
Abstract
Aim:
Utilizing the therapeutic potentials of previously approved medications against a new target or pharmacological response is known as drug repurposing. The health and scientific communities are under continual pressure to discover new compounds with antiviral potential due to the rising reports of viral resistance and the occurrence and re-emergence of viral outbreaks. The use of antiviral peptides has emerged as an intriguing option in this search. Here, this article includes the current United States Food and Drug Administration (FDA)-approved antiviral peptides that might be enforced for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection and carried out docking study of the viral protease inhibitors.
Methods:
In silico techniques like molecular docking was carried out using Autodock Vina software.
Results:
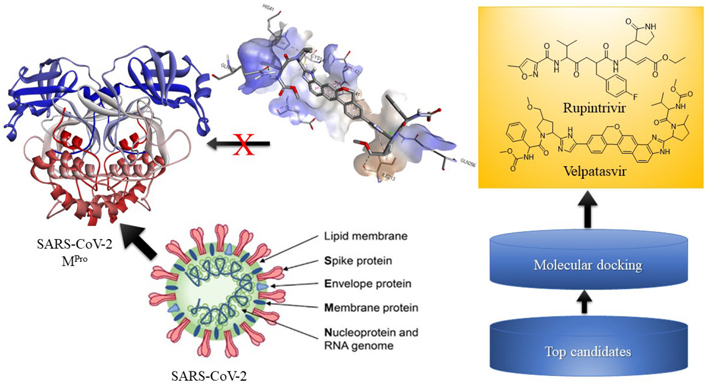
The molecular docking studies of peptide-based antiviral agents against SARS-CoV-2 [Protein Data Bank (PDB) ID: 7P35] using docking software AutoDockTools 1.5.6. Among all the docked ligands, compound velpatasvir showed interaction with residues ILE213, GLN256, LEU141, GLN189, GLU166, HIS41, CYS145, and ASN142, and displayed the highest docking score of –8.2 kcal/mol. This medication could be a novel treatment lead or candidate for treating SARS-CoV-2.
Conclusions:
To conclude, a docking study of peptide based antiviral compounds for their binding mode in the catalytic domain of SARS-CoV-2 receptor is reported. On molecular docking, the compounds have showed remarkable binding affinity with the amino acids of receptor chain A. The compounds occupied the same binding cavity as the reference compound maintaining the interactions with conserved amino acid residues essential for significant inhibitory potential, especially for compound velpatasvir with binding score of –8.2 kcal/mol.
Keywords
Coronavirus disease 2019, severe acute respiratory syndrome coronavirus 2, rupintrivir, ATN 161, velpatasvir, boceprevir, ombitasvirIntroduction
The health, society, and economy of people are seriously threatened by human pathogenic viruses [1]. The possibility of viral resistance and the persistent (re-)emergence of viruses, such as the human immunodeficiency virus type 1 (HIV-1), further complicate the problem like Corona, Ebola, and Zika virus [2]. The coronavirus disease 2019 (COVID-19), which is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), spread quickly around the world [3]. Antivirals that specifically target viruses concentrate on blocking key transcription and replication enzymes, such as proteases and polymerases, or on directly inactivating viral structural proteins [4]. Usually, host-targeting antivirals and antivirals that target viruses are the most prevalent antiviral drug action mechanisms (Figure 1) [5]. Antiviral peptides usually target perturb viral membrane envelopes and restrict numerous steps of viral life cycle. The activity of antiviral peptides can be enhanced by constraining peptides into macrocycles.
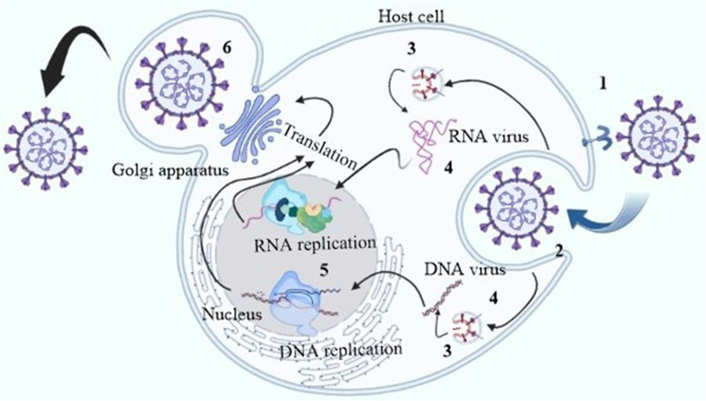
The antiviral peptides blocking sites of certain viral replication cycle. The inhibition sites are 1: viral association; 2: viral penetration; 3: endosomal escape; 4: viral uncoating; 5: viral genome replication; 6: release of mature virions
In clinical studies, the repurposing of various small molecule inhibitors including favipiravir is now being studied. However, the mechanism of treatment for SARS-CoV-2 is still unrecognized [6]. Due to the possibility that peptides show more effectiveness and selectiveness than small molecule medications and will thus be better tolerated, peptide and peptide-based inhibitors are an intriguing substitute for small molecules [7]. Antiviral peptides can be a secure and efficient option for the treatment of infectious illnesses, as demonstrated by the clinical application of the anti-HIV peptide T20 [8]. Bioavailability of antiviral peptides is powerful, stable, and safe in order to be used in therapeutic settings. Peptides are extremely selective and tolerated because of their chemical makeup. Due to their relatively large size and proteolytic breakdown in the gastrointestinal (GI) tract, they may lead to less bioavailable and short half-lives [9]. Moreover, peptides can be altered to increase their activity and stability by simplifying their sequences, altering their amino acid compositions, or fusion of moieties that increase their affinities to the appropriate partner, as frequently determined by in silico modelling or scanning of amino acid [10]. The investigation of different parameters like polarity, charges, and steric effects, as well as mutagenesis studies and biomolecular simulations, are frequently used to simulate binding sites for the development of antiviral peptides [7]. Here, we report the computational-based docking study used for the development of bio-active peptides targeting SARS-CoV-2 entry. There are several microbial peptides that have a broad impact on variety of microbes and pathogens [11]. In silico and in vitro studies on anti-microbial studies against SARS-CoV-2, such as anti-microbial peptide APD3, AP00180 showed the best result with a promising docking score of –6.4 kcal/mol with nucleocapsid receptor. Some important binding interactions were observed, such as Lys85 and Arg13 with a minimum distance of 1.759 Å. Peptide AP00549 also showed the best result against SARS-CoV-2 with a docking score of –3.4 kcal/mol [12]. Rana tagoi, a species of brown frog, secretes some peptides with antiviral activity, and AR-23 is one of them. Different in vitro assay was performed on DNA and RNA containing viruses with or without envelope. Peptide AR23 was proved to be most potent against both DNA herpes simplex virus-1 (HSV-1) and RNA HSV-1 viruses [13]. Another anti-microbial peptide temporin L was collected from amphibian origin. It has strong Gram-positive and Gram-negative activity. The activity was performed in both DNA and RNA viruses. Temporin L showed significant potent inhibitory activity against herpesvirus, paramyxoviruses, influenza virus, coronaviruses, and SARS-CoV-2 [14]. Another anti-microbial peptide LL37 was evaluated by in silico screening. LL37 showed effectiveness against the disruption of SARS-CoV-2 membrane. It also decreased the viral load [15].
Cardiovascular implications are the most prominent problems in SARS-CoV-2. Severe damage includes myocardial damage, acute coronary syndrome, myocardial infarction, and cardiac arrythmia. Increased level of C-reactive protein and markers like troponine-1 may lead to severe inflammation along with hypoxic and respiratory distress [16]. The glycosylated spikes protein of SARS-CoV-2 undergoes an interaction with ACE2 receptor present in lower respiratory track, damages respiratory tissues and causes pneumonia. The reactive oxygen species (ROS) production and the upregulated cytokine level cause cell death and organ damage [17].
Main protease (Mpro) is the main target in this study with significant relevance in viral replication as it plays a crucial role in breakdown of the 12 non-structural proteins including RNA-dependent RNA polymerase (RdRp) and helicase in replication cycle. Inhibition of the Mpro disrupts the replication cycle of virus, although any inhibitor of Mpro is not reported yet [18]. Photo-thermal therapy, another invasive technique to kill virus is also used in cancer treatment. Photothermal active agents upon irradiation cause rise in vibrational energy along with heat, and the temperature above 60°C leads to cell death. In recent studies, second near infrared region (NIR-II, 1,000–1,700 nm) has been proved to be more efficient [19, 20]. The first near infrared region (NIR-I, 620–850 nm) is able to penetrate deeper tissue [21]. Photodynamic therapy (PDT) is another promising non-invasive method clinically used as antiviral therapy. It works with several photo sensitive dyes and drug agents which in excited state produce ROS that causes catalytic oxidation of the viral envelope. Peptide-based antiviral agents, especially the protease and integrase inhibitors, are not synthesized in normal synthetic method. Rather solid and solution phase peptide synthesis techniques are used, utilizing different protocols like Boc and Fmoc, and different linkers and resins as solid phase, to protect both terminals of the peptides. Both acidic and basic medium play an important role in this methodology. Some clinical trial data compiling various combination peptide therapies have been described in Table 1.
Clinical trials on antiviral protease inhibitors
| Compounds used in combined therapy | Chemical structure | Disease | Status | Ref. |
|---|---|---|---|---|
| ASC09F + oseltamivir, Ritonavir + oseltamivir, oseltamivir | 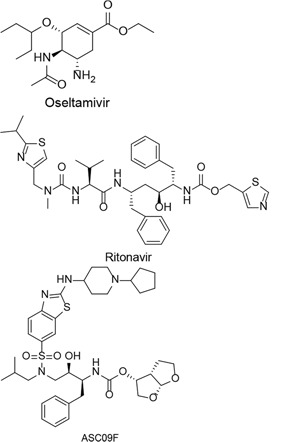 | COVID-19 | Phase-3Started in February 1, 2020Ended in July 1, 2020 | [22] |
| Darunavir + cobicistat, lopinavir + ritonavir | 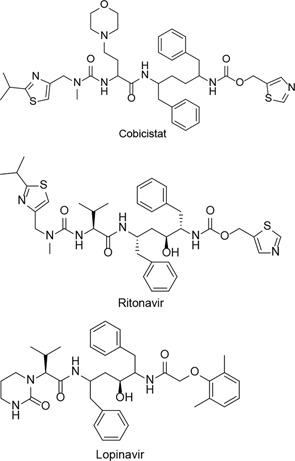 | COVID-19 | Started in March 1, 2020Ended in September 30, 2020 | [23] |
+: combined therapy; Ref.: references
Materials and methods
The recent antiviral marketed drugs were selected, which possess least toxicity and lowered side effects. The selected marketed drugs structures were sketched in ChemDraw and saved in Protein Data Bank (PDB) format. The structures of drugs are shown in Figure 2. Rupintrivir is a 3C cystine protease inhibitor of the picornavirus family. It also inhibits 3C-like protease of SARS-CoV-2 due to its homology. Rupintrivir inhibits Mpro cystine protease which cleaves the catalytic cystine and histidine residues [24], ATN161 (Ac-PHSCN-NH2). It antagonises the integrin α5β1, which plays a crucial role in the inhibition of angiogenesis and metastasis of tumor. Integrin α5β1 antagonist ATN161 was derived from the synergy of the domain [Pro-His-Ser-Arg-Asn (PHSRN)], and it inhibits the tumor growth as well as ocular neovascularization [25], velpatasvir is a direct acting antiviral agent and treats chronic hepatitis C along with HIV infection. It selectively inhibits NS5A in binding domain which consists of amino acids 33–202 at RNA binding site [26], boceprevir reversibly inhibits the NS3 protease, a serine protease enzyme used in the treatment of hepatitis C virus [27]. Ombitasvir inhibits viral replication by inhibiting NS5A protein in hepatitis C viral infection [28].
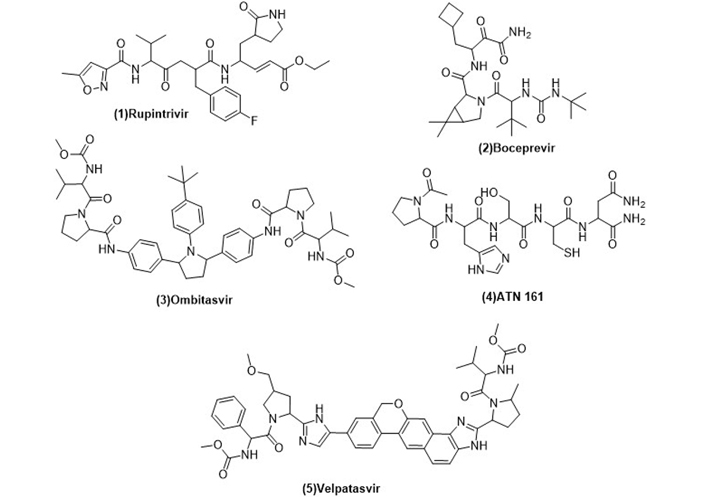
Structures of marketed peptide-based antiviral drugs utilized for molecular docking against SARS-CoV-2 Mpro
Selection and preparation of protein
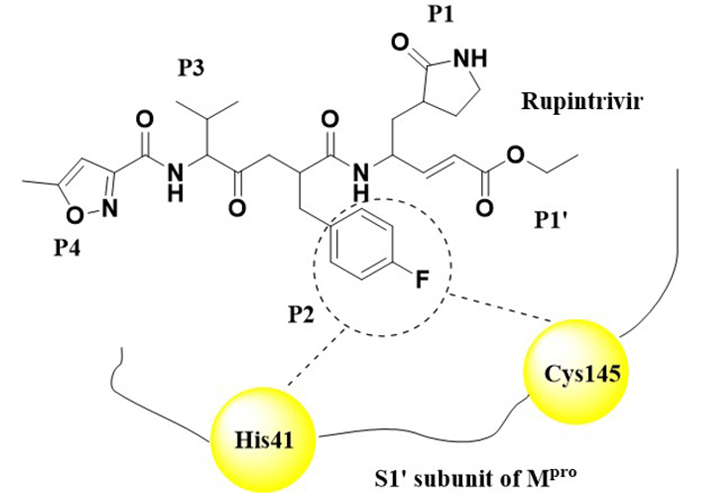
The viral protein obtained from SARS-COV-2 was selected for docking. The protein target was fetched from database PDB. The crystal structure of SARS-CoV-2 Mpro (PDB ID: 7P35) [29] possesses a resolution of 2.26 Å . Rupintrivir was co-crystallized in this receptor. There are hydrogen-bonding interactions between Gly143, His164, Phe140, and Glu146. However, several Pi-pi interactions were also observed with Ala191, Pro168, and Leu27 is shown in Figure 3. SARS-CoV-2 Mpro was complexed with Rupintrivir. Rupintrivir breaks down the catalytic activity of this cystine protease. The P2 subunit was interacting with the two residues of the catalytic thread His41 and Cys145 which undergoes catalytic breakdown of the enzyme.
Selection and preparation of the ligand
All the compounds were sketched in ChemDraw. All the compounds are then converted into PDB file. The parameters such as hydrogen atoms were added, correct bond types were defined, charges were assigned to each atom, and finally, the energy minimization of structures was done using force field molecular mechanics-2 (MM2) with minimum root mean square (RMS) gradient.
Protein preparation
Protein was prepared in MGLTools 1.5.6 [30]. In the first step, protein was inserted in MGLTools graphical user interface (GUI) and all the water molecules were removed, partial Kollman charges were added along with polar hydrogen addition and finally it was saved into PDB, Partial Charge (Q) & Atom Type (T) (PDBQT) format. Further, the PDBQT file of protein was taken for grid generation process to optimize same binding pocket as the co-crystallized ligand occupied and define three coordinates of the pocket with the size of 1 Å.
Molecular docking
Prepared ligand and receptor were docked by using Autodock Vina [31] module in command prompt interface. The docking was performed in the specific binding pocket of the putative receptor, as described in previous research works. After docking, the conformation of docked ligands were visualized via BIOVIA discovery studio [32] in both 3D and 2D to obtain the receptor ligand interactions.
Results
Result of molecular docking studies
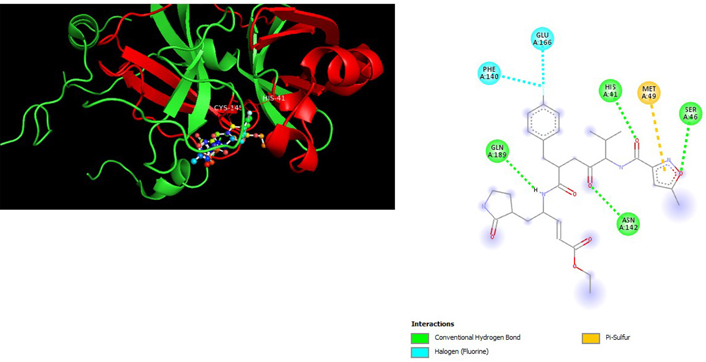
A molecular docking study of the compounds against SARS-CoV-2 Mpro (PDB ID: 7P35) displayed significant interaction via a number of conventional hydrogen bonds, pi-sulfur, pi-anion bond, van der Waals forces, amide interaction, halogen interaction. Ligand rupintrivir showed interactions with residues Glu166, Phe140, Gln189, Asn142, Ser46, Met49, and His41. The conformation binds the protein with docking score of –7.1 kcal/mol (Figure 3). Similarly, ligand ATN 161 interacted with residues Cys145, Ser144, His41, His164, Pro168, and Glu166, and possessed docking score of –6.8 kcal/mol (Figure 4). Furthermore, ligand velpatasvir which showed the highest binding affinity by interaction with residues Ile213, Gln256, Leu141, Gln189, Glu166, His41, Cys145, Asn142 and displayed the docking score of –8.2 kcal/mol (Figure 5). Ligand boceprevir showed the lowest binding affinity of the series by interacting with amino acid interaction with Gln189, Ser144, Phe140, Glu166, Met49, Met165, His41 with a docking score of –6.7 kcal/mol (Figure 6). Moreover, ligand ombitasvir showed the amino acid interaction with Gln189, Glu166, Cys145, His164, His41, and Ser46 with a docking score of –7.5 kcal/mol (Figure 7). The docking score and root means square deviation (RMSD) of chosen compound are displayed in Table 2.
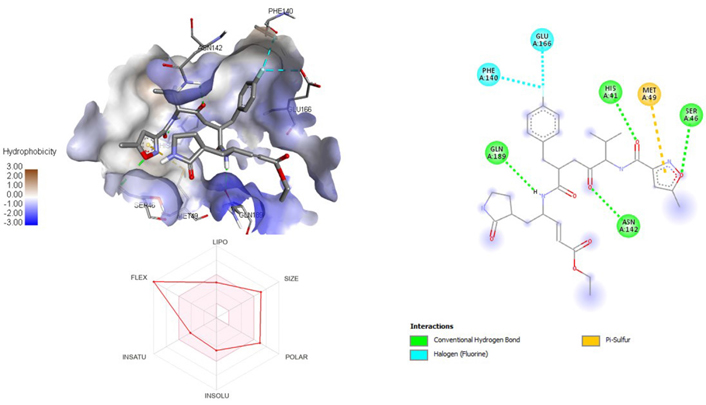
3D and 2D interaction of ligand rupintrivir with SARS-CoV-2 protein (PDB ID: 7P35) along with in silico absorption, distribution, metabolism, excretion, and toxicity (ADMET) representation
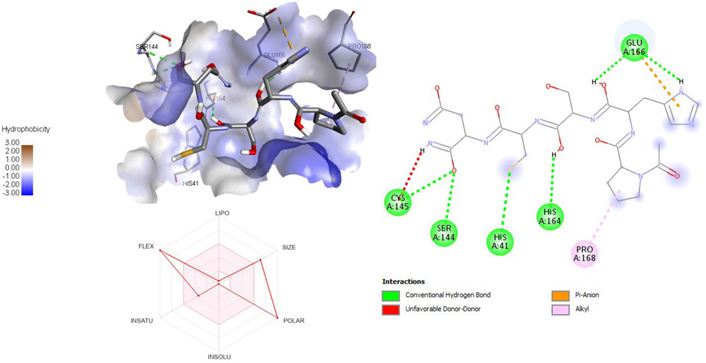
3D and 2D interaction of ligand ATN161 with SARS-CoV-2 protein (PDB ID: 7P35) along with in silico ADMET representation
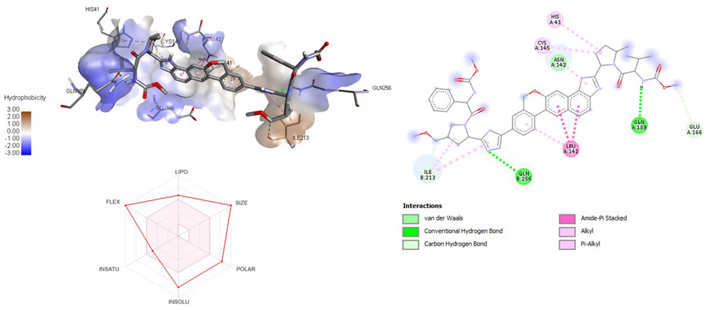
3D and 2D interaction of ligand velpatasvir with SARS-CoV-2 protein (PDB ID: 7P35) along with in silico ADMET representation
Binding score of drugs against PDB ID 7P35
| Sl. No. | Drugs | Binding score (kcal/mol) |
|---|---|---|
| 1 | Rupintrivir | –7.1 |
| 2 | ATN 161 | –6.8 |
| 3 | Velpatasvir | –8.2 |
| 4 | Boceprevir | –6.7 |
| 5 | Ombitasvir | –7.5 |
Sl.: serial number
Drug-likeness study
Drug-likeness study was performed in SwissADME, a freeware webserver. Several parameters like molecular weight (wt), hydrogen bond acceptor (HBA), hydrogen bond donor (HBD), topological polar surface area (TPSA) in (Å)2, lipophilicity (log P), molar refractivity (MR), rotatable bond (RB), solubility (Log S), no. of sp3 hybridised carbon bioavailability and no. of Lipinski violations are compiled in Table 3. There were several violations found due to its peptide nature, the molecular wt was greater than 500 g/mol. Compound 5 (ombitasvir) showed two violations, such as, molecular wt > 500 and HBA > 10. Compound 4 (boceprevir) showed three violations such as, molecular wt > 500, HBD > 5, and compound 3 (velpatasvir) showed two violations with molecular wt > 500, log P > 5. A radar graph projection of the chosen ligand was displayed along with docking pose in Figures 5–9. Parameters like INSATU [ratio of sp3 carbon in total carbon count (Csp3)], Flex (RB), INSOLU (Log ESOL), Lipo (Log P), Polar (TPSA) and size (molecular volume) of the ligand are described in graph. According to Veber’s rule no. (number) RB > 10, in this criterion all compounds failed to be eligible for the drug like property. All compounds showed greater fraction Csp3 (standard Csp3 < 0.25), as compound should be less than equal 130 (Å)2 so all compound fails but all the drugs are not following oral drug route administration that’s why this oral drug-likeness screening is less necessary in this whole study.
Drug-likeness profile of ligands
| Sl. No. | Molecular wt. (g/mol) | HBA | HBD | TPSA (Å)2 | Log P | Bioavailability | Log S | Csp3 | RB | MR | Lipinski violations |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 598.66 | 9 | 3 | 156.7 | 3.46 | 0.17 | –4.5 | 0.48 | 18 | 159.06 | 2 |
| 2 | 519.68 | 5 | 4 | 150.7 | 2.06 | 0.55 | –4.12 | 0.81 | 14 | 144.39 | 1 |
| 3 | 894.11 | 8 | 4 | 178.72 | 5.63 | 0.17 | –9.14 | 0.52 | 22 | 262.84 | 2 |
| 4 | 597.64 | 9 | 8 | 310.6 | –3.17 | 0.17 | 0.65 | 0.57 | 20 | 146.7 | 3 |
| 5 | 883 | 10 | 4 | 193.1 | 5.05 | 0.17 | –8.41 | 0.39 | 17 | 251.54 | 2 |
Sl.: serial number
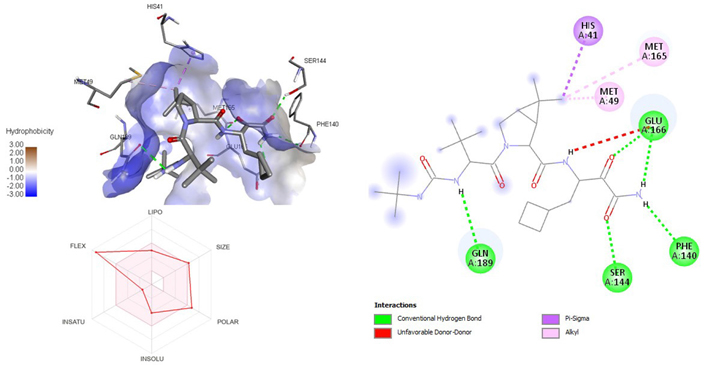
3D and 2D interaction of ligand boceprevir with SARS-CoV-2 protein (PDB ID: 7P35) along with in silicoADMET representation
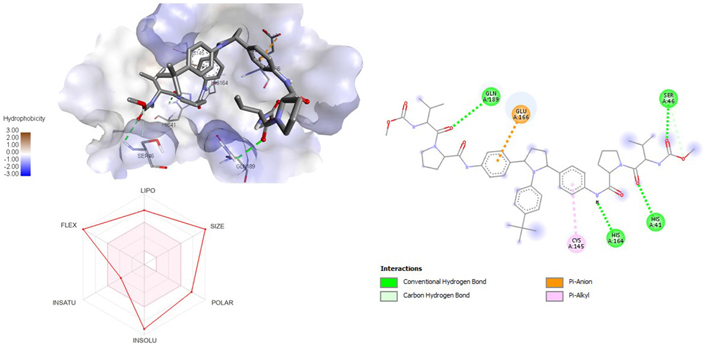
3D and 2D interaction of ligand ombitasvir with SARS-CoV-2 protein (PDB ID: 7P35) along with in silicoADMET representation
Discussion
To conclude, we have reported docking study of peptide based antiviral compounds for their binding mode in the catalytic domain of SARS-CoV-2 receptor. On molecular docking, the compounds have showed remarkable binding affinity with the amino acids of receptor chain A. The compounds occupied the same binding cavity as the reference compound maintaining the interactions with conserved amino acid residues essential for significant inhibitory potential, especially for compound velpatasvir with binding score of –8.2 kcal/mol. This analysis provided a mechanistic justification for the antiviral potential of the compounds under study. In vivo and in vitro studies are predicted to be desirable in the future to empirically confirm the SARS-CoV-2 inhibitory efficacy of these medicines. Significantly, the therapeutic efficacy of velpatasvir suggests that prompt treatment of individuals with mild to intermediate COVID-19 may be safe and efficient for eradicating SARS-CoV-2 more quickly and for preventing the progression and consequences of COVID-19. The fact that velpatasvir may be used to treat early infection in the outpatient context must be highlighted since doing so may be crucial for preventing the spread of infection, hospitalization, and disease progression. Further structural modifications of these compounds can be done for the generation of potent analogs. There are so many violations in drug-likeness study but as these compounds not followed oral route that’s why the drug-likeness study was less important.
Abbreviations
| ADMET: | absorption, distribution, metabolism, excretion, and toxicity |
| COVID-19: | coronavirus disease 2019 |
| HBA: | hydrogen bond acceptor |
| HBD: | hydrogen bond donor |
| HIV-1: | human immunodeficiency virus type 1 |
| Log P: | lipophilicity |
| Mpro: | main protease |
| PDB: | Protein Data Bank |
| RB: | rotatable bond |
| SARS-CoV-2: | severe acute respiratory syndrome coronavirus 2 |
| TPSA: | topological polar surface area |
Declarations
Acknowledgments
The authors are heartily thankful to the management of ISF College of Pharmacy for constant encouragement, support, and motivation.
Author contributions
AC: Data curation. AS: Investigation. PAC: Writing—original article, Writing—editing and review.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not Applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2023.