result; adding both VEGF and C1q, the effect was additive. The function of C1q was then evaluated in vitro, where endothelial cells growing into Matrigel formed tubes; ex vivo, where vessel sprouting was observed around aortic ring assays in rats; in vivo, where skin samples of wounds collected from C1q deficient mice showed a lower number of newly formed vessels compared to skin samples derived from wild type mice. Interestingly, the angiogenesis was restored when C1q was added locally. Thus, C1q is also important for a correct placentation process as it favors the active angiogenesis in the developing decidua; reduced angiogenesis could favor the development of PE [8].
Maduhukaran et al. [78] in 2015 investigated the transcriptional regulation C1q production in the developing placenta. As in macrophages and dendritic cells, the transcription factor purine rich box-1 (PU.1) was found to be involved in the regulation of C1q expression in transforming trophoblasts into EVTs and in endothelial cells undergoing the remodelling of spiral arteries [78].
C1q deficiency is associated with PE-like symptoms in mice
The importance of C1q in the placentation process has been highlighted by pregnant C1qa−/− mice that showed a reduction in labyrinth development, decreased trophoblast invasion, and impaired vascular remodelling at the implantation site, compared to wild-type (WT) mice of the same gestational age [6]. Singh et al. [5] in 2011 reported that pregnant C1q-deficient mice recapitulated the key features of human PE, such as hypertension, albuminuria, endotheliosis, decreased placental VEGF and elevated levels of sFlt-1, which correlated with increased fetal death. In addition to confirming these data, Sutton and co-workers [79] showed that the pregnancy-specific phenotype, driven by the heterozygous C1q+/− placenta, resulted in systemic maternal endothelial-dependent and -independent vascular dysfunction that persisted postpartum. Strikingly, C1q sufficient female mice, which were mated with C1q deficient male mice, showed the symptoms of abnormal placentation. Since the paternally expressed genes are dominant in the placenta, this observation supports the notion that trophoblast-derived C1q is essential for a normal pregnancy.
A recent study examined the decidua of female C1qa−/− mice mated with either C1q sufficient or C1q deficient male mice and compared the results with those observed in the decidua of WT mice [46]. As already shown, the percentage of remodelled decidual vessels in WT mice was significantly higher than that of unchanged vessels, whereas unremodelled vessels were prevalent in C1qa−/− mice. Interestingly, the percentages of changed and unchanged vessels in heterozygous C1qa+/− mice were essentially similar to those observed in WT mice [46].
C1q as a trigger for the classical C activation in the placenta
During the process of placentation, the tissue is remodelled, forming apoptotic debris, which together with natural IgG present in the placenta, are recognized by C1q that activates the classical pathway at the fetal-maternal interface. C1q helps to clear the apoptotic bodies and in this way helps maintain the fetal-maternal tolerance. The process is well balanced in healthy pregnancies by C regulatory proteins in order to avoid hyperactivation from the maternal immune system. Excessive C activation leads to oxidative stress, improper remodelling and placentation, causing a pathologic pregnancy, such as PE [80].
Localization of C1q in pre-eclamptic placenta
The analysis of the distribution of C1q in pre-eclamptic placentae can be compounded by study variations such as the type of PE (early-onset vs. late-onset) or the population studied, or the section of the placenta examined. Sinha et al. [81] found C1q to be present more on chorionic villi in PE compared to healthy placentae. Buurma et al. [47] observed C1q deposition in syncytiotrophoblast layers both in pre-eclamptic and control placentas. However, Lokki et al. [82] observed fewer C1q in the external multinucleated syncytiotrophoblast layer of the villi in late-onset pre-eclamptic patients compared to controls. The same group described less C1q in the walls of placental vessels in late-onset PE. The area stained with C1q showed high levels of necrosis. It is possible that in late-onset PE, where less C1q is detected, the ability to remove apoptotic bodies can be limited. C1q levels were higher in early-onset PE, suggesting a different pathology between the two groups. C1q deposition was not accompanied by terminal pathway products of the C system, suggesting a non-C role of C1q. C1q could bind to structures exposed to turbulent blood flow in PE [82]. Analysis of perivascular trophoblasts by Belmonte and colleagues [46] revealed the presence of C1q in cells surrounding remodelled vessels, in contrast to trophoblasts localized around unremodelled vessels that failed to express C1q.
Since C1q is essential in the placentation process (Figure 2) and C1q deficient mice show PE-like symptoms, Agostinis et al. [12] examined whether the circulating levels of C1q could correlate with the C1q levels in the placenta, therefore can be considered as a possible predictive marker for the development of PE. C1q levels in pre-eclamptic patients were compared with healthy pregnant women matched for age, parity, and gestational age. C1q levels were measured in the three trimesters of a healthy pregnancy and correlated with the C1q levels obtained from non-pregnant controls. The circulatory C1q values remained stable throughout pregnancy and were similar to non-pregnant women. However, the C1q levels in pre-eclamptic patients were reduced compared to healthy pregnancy controls. Although C4 showed a significantly lower level, the activation products C5a and downstream sC5b9 were not increased. One explanation is that C components can be reduced due to their binding to circulating macrovesicles such as syncytiotrophoblast extracellular vesicles (STBM) [83]. STBM are released from the placenta into the maternal circulation starting from the end of the first trimester. C1q and C4 were indeed present on STBM at similar concentrations between PE and healthy patients; the levels of C1q were reduced in the serum of PE women because more STBM are released from the dysfunctional placenta [12]. An alternative argument for a reduced expression of C1q can be a decreased production, but it is more plausible that C1q is consumed after C activation. In addition to STBM, C1q can also bind to immune complexes or apoptotic and necrotic bodies, all of which are increased in PE. However, neither the levels of C1q nor C3 and C4 were found to be statistically different between pre-eclamptic patients and controls at 11–13 weeks [12].
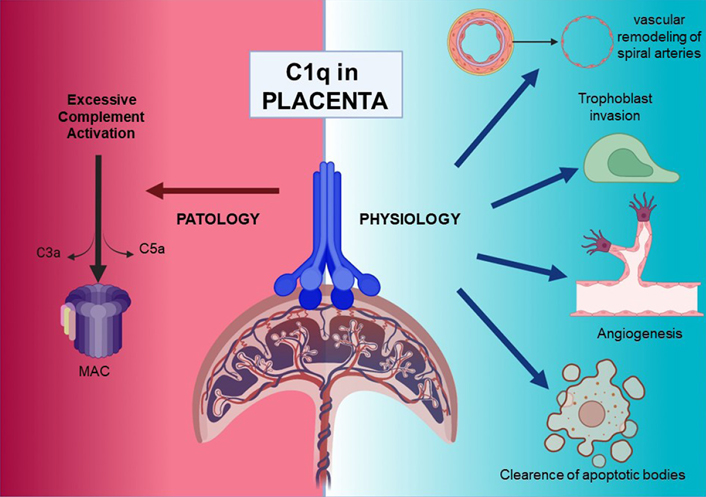
Overview of C1q roles in pathological and physiological placentae. Figure was created with BioRender.com
C1q mRNA expression in the pre-eclamptic placenta
In a study involving placental tissues from 7 pre-eclamptic patients and 6 matched normotensive controls, the mRNA expression levels of the three C1q chain genes, C1qA, C1qB and C1qC, were found to be much lower in PE [53]. In order to ascertain the cause of the reduced level of C1q in PE, isolated trophoblasts were stimulated with sera of both pre-eclamptic and control patients; PE samples induced a lower C1q expression. Inflammatory cytokines such as interleukin 6 (IL-6), IL-8, and tumor necrosis factor-α (TNF-α) were tested, which are increased in the sera of pre-eclamptic patients, and can also reduce C1q expression [53].
C1q–MBL interaction in PE
As explained earlier, the spiral arteries undergo physiologic changes during pregnancy, and the failure of this process leads to PE onset. Data indicate that DECs acquire the ability to synthesize C1q, which acts as a link between endovascular trophoblast and DECs favoring vascular remodelling. Sera obtained from pre-eclamptic patients strongly inhibited the interaction between EVT and DECs, preventing endovascular invasion of trophoblast cells [84]. MBL, one of the C components increased in pre-eclamptic patient sera, is able to block the interaction between C1q and EVT interfering with the process of EVT adhesion to and migration through DECs. These data suggest that the increased level of MBL in PE may contribute to the failure of the endovascular invasion of trophoblast cells.
Conclusions
Several studies have demonstrated that there is clearly a role for the C system and in particular for C1q in the pathogenesis of PE. C1q is physiologically involved in the trophoblast invasion process, the remodelling of the spiral arteries and neoangiogenesis. In PE, the local production of C1q is downregulated and strong deposition of C can be observed. Unidentified factors present in the sera of PE women could downregulate the expression of C1q in the placenta and impaired placentation then causes the release of other pro-inflammatory mediators that cause the clinical symptoms of PE. C1q is also slightly reduced in the circulation of pre-eclamptic patients. Possible explanations for this decrease could be that C1q binds to apoptotic cells of the damaged placenta, which are increased in PE; C1q binds to STBM, released from the hypoxic placenta in larger quantities; C1q binds to circulating immunocomplexes in pre-eclamptic patients; C1q is consumed in the damaged kidney during PE, because of the activation of the classical pathway [53]. All these classical and non-classical roles of C1q in the pathogenesis of PE need further investigation.
Abbreviations
| C: | complement |
| C1q: | complement component 1q |
| Crry: | C receptor 1 related protein y |
| DECs: | decidual endothelial cells |
| ECM: | extracellular matrix |
| EVT: | extravillous trophoblast |
| IgG: | immunoglobulin G |
| MAC: | membrane attack complex |
| MASP: | mannose-binding lectin-associated serine protease |
| MBL: | mannose-binding lectin |
| mRNA: | messenger RNA |
| PlGF: | placental growth factor |
| PE: | pre-eclampsia |
| sFLt-1: | soluble fms-like tyrosine kinase-1 |
| SLE: | systemic lupus erythematosus |
| STBM: | syncytiotrophoblast extracellular vesicles |
| TCC: | terminal C complex |
| TMA: | thrombotic microangiopathy |
| VEGF: | vascular endothelial growth factor |
| WT: | wild-type |
Declarations
Acknowledgments
We thank Gabriella Zito and Erica Curtolo for their intellectual assistance.
Author contributions
CA, AM, AB, UK, and RB reviewed the literature and wrote sections of the review article. CA created figures. UK critically reviewed the entire manuscript. All authors contributed to the article and approved the submitted version.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2022.