 Review
Review

Affiliation:
1Department of Immunology, Graduate School of Pharmaceutical Sciences, Hokkaido University, Sapporo 060-0812, Japan
ORCID: https://orcid.org/0000-0002-7474-493X
Affiliation:
2Department of Hematology, International University of Health and Welfare, Narita 286-8686, Japan
ORCID: https://orcid.org/0000-0002-5571-2457
Affiliation:
1Department of Immunology, Graduate School of Pharmaceutical Sciences, Hokkaido University, Sapporo 060-0812, Japan
Email: tmatsuda@pharm.hokudai.ac.jp
ORCID: https://orcid.org/0000-0002-3089-3757
Explor Immunol. 2022;2:760–770 DOI: https://doi.org/10.37349/ei.2022.00081
Received: July 28, 2022 Accepted: October 31, 2022 Published: December 27, 2022
Academic Editor: Masutaka Furue, Kyushu University, Japan
The article belongs to the special issue Cytokines and Skin Diseases
Psoriasis is a skin disease characterized by scaly erythema, parakeratosis, and epidermal hyperplasia. Application of imiquimod (IMQ), a ligand for Toll-like receptor 7, produces a mouse model for psoriasis. IMQ application induces scaling, erythema, and thickness in skin lesions, and the symptoms are milder in interleukin-23 p19 (Il23p19)-deficient and Il17a-deficient mice than in wild-type mice; this suggests that the interleukin-23 (IL-23)/T helper 17 (Th17) axis and Th17 cell-secreting cytokines play essential roles in the IMQ-induced psoriasis model. It is notable that a genome-wide association study identified the human tyrosine kinase 2 (TYK2) gene within the psoriasis susceptibility locus. After IMQ application, mice lacking Tyk2, a mouse homologue of the human TYK2 gene, exhibited significantly lower symptom scores of psoriasis and diminished inflammatory cell infiltration in the skin lesions. Tyk2-deficient mice also failed to increase CD4+IL-17+ or CD4+ interferon-γ+ (IFN-γ+) T cells in the draining lymph nodes or to produce Th17 cell-related cytokines after IMQ application. Furthermore, Tyk2 deficiency led to diminished skin inflammation induced by IL-23 and IL-22 injections. These results indicate that Tyk2-mediated signals in mice contribute to multiple steps of immune and inflammatory responses during the development of psoriasis; therefore, TYK2 targeting may be a promising strategy to treat patients with psoriasis. Recent clinical trials have shown that TYK2 inhibitors have a high overall response rate with good tolerability in the management of psoriasis. This review describes the fundamental mechanisms of Tyk2 inhibition in immune/inflammatory diseases.
Experiments using murine tyrosine kinase 2 (Tyk2)-deficient models have revealed the possibility of human TYK2 involvement in several human diseases [1–3] (Figure 1). Tyk2-deficient macrophages cannot constitutively produce type I interferon (IFN-I), which regulates physiological cellular functions in vivo [4]. In addition, upon CpG oligodeoxynucleotide stimulation, Tyk2-deficient dendritic cells (DCs) lose some efficacy to produce interleukin 12 (IL-12) and IL-23 and fail to induce T helper 1 (Th1) differentiation [5]. In murine rheumatoid arthritis models, Tyk2 deficiency confers great resistance to the development of arthritis [6]. Murine multiple sclerosis models of Tyk2-deficient mice show lower clinical scores and fewer lymphocyte infiltration into the inflamed central nervous system [7]. Murine Crohn’s disease or ulcerative colitis models of Tyk2-deficient mice show slower and milder clinical symptoms, such as body weight loss and bloody stool than wild-type (WT) mice [8]. In an acute inflammatory phase of granuloma-forming models treated with heat-killed Propionibacterium acnes, Tyk2 deficiency decreases neutrophil infiltration and upregulates anti-inflammatory IL-10 production [9]. Tyk2-deficient mice of a murine-delayed hypersensitivity model exhibit only slight footpad swelling [8]. In addition, human patients with a TYK2 mutation develop an autosomal recessive-type hereditary disease characterized by hyper IgE syndrome and dysregulation of the IL-23, IL-10, and IL-6 signal transduction pathways [10]. Herein, this review describes the knowledge of the role of TYK2-mediated signals in psoriasis and discusses possible clinical applications of TYK2 inhibitors.
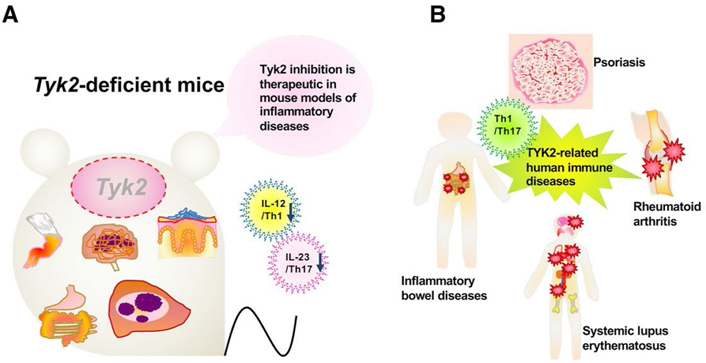
Tyk2 in various immune responses and its pathological significance. A. A schematic illustration of participation of Tyk2 in various experimental models utilizing Tyk2-deficient mice; B. a schematic diagram of participation of TYK2 in its pathological significance in human
Psoriasis is an immune and/or inflammatory skin disease caused by the dysregulation of complex interactions among immune cells [11, 12]. There are two classes of psoriasis: cutaneous and systemic. Lesions of the skin in the most common plaque psoriasis, also known as psoriasis vulgaris, are observed mainly at the extensors of elbows and knees, the scalp, and the sacral region. In addition, psoriasis patients often seem to experience health disturbances, such as arthritis, metabolic/cardiovascular diseases, and some types of cancers. Psoriatic arthritis affects approximately 30% of psoriasis patients [13]. Clinically, skin lesions of plaque psoriasis characteristically exhibit round to oval erythematous plaques, accompanied by thick and silvery adherent scales. Epidermal hyperplasia (acanthosis) and elongation of papillary ridges result from early maturation of keratinocytes whose shape is characterized by an incomplete cornification with the retention of nuclei in the stratum corneum. The histological examination would reveal infiltration of inflammatory cells, especially neutrophils and T cells in the epidermis and DCs, macrophages, and T cells in the dermis.
Based on the pathological characteristics, which are related to skin symptoms, the onset and development of psoriasis result from overactivated immune systems, in which components of the innate and adaptive immune systems interplay [12, 14]. IFN-α and cytokines produced by innate immune cells [IL-1, IL-6, tumor necrosis factor-α (TNF-α), and IFN-γ] induce the activation and maturation of DCs to link innate and adaptive immune systems. DCs also express Toll-like receptors (TLRs), through which they can directly recognize the self-RNA-LL-37 complex, leading to the production of pro-inflammatory cytokines. Mature DCs then migrate into the lymph nodes, in which they promote the differentiation of naive CD4+ T cells (Th0) into effector Th17, Th1, or Th22 cells in the presence of the cytokines they produce. In psoriasis, the differentiated CD4+ T cells migrate into the dermis, in which they interact with innate immune system-related cells. Th17 cells produce IL-6, IL-21, IL-22, and TNF-α, as well as IL-17; Th1 cells produce IFN-γ and TNF-α, and Th22 cells produce IL-22 and TNF-α. The initial inflammatory processes are mainly promoted by Th17 cells, and the contributions subsequently shift to Th1 cells, which produce IFN-γ. In psoriasis, these cytokines promote the hyperproliferation of keratinocytes and the production of additional cytokines, chemokines, and antimicrobial peptides, which in turn recruit immune cells into inflammatory lesions, perpetuating inflammatory processes (Figure 2). Recently, pro-inflammatory IL-36 family cytokines (IL-36α, IL-36β, and IL-36γ) have been proposed as critical drivers in psoriasis pathogenesis [15, 16]. IL-36 members are overexpressed in psoriasis skin lesions. In addition, IL-36α transgenic mice present psoriasis-like symptoms. The elevated levels of IL-36 members in skin lesions correlate well with key cytokines of the pathology of psoriasis. IL-36 prominently synergizes the IL-23/Th17 axis, which causes psoriasis symptoms. In psoriasis immunopathogenesis, TNF-α induces the activation and maturation of DCs to secrete a large amount of IL-23. Regulatory T cells (Tregs) play an essential role in immune tissue homeostasis and self-tolerance because they can stop the activation and proliferation of effector T cells via IL-10 production, thus regulating inflammatory processes in the body. Several experiments have indicated a malfunction of and/or a reduced number of Tregs in psoriatic lesions [12].
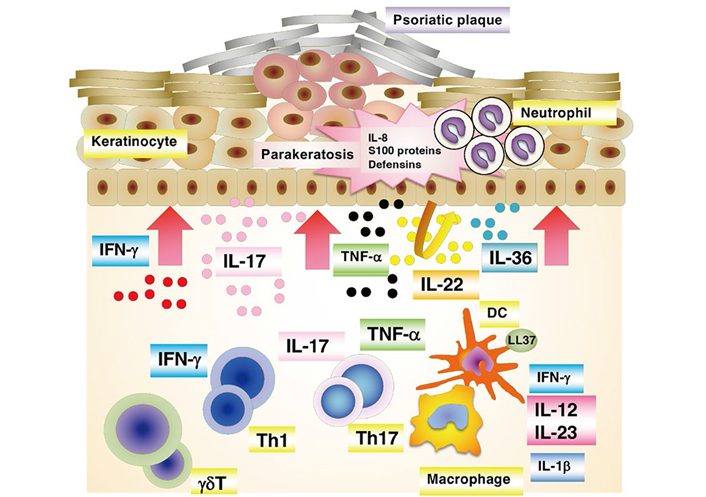
A schematic presentation of immunopathogenesis of psoriasis. At the psoriatic site, a cascade of events start with the activation of a variety of immune cells including keratinocytes. These activated cells secrete cytokines, which in turn activate myeloid DCs. DCs are central to provide a link between innate and adaptive immune responses. The activated myeloid DCs trigger differentiation into effector T cells (Th1 and Th17), leading to their migration into the skin tissue. The secreted mediators [e.g., IL-17 subunit alpha (IL-17A), IL-17F, IL-22 from Th17 cells, and IFN-γ and TNF-α from Th1 cells] then stimulate keratinocyte activation and proliferation, leading to plaque formation. Activated keratinocytes produce antimicrobial peptides and various chemokines that feedback into the proinflammatory cycle, resulting in the continued immunopathologic progression of psoriasis
Genetic studies have revealed several susceptibility loci, some of which contain immune-related genes, and have provided meaningful insights into the pathogenic processes of psoriasis [17–20]. Genetic linkage analysis of familial psoriasis identified major histocompatibility complex alleles, mainly human leukocyte antigen (HLA)-C*06:02. To date, targeted candidate gene screening approaches have suggested more than 40 single nucleotide polymorphisms possibly associated with psoriasis, and many of them are situated near genes encoding innate/adaptive immunity-related molecules, such as those involved in nuclear factor-kappa B (NF-κB) signaling [e.g., NF-κB inhibitor alpha (NFKBIA), REL proto-oncogene, NF-κB subunit (REL), TNF-α induced protein 3 interacting protein 1 (TNIP1), and caspase recruitment domain family member 14 (CARD14)], IFN signaling [e.g., TYK2 and IL-28 receptor alpha (IL-28RA)], T-cell regulation [e.g., runt related transcription factor 3 (RUNX3), ETS proto-oncogene 1, transcription factor (ETS1), methyl-CpG-binding domain protein 2 (MBD2), T cell activation RhoGTPase activating protein (TAGAP), and IL-13], and antiviral signaling [e.g., ring finger protein 114 (RNF114), DExD/H-box helicase 58 (DDX58), and interferon induced with helicase C domain 1 (IFIH1)]. Many psoriasis-associated loci also contain genes involved in the IL-23 pathway [e.g., IL-12 subunit beta (IL-12B), IL-23 subunit alpha (IL-23A), IL-23 receptor (IL-23R), TNF receptor-associated factor 3 interacting protein 2 (TRAF3IP2), TNF-α induced protein 3 (TNFAIP3), and signal transducer and activator of transcription 3 (STAT3)]. It is notable that genome-wide association studies have identified TYK2 as a psoriasis susceptibility gene [20]. In addition, genetic variation in TYK2 correlates with other autoimmune diseases such as systemic lupus erythematosus, multiple sclerosis, and Type 1 diabetes [21].
Several effective options are available for the treatment of psoriasis [22]. Topical therapy is a standard of care for mild to moderate cases. A vitamin D3 analog, calcipotriol, is a first-line topical agent to treat patients with plaque or scalp psoriasis. Calcipotriol ameliorates clinical symptoms by inhibiting the proliferation and differentiation of keratinocytes and suppressing T cell functions. Corticosteroids, a cornerstone of topical treatment, are effective and well-tolerated in the treatment of mild psoriasis. Phototherapy treatment, a systemic therapy, is effective for moderate to severe psoriasis that is unresponsive to topical agents. Methotrexate, an inhibitor of folate biosynthesis, exhibits cytostatic and anti-inflammatory effects in patients with moderate to severe psoriasis, as well as psoriatic arthritis. Although this drug is used in clinical settings, its efficacy and safety benefits are limited. Cyclosporine, a calcineurin inhibitor, is also used for patients with moderate to severe psoriasis, as well as psoriatic arthritis. This drug is superior to other systemic agents with regard to rapid action and myelosuppression/hepatotoxicity although it has some adverse effects, such as nephrotoxicity, numerous drug interactions, and association with malignancies. Biologics are expected to have a high potential for treating patients, for whom traditional systemic therapies have failed to produce an effective response or could not be tolerated due to adverse effects [23–25]. Anti-TNF drugs are adaptive in cases of therapeutic failure or contraindication of methotrexate. Therefore, systemic treatment options for psoriasis are mainly applied to suppress immune responses and/or inflammation, and thereby improve psoriatic lesions. Several immunobiological drugs are now approved for psoriasis: anti-TNF drugs (adalimumab, infliximab, and etanercept) and IL-23 inhibitors (ustekinumab, guselkumab, tildrakizumab, and risankizumab). In addition, biological agents that neutralize (i.e., secukinumab and ixekizumab) or antagonize (i.e., brodalumab) the IL-17R have excellent efficacy for the treatment of psoriasis. There is no consensus as to when and how these biologics should be initiated or switched, and the choice of treatment option is highly dependent on clinical efficiency, benefits and risks, patient requests, and cost. In this regard, many clinical studies on biological therapy have reported no association with increased risks of malignancy or serious infection.
The TYK2 Janus kinase (JAK) is activated in response to various cytokines [3]. For example, the binding of IL-12 or IL-23 to their receptors triggers the dimerization of IL-12 receptor subunit beta1 (IL-12Rβ1)/IL-12Rβ2 and IL-12Rβ1/IL-23R, respectively. TYK2 associates with IL-12Rβ1 and JAK2 associates with IL-12Rβ2 and IL-23R subunits. In T cells, IL-12 promotes IFN-γ production in a TYK2-dependent manner. IL-23 enhances activated T cell differentiation into Th17 cells in a TYK2- and JAK2-dependent manner. TYK2 is also essential for IFN-α/β-mediated signaling to suppress hematopoietic cell growth. Thus, TYK2-mediated signals have different roles in cytokine stimulation. It is notable that clinical information on patients lacking TYK2 indicates the physiological roles of this enzyme in humans. Such patients often and repeatedly experience severe viral and/or mycobacterial infections; therefore, TYK2 is important to regulate human immune systems. As described below, TYK2 also mediates IL-23-induced signals to promote the production of pro-inflammatory cytokines, macrophage/neutrophil-attracting chemokines, antimicrobial peptides, and a keratinocyte proliferation marker. TYK2 is also involved in IL-22-induced signals to promote the production of IL-22 and IL-17A, antimicrobial peptides, and a keratinocyte proliferation marker. Thus, TYK2 affects multiple events of immune and/or inflammatory responses during the development of psoriasis.
Topical application of imiquimod (IMQ), a ligand of TLR7, on mouse skin induces psoriasis-like dermatitis [26]. The IMQ-induced model has helped elucidate the underlying mechanisms of psoriasis development and evaluate new therapies against psoriasis. Symptoms after IMQ application are similar to human plaque-type psoriasis because of skin erythema, thickening, scaling, epidermal alterations (acanthosis and parakeratosis), and neo-angiogenesis as a result of inflammatory cell infiltration.
We analyzed skin inflammation after IMQ application in Tyk2-deficient mice [8]. Tyk2 deficiency significantly inhibited IMQ-induced ear thickness and ear tissue weight elevation and reduced inflammatory cell infiltration into the skin. The expression of Th17-related cytokines in the skin decreased in Tyk2-deficient mice after IMQ application. In addition, IL-12 (p35) expression, as well as the number of both CD4+IL-17+ and CD4+IFN-γ+ T cells, reduced after IMQ application in Tyk2-deficient mice. Therefore, the skin inflammation induced by IMQ is highly dependent on Tyk2 expression. In a genome- wide association study, the Tyk2 gene was detected in loci with psoriasis susceptibility. Our data obtained using Tyk2-deficient mice in the IMQ-applied model indicated the possible involvement of Tyk2-mediated signals in psoriasis-like skin inflammation.
In the IMQ-induced model, IL-23 and its inducible cytokines, such as IL-17 and IL-22, are believed to be the key players in the onset and development of psoriasis [27]. To further analyze Tyk2 involvement, we employed IL-23 injection as a more direct inducer of psoriasis-like skin inflammation [28]. Compared with WT mice, Tyk2-deficient mice showed milder ear swelling and smaller increases in ear tissue weight, as well as reduced inflammatory cell infiltration into the skin, during the observation period. Hence, Tyk2-mediated signals seem to be essential for IL-23-induced psoriasis-like skin inflammation.
In the IL-23-induced model, the numbers of IL-17- or IL-22-secreting CD4+ infiltrating cells in Tyk2-deficient mice decreased, compared with those in WT mice [28]. However, the γδT-cells, but not αβT-cells, similarly recruited and expanded in the ear skin in both Tyk2-deficient and WT mice. Therefore, Tyk2 has different roles in the infiltration of immune/inflammatory cells, such as αβT-cells and γδT-cells, into ear skin after IL-23 injection. The expression levels of macrophage/neutrophil-attracting chemokines, such as C-C motif chemokine ligand 2 (CCL2), CCL20, and C-X-C motif chemokine ligand 1 (Cxcl1), in the ear skin, were highly upregulated by IL-23 injection in WT but not Tyk2-deficient mice.
Tyk2 deficiency also reduced the induction of pro-inflammatory cytokines, such as IL-17A, IL-22, IFN-γ, IL-1β, IL-6, IL-12A (p35), and IL-12B (p40), upon IL-23 injection [28]. However, TNF induction was similar in WT and Tyk2-deficient mice. Therefore, Tyk2 is selectively involved in cytokine production after IL-23 injection. High levels of antimicrobial peptide production have been detected in psoriatic lesions, and the production levels well correlate with the severity of inflammation. After IL-23 injection, high levels of gene expression of defensin and S100A8, as well as elevated production of a keratinocyte proliferation marker, keratin16 (K16), were detected in ear skin samples of WT mice but not in those of Tyk2-deficient mice [28]. Hence, antimicrobial protein production in ear skin after IL-23 injection is highly dependent on Tyk2-mediated signals. In addition, pretreatment of mice with anti-IL-22 or anti-IL-17 antibody, but not anti-IFN-γ antibody, significantly inhibited IL-23-induced ear swelling [28]. Because IL-22 receptors are expressed on keratinocytes but not on lymphocytes, keratinocytes are the direct target cells of IL-22 injection. We analyzed IL-22-induced skin inflammation [28]. Although IL-22 injection promoted only limited ear swelling, IL-22 injection significantly induced IL-22 and IL-17A expressions in the ear skin of WT mice but not in those of Tyk2-deficient mice. After IL-22 injection, elevated gene expression of defensin and S100A8, as well as K16, were detected in ear skin samples of WT mice but not in those of Tyk2-deficient mice. Thus, IL-22 is likely to act as one of the important downstream mediators during IL-23-induced skin inflammation, and Tyk2 plays a crucial role in both IL-23- and IL-22-mediated signaling. Notably, TYK2 is directly involved in IL-22-dependent events because human keratinocyte (HaCaT) cells with TYK2-knockdown failed to phosphorylate STAT3 after IL-22 stimulation.
The inhibitor of NF-κB kinase-ζ (IκB-ζ) is a nuclear inhibitor of NF-κB family protein that positively or negatively regulates NF-κB transcriptional activities [29–31]. IκB-ζ is highly expressed by epidermal keratinocytes in psoriatic lesions [32], and the NFKBIZ gene has been identified as a psoriasis susceptibility locus [33]. IκB-ζ gene expression is induced by the transcription factors, NF-κB, STAT3, and CCAAT/enhancer binding protein β (C/EBPβ), and its expression depends on psoriasis-related cytokines, such as IL-17, IL-1β, and IL-36 [34–38]. Interestingly, in the IMQ-induced skin inflammation model, Tyk2-deficient mice, which showed only slight inflammation, exhibited low IκB-ζ messenger RNA (mRNA) expression [39]. The catalytic activity of TYK2 and STAT3 is an effective promoter of IκB-ζ in HaCaT cells. However, STAT3 activation was not enhanced in HaCaT cells upon IL-17 stimulation [39].
The signaling pathway induced by IL-17 stimulation regulates mRNA stability, which also controls inflammatory responses [40–42]. Regulatory RNase 1 (Regnase-1; also known as zinc finger Cys-Cys-Cys-His (CCCH)-type containing 12A or monocyte chemotactic protein-induced protein-1) is an IL-17-inducible protein with endoribonuclease activity and acts as a negative regulator for inflammatory signaling promoted by various pro-inflammatory cytokines [43]. Regnase-1 is rapidly degraded through the ubiquitin-proteasomal pathway in IL-1β-, IL-17-, or IL-36α-stimulated keratinocytes [44–46]. It is notable that the 3’-untranslated region (3’-UTR) of IκB-ζ mRNA is essential for recognition and degradation by Regnase-1 [39, 47]. Thus, IL-17 stimulation post-transcriptionally stabilizes IκB-ζ mRNA via Regnase-1 inhibition.
NF-κB activator 1 (ACT1) is adaptor protein that is critical for IL-17-induced signaling events, such as the activation of NF-κB and C/EBPβ, as well as the post-transcriptional stabilization of mRNA in autoimmune and inflammatory diseases [42]. TANK-binding kinase 1 (TBK1) and IκB kinase ε (IKKε) are activated dependent on direct interaction with ACT1 after IL-17 stimulation. IL-17 can induce Regnase-1 phosphorylation in an ACT1-TBK1/IKKε-dependent manner, and phosphorylated Regnase-1 in turn loses its functional activity to degrade mRNA, leading to the expression of IL-17 target gene mRNA [46, 48]. IL-17/ACT1 signaling counteracts the constitutively occurring degradation of IκB-ζ mRNA by Regnase-1.
In summary, TYK2 is involved in the development of psoriasis through not only the enhancement of IL-22- and IL-23-mediated signals but also the control of IκB-ζ in cooperation with the IL-17/Regnase-1, IL-17/ACT1, and/or TBK/Regnase-1 axes (Figure 3).
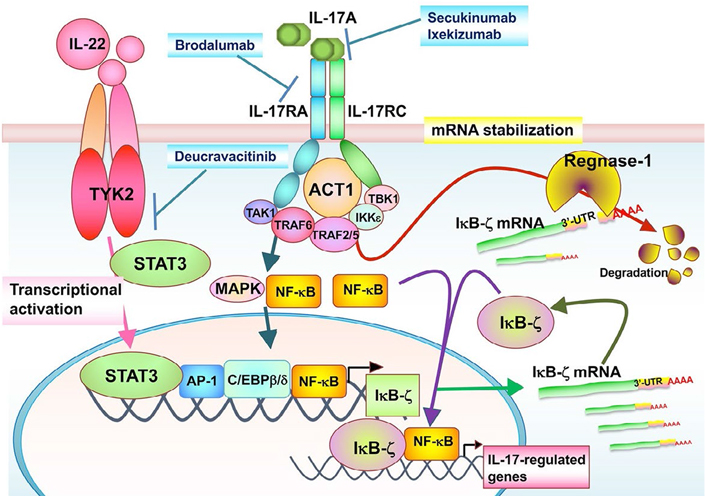
The TYK2/IL-17 axis-mediated signaling in IκB-ζ expression. The STAT3-activating cytokines, such as IL-22, promote TYK2/STAT3 signals in keratinocytes. TYK2/STAT3 signals directly enhance IκB-ζ promoter activity. IL-17A binds to IL-17RA and IL-17RC subunits, whose intracellular domains interact with the adaptor ACT1. ACT1 associates with TRAF family proteins, TBK1 and IKKε. These IL-17 signals posttranscriptionally stabilize IκB-ζ mRNA through inhibition of an endonuclease Regnase-1 independently of TYK2. The TYK2/IL-17 axis is regulated independently of each other, and both of them are required for IL-17-induced IκB-ζ upregulation and further IL-17-regulated gene expression. TAK1: transforming growth factor β-activated kinase 1; MAPK: mitogen-activated protein kinase; AP-1: activating protein-1
Although therapeutic strategies to treat patients with severe psoriasis have been progressing, a safe and highly effective systemic therapy is desired [22, 23]. Treatment with an IL-23 neutralizing antibody has a high response rate for ameliorating psoriatic plaques, pruritus, and nail psoriasis [25, 49]. These facts support the possibility that IL-12 and IL-23, as well as TYK2-mediated signals, may play essential roles in the onset and development of human psoriasis. JAK inhibitors confer great therapeutic benefits by controlling disease conditions in patients with autoimmune diseases with high levels of circulating cytokines. It has been shown that the IL-23 blockage might be more effective than the IL-12 blockage in the treatment of psoriasis patients [25]. Furthermore, the manufacturing of JAK inhibitors is relatively easier and cheaper than that of biologics, therefore increasing the number of compounds being formulated and tested for clinical use. Thus, combined inhibition of broader cytokines may increase clinical efficacy. In this regard, TYK2 inhibition seems to be suitable, because TYK2 is involved in signals induced by multiple psoriasis-related cytokines, including IL-12, IL-22, and IL-23 [8, 28]. Several available JAK inhibitors show some characteristic differences in their JAK selectivity [50]. A JAK3 inhibitor impedes signals induced by γc cytokines (e.g., IL-2, IL-7, and IL-15), leading to the inhibition of immune cell function. However, such agents also inhibit Tregs, which negatively regulate immune responses. A JAK2 inhibitor impedes IL-23- and erythropoietin-induced signals. JAK1 inhibitors may require much attention to their adverse effects because JAK1-deficient mice show severe phenotypes. Tyrphostin A1, a TYK2 inhibitor, can impede IL-23-induced skin inflammation and Th17 cell cytokine production [28]. TYK2 inhibitors can directly inhibit IL-22 signals; in addition, they do not influence the differentiation of Tregs. Thus, selective TYK2 inhibitors, which mainly inhibit IL-22 and IL-23, have unique inhibitory profiles, which cannot be achieved by the other JAK inhibitors. In a clinical study, JAK inhibitors displayed general safety risks, which are dependent on on- or off-target effects, including anemia and neutropenia, as well as secondary malignancies. Because Tyk2-deficient mice grow normally without any immune confusion under steady state conditions, drugs to specifically inhibit TYK2 activity are likely to be used to suppress immune systems without severe adverse effects in clinical settings. In summary, the aforementioned results along with our data obtained using Tyk2-deficient mice and cells indicate that TYK2 will be a promising and suitable drug target for the treatment of patients with psoriasis [51].
The onset and development of psoriasis are dependent on functional effector T cells, and Th1 and Th17 cells, as well as the cytokines, IL-12, IL-17, IL-22, and IL-23. Under stimulation by these cytokines, IκB-ζ protein expression and Regnase-1 activity control immune and inflammatory responses. Our data obtained using Tyk2-deficient mice and cells indicate that TYK2-mediated signals directly and/or indirectly contribute to these molecular events, suggesting the fundamental and functional roles of TYK2 in the appearance of psoriasis symptoms. Recently, highly selective TYK2 inhibitors, deucravacitinib (BMS-986165) and PF-06826647, have been developed and clinically tested for the treatment of psoriasis patients [52–55]. As expected, TYK2 inhibitors have a significant clinical impact with high efficacy and low risks. The data presented in this review hopefully constitute useful information about the management of psoriasis using novel TYK2 inhibitors.
ACT1: NF-κB activator 1
DCs: dendritic cells
IFN: interferon
IL-12Rβ: interleukin-12 receptor beta
IL-23: interleukin 23
IMQ: imiquimod
IκB-ζ: inhibitor of nuclear factor kappa B kinase-ζ
JAK: Janus kinase
mRNA: messenger RNA
NF-κB: nuclear factor-kappa B
Th: T helper
TNF: tumor necrosis factor
Tregs: regulatory T cells
TYK2: tyrosine kinase 2
WT: wild-type
We would like to thank Editage (www.editage.com) for English language editing.
RM, KO and TM wrote the first and final draft of the manuscript and the sections of the paper. They also contributed to the conception and design of the paper. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This study was supported in part by Grant-in-Aid for scientific research 19H03364 (T.M.) and 20K07010 (R. M.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Masanori Fujii ... Susumu Ohya
Aparna Mukhopadhyay ... Debprasad Chattopadhyay
Kalthoum Abid ... Hamouda Boussen
