Review
Review

Affiliation:
1School of Medical and Allied Sciences, K.R. Mangalam University, Gurugram 122103, Haryana, India
Affiliation:
1School of Medical and Allied Sciences, K.R. Mangalam University, Gurugram 122103, Haryana, India
Affiliation:
2University School of Biotechnology, Guru Gobind Singh Indraprastha University, Delhi, 110078, India
ORCID: https://orcid.org/0000-0001-5993-7576
Affiliation:
3Department of Pharmacology, NIMS Institute of Pharmacy, NIMS University, Jaipur 303121, Rajasthan, India
ORCID: https://orcid.org/0000-0003-3807-3953
Affiliation:
4Ram Gopal College of Pharmacy, Gurugram 122506, Haryana, India
ORCID: https://orcid.org/0000-0002-5159-5409
Affiliation:
5SGT College of Pharmacy, SGT University, Gurugram 122505, Haryana, India
Email: mdsabiralam86@gmail.com
ORCID: https://orcid.org/0000-0002-2845-0156
Explor Med. 2023;4:85–106 DOI: https://doi.org/10.37349/emed.2023.00126
Received: June 28, 2022 Accepted: October 12, 2022 Published: February 28, 2023
Academic Editor: Lindsay A. Farrer, Boston University School of Medicine, USA
The article belongs to the special issue Techniques in Repurposing and Targeted Delivery: Bringing a New Life to Shelved Drugs
Neuropathic pain (NP) remains maltreated for a wide number of patients by the currently available treatments and little research has been done in finding new drugs for treating NP. Ziconotide (PrialtTM) had been developed as the new drug, which belongs to the class of ω-conotoxin MVIIA. It inhibits N-type calcium channels. Ziconotide is under the last phase of the clinical trial, a new non-narcotic drug for the management of NP. Synthetically it has shown the similarities with ω-conotoxin MVIIA, a constituent of poison found in fish hunting snails (Conus magus). Ziconotide acts by selectively blocking neural N-type voltage-sensitized Ca2+ channels (NVSCCs). Certain herbal drugs also have been studied but no clinical result is there and the study is only limited to preclinical data. This review emphasizes the N-type calcium channel inhibitors, and their mechanisms for blocking calcium channels with their remedial prospects for treating chronic NP.
Neuropathic pain (NP) is the pain triggered by primary laceration or somatosensory disfunction as defined by International Association for the Study of Pain [1]. Throughout the world, approximate 9% population is affected by this pain [2]. This pain is mostly divided into two categories: one is peripheral which is the most common, while the other is central. The peripheral pain is the result of nerve injury, neuralgia, or radiculopathy. The reason for the central pain may be the brain injury, spinal injury, brain stroke, or multiple sclerosis [3]. NP is recognized by several symptoms such as spontaneous shooting, burning pain, and allodynia. It affects the life quality of a patient [4, 5], leading to depression, certain other mental and physical health issues [6, 7]. The explanation of NP is not much clear but rather quite confusing resulting in lack of fixed treatment strategy. So, certain invasive therapies are used for mitigation of pain due to a lack of treatment. Treatment is totally a literature-based outcome. Certain antidepressant and antiepileptic drugs are used to cure the symptoms although these methods are associated with certain side effects such as addiction. Beyond these, surgical methods are also used. So, there is much space for the development of new medication in this field, which should be specific with less side effects. The International Association for the Study of Pain (IASP) defines NP as pain initiated or caused by a primary lesion or due to dysfunction within the nervous system [8]. This condition arises as the outcome of sequence of several pathological mechanisms which are frequently described on the basis of anatomic localization [9]. Neuropathic syndromes are typically characterized with several intricate neuronal episodes that incorporate all types of allodynia, hyperalgesia, paraesthesia, and dysesthesias [10]. These signs are typically followed by depression, anxiety, and sleep disturbances [11]. Large numbers of neuropathic patients do not attain satisfactory relief from popular treatment methods [12–14]. The failure of present treatment methods highlights the crucial requirement for brand new categories of drugs and improved use of available treatments. Medicines presently in use for treating NP are mentioned in Table 1 [15–17]. Certain herbal drugs also have been preclinically studied for the treatment of NP such as Aconiti tuber, Curcuma longa, Ocimum sanctum, but with no clinical application yet.
Pharmacotherapy for nerve pain
| Therapy | Drug class | Drugs | Mechanism of action | Adverse effects |
|---|---|---|---|---|
| First-line therapy | Calcium channel blockers | Gabapentin | It inhibits α2δ subunit of VGCCs. | Peripheral swelling |
| Pregabalin | It inhibits α2δ subunit of VGCCs. | Peripheral swelling and increased bodyweight | ||
| TCAs | Amitriptyline | It inhibits the reuptake of serotonin and norepinephrine. | Arrhythmia and suicide risk | |
| SNRI | Duloxetine, venlafaxine | These act by inhibiting the reuptake of serotonin and norepinephrine. | Ataxia, hyperhidrosis and hypertension | |
| Second-line therapy | Opioids | Tramadol | It acts by binding to opioid receptors. | Seizures and ataxia |
| Tapentadol | It inhibits the reuptake of serotonin and norepinephrine. | Seizures and ataxia | ||
| Topical treatments | Lidocaine | It acts by binding to sodium channels. | Local erythema, itching and pain | |
| Capsaicin | It interacts chemically with sensory neurons for its action. | Pain and itching | ||
| Third-line therapy | Strong opioids | Morphine | It acts by binding to opioid receptors. | Dizziness and lethargy |
| Neurotoxins | Botulinum toxin | It acts by decreasing the release of acetylcholine. | Pain at injection site |
SNRI: serotonin-norepinephrine reuptake inhibitors; TCAs: tricyclic antidepressants; VGCCs: voltage-gated calcium channels
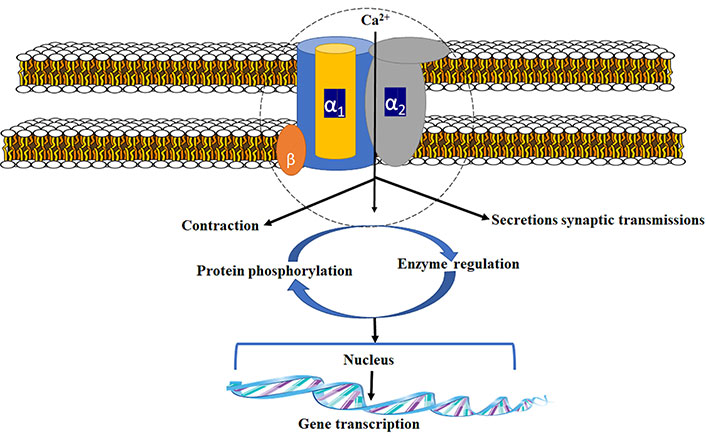
Because of the crucial side effect offered by current treatments, novel approach towards management of nerve pain is desperately required [18]. Ziconotide (PrialtTM), which was studied as N-type calcium channel blocker, was developed on account of the new drug in the class of ω-conotoxins. Synthetically it was similar to conotoxin MVIIA, an N-type calcium channel inhibitor permitted for NP [19]. The calcium channel is made up of three different subunits (Figure 1). For channel activity α-1 subunit is important. Gabapentin/pregabalin binds at a site in the extracellular region of α2 subunit [20]. N-type voltage-dependent calcium channel (VDCC) controls the discharge of neurotransmitters towards synaptic cleft [21, 22]. These neurochemicals carry nociceptive signals across the afferent nerve, and the pain signals get suppressed due to antagonism at the channel. Structure of voltage-gated Ca2+ channel is shown in these articles [23–25].
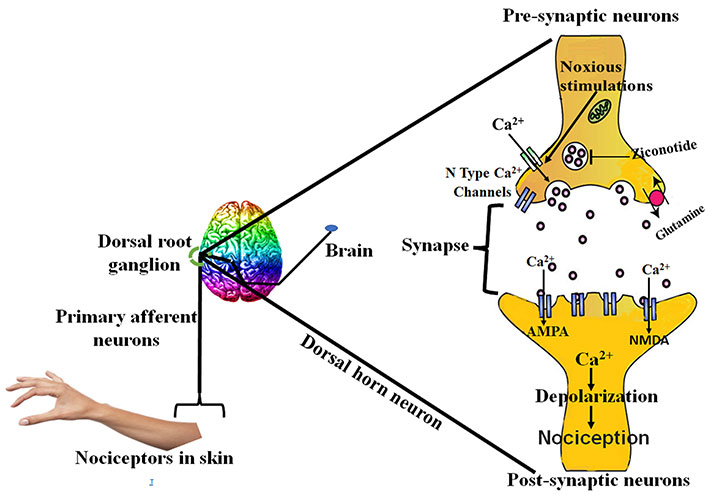
Peripheral nerve lesion causes aberrant regeneration which unusually sensitizes the neurons that cause abnormal excitability [26]. This process is recognized as superficial stimulation, which results in NP. Peripheral nervous system injury causes the release of certain mediators of inflammation like histamine, cytokines, potassium, and neuropeptides [27]. These mediators cause the change in the quantity and location of sodium ion channels in the damaged nerve fibres [27, 28]. This results in the depression of depolarization threshold and ectopic discharge resulting in the higher nociceptor response to any type of external stimuli known as peripheral sensitization [28, 29]. Ectopic discharge results from the demyelinated nerve fibres due to shrivelled blood supply [30, 31]. Due to injury, the chemical mediated electrical connections are developed between adjoining neurons called ephaptic conductions resulting in pain in a normal calm nociceptor [27, 31, 32]. Hyperalgesia and burning sensation mediated through uninterrupted excretion in C-fibres [33]. Dysesthesias and paraesthesias are the results of sporadic spontaneous discharges in A-δ or A-β fibres, all due to peripheral sensitization [34, 35]. The pathophysiology of peripheral NP is stated below in Figure 2.
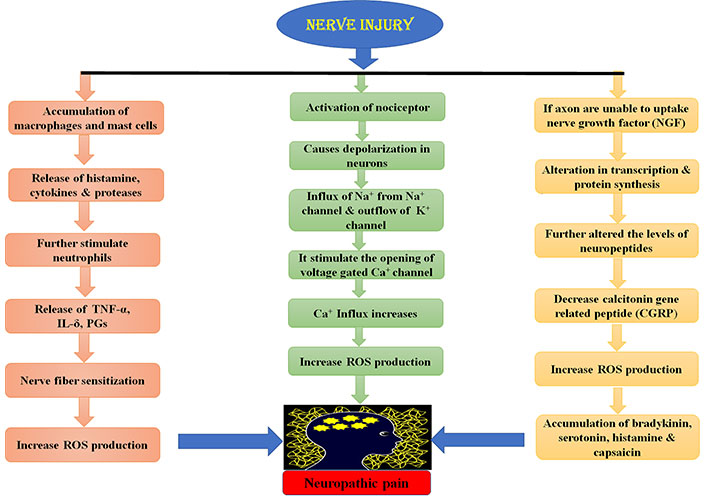
Physiology of peripheral NP. TNF-α: tumor necrosis factor α; IL-δ: interleukin δ; PGs: prostaglandins; ROS: reactive oxygen species
The tachykinins and neurotransmitters released as a result of peripheral injury causes the hype in the excitability of central nociceptor receptors present in the spinal cord [36]. Overexcitation of these receptors causes the hyperexcitation of N-methyl-D-aspartate (NMDA) receptor, rising intracellular calcium levels [37] through N-type calcium channels [38], considered imperative for the maintenance of central sensitization [38, 39]. The hyperactivity results in the biochemical abnormalities in the dorsal horn neurons of the spinal cord, which lowers the threshold for activation and thus response to stimuli is enhanced with enlarged receptive field [36]. Another mechanism responsible for central NP is suppression of central inhibitory control, resulting in overexpression of excitatory mechanism [39, 40]. All these mechanisms collectively lead to allodynia, which is a clinical state in NP in which the condition is induced by stimulus not usually evoking any pain [36, 41]. The drugs which are used to treat central NP act by affecting the levels of calcium, 5-hydroxytryptamine (5HT) and noradrenaline etc. Ziconotide, gabapentin and anticonvulsants drugs act by antagonizing N-type calcium channel, and oxcarbazepine, lamotrigine and lidocaine act by modulating the sodium channels [42–44]. Tricyclic antidepressants act both peripherally and centrally. Peripherally by modulating sodium channels and centrally by modulating neurotransmitter levels (e.g., 5HT, NE). Opioids act by closure of calcium channels, and thus further reducing the release of key neurotransmitters involved in the pain transmission [36].
The major ascending nociceptive pathway constituted by the spinothalamic tract (STT) forms the neurons of the spinal cord [45]. Due to the rise in the spontaneous activity in the periphery, the background activity of STT neurons increases, the receptive fields became enlarged and the response to afferent impulse increases [46]. The stated event is called central sensitization [47]. The pathophysiology of central NP is stated below in Figure 3.
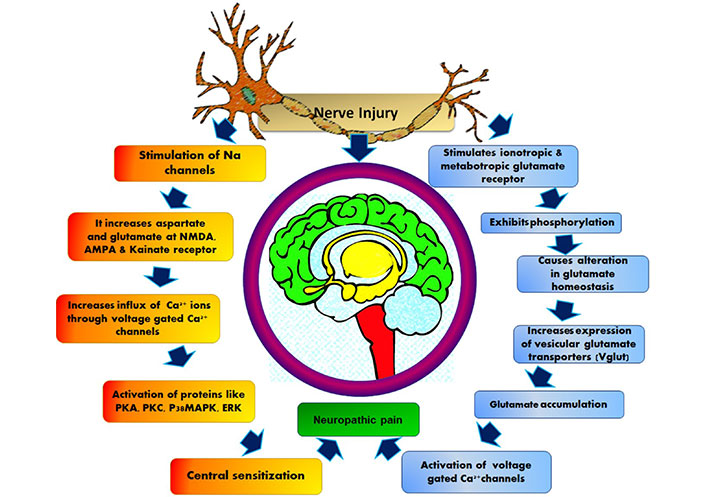
Pathophysiology of central nervous system NP. AMPA: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; PKA: protein kinase A; p38MAPK: p38 mitogen-activated protein kinase; ERK: extracellular signal-regulated protein kinase
The detail peripheral and central pathophysiology depends on changes that occur at cellular and molecular levels [48]. Functional changes occur due to change in sodium and calcium channel subunit expression related to NP. During a nerve injury, the sodium and calcium channel subunits reshuffle, which results in the sudden firing of neurotransmitters [49].
Pain, a complicated medical condition that has considerable unsatisfied clinical requirements. The opioid treatment remains standard despite of their side effects like tolerance and respiratory depression bounded to their continuous use. The foundation for pain comprises different pathways from dorsal root ganglion to brainstem [50–52]. The downward pathways vigorously regulate the upward pain pathways [53]. G protein-coupled opioid receptor is the main target of opioid analgesics, and the neuronal excitability decreases when these receptors are activated [54–57].
These channels are strongly blocked by numerous peptides obtained from the toxin of fish-trapping cone snails from Conus species, along with GVIA, MVIIA and CVID [58–60]. At elevated concentrations the peptides even can block 0 channel subtypes [61, 62]. Along with MVIIA, a specific conotoxin Cav2.2 blocker is approved for managing neuronal pain. Medicinal prospective of ω-conotoxins specific for Cav2.2 for pain management has been recognized as the Cav2.2 channel, which plays a vital role in the transmission of pain [63–65].
The GVIA conotoxin obtained from Conus geographus permanently blocks Cav2.2 channels in the nanomolar range [61, 66]. GVIA has more potency in vivo, compared to other structurally similar peptides [67, 68]. GVIA is three to four folds more effective than other peptides, and almost 40 times more effective compared to morphine, when given to rats intrathecally. Because of the permanent blockage of the Cav2.2 channel, it is possibly difficult to decide the safe dose in an experimental setting [69].
MVIIA obtained from Conus magus, inhibits the Cav2.2 channels. The synthetic form (ziconotide) has been developed, which is injected intrathecally for treatment of NP clinically [70–72]. The USA and Europe permitted MVIIA for managing NP [73, 74]. This peptide shows appreciable relief from pain. Like morphine, there is no adverse effect of tolerance and addiction in the case of MVIIA [75, 76].
CVID is obtained from Conus catus, the major selective antagonist of Cav2.2 channels among all peptides [77, 78]. It is presently under clinical trials by the name of AM336 [79]. In the previous studies, it has been proved that CVID is a highly selective antagonist for Cav2.2 rather than Cav2.1. Different Conus species [80] with their actions and the biological sources are shown in Table 2.
Different Conus species with their action and biological source
| Name of peptide inhibitor | Biological source | Mechanism of action |
|---|---|---|
| GVIA | Conus geographus | Irreversible inhibitor of Cav2.2 |
| MVIIA | Conus magus | Potent inhibitor of Cav2.2 |
| CVID | Conus catus | Selective inhibitor of Cav2.2 |
Ziconotide has a neuroprotective activity and is derived from the venom of cone snail. However, it is the synthetic peptide that has similarities with ω-conopeptide (MVIIA) obtained from Conus magus. The pharmacodynamics and pharmacokinetics affect living cells, and their dosage regimens have been reviewed and studied [81]. This drug is in the final stage of human testing to confirm its efficacy against NP and is one of the non-opioid neuroprotective agents [82]. Moreover, it acts by inhibiting Cav2.2 channels [83, 84]. The neuroactive peptides obtained from the venom of Conus snail have the unique property of high potency and specificity as this ziconotide is epitomizing as an excellent option for the management of nerve pain. Clinical studies have been done as proofs for the therapeutic potency of ziconotide over approximate 2,000 human subjects [85].
(1) Protection of people against cerebral ischemia.
(2) Study the surgical cases with bypass procedures used to treat coronary heart failure.
(3) Pain relief in the patient having chronic pain disorder.
The biological activity of ziconotide was proved by administering intracranial injections in mice that further produced shaking behaviour; this demonstrates its neuroactive property in mammals, which differentiates it from the other peptides obtained from the venom of Conus magus [86]. The Conus peptide belongs to the “Omega” class as it shares properties similar to the omega family [87]. One of the preclinical studies performed on isolated embryonic chick sympathetic ganglia shows that ziconotide blocks the electrical transmission of calcium along the synaptic end giving the evidence that it inhibits the voltage sensitive calcium channels present on presynaptic nerve ending [88, 89]. It has been observed that the pharmacodynamics and pharmacokinetics of ziconotide resemble ω-GVIA, one of the Conus peptides [90, 91].
Ziconotide is the selective antagonist of voltage-gated selective Ca2+ channels. The predominant position of its activity is the nociceptive receptor within the neurons of the spinal cord [92–95]. The analgesic potential of ziconotide is comparable to opiate and is successful against the NP in patients developed tolerance for opiate [96]. Analgesic effects of opiate are due to its binding to μ-opiate receptor resulting in its inactivation through the G protein-coupled receptor (GPCR) pathway that further blocks inflow of calcium in the cell [97]. Moreover, ziconotide directly antagonizes voltage-gated Ca2+ channels [98] as shown in Figure 4. However, ziconotide achieves complete blockade of channels as compared to opiates which only lowers the potential opening of voltage-gated Ca2+ channels. On the other hand, ziconotide also modulates other voltage-gated ion channels like potassium channels and glutamate receptors [99]. Ziconotide is considered as a preferred treatment against NP because of its following properties:
(1) It’s effectiveness in opiate tolerant patients.
(2) Patients do not develop tolerance against ziconotide.
Therefore, it is considered as a novel treatment against the NP mechanism of ziconotide [100] as shown in Figure 4.
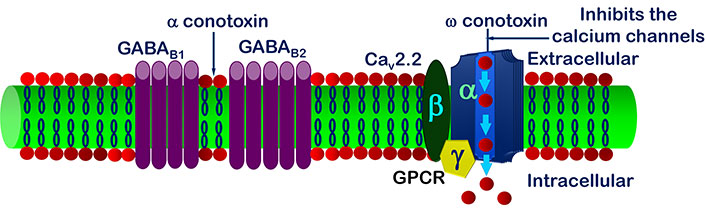
It has been reviewed that inside cell the signal transduction modulates N-type VGCCs regulated by GPCR [101]. Inhibition of VGCCs decreases the span of signal transmission in spinal root ganglia nerve cells [102]. The observation shows the stimulation of γ-aminobutyric acid type B (GABAB) channels leads up the inhibition of VGCCs current [103–107] (Figure 5). GABA can act either through ligand ion channels (GABAA) or by membrane receptor (GABAB) [108, 109]. GABAB receptor is made up of 2 subunits, GABAB1 and GABAB2 [110, 111], out of which GABAB1 is inotropic and GABAB2 is a metabotropic receptor [112–117]. Stimulation of GABAB blocks the inflow of Ca2+ through VGCCs; this is only possible by activation of GPCR [118–120] as shown in Figure 5.
U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) authorized ziconotide (Prialt) ω-conotoxin MVIIA is administered in the management of extreme persistent pain related to malignancy, AIDS, and neuritis as the alternative medication for current pain therapy. The major problem is only with its administration through the intrathecal route to the patients [121]. However, it acts by the blockade of N-type VGCCs; therefore, it represents the new target for pain therapy with its evidence in knockout mice [122, 123]. The ability of binding Prialt with N-type VGCCs presents at A-d and C fibres of dorsal root ganglia (DRG) of the spinal cord only after its intrathecal administration [124]. It is the potent antagonist of sensitive signalling and thus has the potential to treat the pain. MVIIA and CVID during its in vitro testing show that these drugs have the fastest onset and offset of action. Intrathecal delivery is the only novel route for the administration of conotoxins [125–127].
The concept of topical therapies is consistent with current theory of mechanism-oriented pain treatment; thus, they may increase pain management quality and patient satisfaction with treatment. There are numerous preclinical trials in the literature demonstrating the antinociceptive impact of drugs applied topically in inflammatory and NP animal models. They are not currently used in clinical practise and so are not included in this evaluation. The topical agents act by either excitatory or inhibitory mechanism but through different receptor ion channel enzyme, many are currently being utilized in clinical practice (Table 3).
Mechanisms of action of topical drugs for the therapy of NP for excitatory and inhibitory effects
| Mechanism of action of topical agents | Receptor ion channel enzyme | Topical agent utilized in clinical practice | Possible site of action | Reference |
|---|---|---|---|---|
| Mechanism for excitatory effect of drugs | Voltage-gated sodium channels (NaVs) | Lidocaine, amitriptyline, doxepin, phenytoin, ambroxol, TV-45070, opioids, NSAIDs, clonidine | Neurons, keratinocytes | [128–132] |
| TRPV1 | Capsaicin, NSAIDs | Neurons, keratinocytes, immune cells | [133–135] | |
| VGCC | Gabapentin, lidocaine | Neurons, keratinocytes | [136–138] | |
| NMDAR | Ketamine, amitriptyline, diclofenac | Neurons, keratinocytes, immune cells | [139–141] | |
| COX-2 | NSAIDs | Neurons, immune cells, Schwann cells | [142–144] | |
| Mechanism for inhibitory effect of drugs | GABAR | Amitriptyline | Neurons, keratinocytes, immune cells | [145–148] |
| GABAAR | Ketamine, phenytoin | Neurons, keratinocytes, immune cells | [149, 150] | |
| GABABR | Baclofen | Neurons, keratinocytes, immune cells | [151–154] | |
| α2-AR | Clonidine | Neurons | [155–157] | |
| OR | Opioids | Neurons, keratinocytes, immune cells | [158–160] | |
| CR | Cannabinoids | Neurons, keratinocytes, immune cells | [161–163] |
α2-AR: α2 adreno receptors; COX-2: cyclooxygenase-2; CR: cannabinoid receptors; GABAR: γ-aminobutyric acid receptors; GABAAR: γ-aminobutyric acid type A receptors; TV-45070: topical sodium channel inhibitor; NMDAR: NMDA receptors; NSAIDs: Non-steroidal anti-inflammatory drugs; OR: opioid receptors; TRPV1: transient receptor potential vanilloid 1
Despite the abundance of medications available, there are no curative conventional treatments for NP. In the field of drug research, more emphasis is now being placed on herbal formulation. As a result, we conducted a thorough evaluation of herbal medications and plants that show anti-NP properties [164]. Certain herbal drugs are also used for the therapy of NP but only preclinical studies have been done. The following plants are some of the most popular ones that are used to treat NP: Acorus calamus [165, 166], Artemisia dracunculus [167, 168], Butea monosperma [169, 170], Citrullus colocynthis [171, 172], Curcuma longa [173, 174], Crocus sativus [175, 176], Elaeagnus angustifolia [177, 178], Ginkgo biloba [179, 180], Mitragyna speciosa [181, 182], Momordica charantia [183], and Nigella Sativa [184, 185] (Table 4).
Herbal drugs undergone preclinical studies for NP
| Plant used | Family | Plant part | Reference |
|---|---|---|---|
| Aconiti tuber | Ranunculaceae | Tuber | [186] |
| Acmella oleracea | Asteraceae | Leaves and flowers | [187] |
| Acorus calamus | Araceae | Rhizomes | [188] |
| Alstonia scholaris | Apocyanaceae | Milky juice of plant | [189] |
| Butea monosperma | Fabaceae | Leaves | [190] |
| Crocus sativus | Iridaceae | Flower | [191] |
| Curcuma longa | Zingiberaceae | Roots | [192] |
| Emblica officinalis | Phyllanthaceae | Fruit | [193] |
| Euphorbia tirucalli | Euphor-biaceae | Latex, root, bark | [194] |
| Gelsemium elegans | Loganiaceae | Seeds, stems | [195, 196] |
| Ginkgo biloba | Ginkgoaceae | Herbal extract | [197, 198] |
| Momordica charantia | Cucurbitaceae | Fruit | [199] |
| Nauclea latifolia | Rubiaceae | Roots | [200, 201] |
| Ocimum sanctum | Lamiaceae | Leaves | [202] |
| Olea europaea | Oleaceae | Oil and leaves | [203] |
| Paederia scandens | Rubiaceae | Roots, leaves, bark, and fruits | [204] |
| Punica granatum | Punicaceae | The juice, whole fruit, flowers, and roots | [205] |
| Senna spectabilis | Fabaceae | Flowers, fruits and leaves | [206] |
| Thymus caramanicus Jalas | Lamiaceae | Aerial parts | [207] |
| Tribulus terristris | Zygophylla-Ceae | Complete herb | [208] |
| Trigonella foenum-graecum | Fabaceae | Seeds | [209] |
| Vernonia cinerea | Asteraceae | Whole plant | [210] |
Certain limitations associated with the existing N-type VGCCs inhibitors provide a great opportunity for the development of better and new drugs with targeted therapeutic outcomes and lesser side effects. The peptide currently used has a drawback of narrow therapeutic window [211, 212]. Further, ziconotide shows peripheral interaction, so oral and intravenous administration are not possible, thus leaving with only choice of intrathecal route [213, 214]. The future research can be focused on development of novel ω-conotoxins with improved selectivity as inhibitor for N-type VGCCs, without any peripheral interactions, thus reducing the associated adverse effects. Although ω-conotoxin therapeutics currently is under clinical and preclinical development, there is still vast opportunity for the new research drug candidate with respect to NP. There are also several non-peptide small molecules under preclinical and clinical trials but till no successful outcome has been noticed. But of course, because of the small size and non-peptide nature they can lead to the development of molecule that can follow an alternate route with fewer side effects and more specific to the receptor [215–221]. In recent years, lots of review and research have been published on recent advancements of nanotechnology and their application on neurological disease as well as NP [222–233].
The existing pharmacotherapy for the NP is very challenging being non-specific in action. The therapies are non-invasive associated with several side effects and drug-drug interactions. There is a scope for the development of therapy which is target specific with lesser side effects. Ziconotide acts as N-type VGCC antagonist the only approved drug administered through intrathecal route. Certain herbal drugs also have been studied but no clinical result is there and the study is only limited to preclinical data. New research is being carried out on ω-conotoxins and small peptides have been obtained from Conus species. The vast number of Conus species and wide variety of conotoxin availability open a path for further research on more target specific and least side effect molecules.
GABA: γ-aminobutyric acid
GABAB: γ-aminobutyric acid type B
GPCR: G protein-coupled receptor
NP: neuropathic pain
VGCCs: voltage-gated calcium channels
Our sincere thanks to Paramveer Singh, Shadab Husain and Jamilur Rahman Ansari for editing the language of this manuscript. The authors gratefully acknowledge the contributions of the collaborators and co-workers mentioned in the cited reference. SC, R Kaur, and AG acknowledges K.R. Mangalam University, AW acknowledges Guru Gobind Singh Indraprastha University (GGSIPU). R Kadian gratefully acknowledges the support and cooperation from the Director of Ram Gopal College, Gurugram. MSA gratefully acknowledges the Vice Chancellor and Dean of Pharmacy, Shree Guru Gobind Singh Tricentenary University (SGTU) for their kind cooperation and support.
SC, AW, R Kaur, and AG: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. R Kadian and MSA: Validation, Writing—review & editing, Supervision.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Neha Minocha ... Parijat Pandey
Monu Yadav ... Anil Kumar
Sangeet Kumar Mall ... Md Sabir Alam
Rouchan Ali ... Pooja A. Chawla
Rosy Yesela Mancilla Santa Cruz ... María Segunda Aurora Prado
Tausif Alam
Abhishek Chaurasiya ... Pooja A. Chawla
Dilpreet Singh ... Pooja A. Chawla