
Abstract
During cellular stress, the master regulators of intrinsic self-death (apoptosis) are BCL-2 family proteins. The BCL-2 family proteins play a key role in apoptosis and are tightly regulated via other BCL-2 family proteins, non-BCL-2 protein suppressors, and epigenetic modifications. As the name implies, these proteins possess one or two of the four BCL-2 homology domains (BH1–BH4). According to their roles, they are classified as pro-apoptotic or pro-survival proteins. BH-3-only proteins possess a single BH3 domain and are specific/key effector proteins for intracellular death commitment, particularly in the context of cell survival and programmed cell death. This delicate interplay among the BCL-2 family members is essential for maintaining the primary hemostasis, or balance, of cell fate. The anti-apoptotic proteins, such as BCL-2 and BCL-XL, promote cell survival by inhibiting apoptosis. On the other hand, the pro-apoptotic proteins, such as BAX and BAK, drive apoptosis. It ensures that cells are able to respond appropriately to various internal and external signals, ultimately determining whether a cell survives or undergoes programmed cell death. Understanding and targeting this delicate balance is a promising avenue for developing therapeutic strategies to modulate cell fate and treat various diseases. The molecular pathogenesis of BCL-2 family proteins in blood disorders involves differential expression of these components resulting in the dysregulation of the pathway contributing to cell survival and resistance to apoptosis as observed in follicular lymphoma, diffuse large B-cell lymphoma, acute lymphoblastic leukemia, and acute myeloid leukemia. Such dysregulation is a major impediment to standard therapies and aids in chemo resistance. Studies show some promising clinical outcomes with antineoplastic agent venetoclax either as a monotherapy or in combination with other agents. This review discusses recent studies on the regulation of BCL-2 family proteins which might provide a molecular landscape for their clinical implications in blood disorders.
Keywords
BCL-2 family proteins, apoptosis, cellular stress, BH3-only proteins, blood neoplasm, therapeutic strategies, hemostasisIntroduction
BCL-2 family proteins play a pivotal role in influencing the delicate balance between the states of cell survival and cell death, thereby determining the cellular fate [1–4]. Their activity is intricately regulated by the cell to maintain cellular homeostasis, determining whether to support cell survival or initiate cell death (apoptosis). Moreover, post-translational modifications (PTMs) introduce an additional layer of complexity to cellular balance, particularly during periods of cell stress. In disease states, such as blood malignancies, it is believed that the regulation of BCL-2 cellular balance is disrupted, leading to resistance to therapy. Nevertheless, venetoclax, an anti-BCL-2 protein agent, exhibits promising therapeutic responses in the treatment of blood cancers. Venetoclax can also be combined with other drugs as an adjunctive approach for challenging cases. Laboratory investigations provide insights into the effectiveness of anti-BCL-2 protein agent treatment. This review explores the regulation of intrinsic apoptosis, the role of PTMs, the imbalance of BCL-2 in blood cancers, the treatment strategy involving venetoclax, and laboratory testing of the response to anti-BCL-2 protein agent treatment.
Unraveling the secrets of cell death: the intriguing role of BCL-2 family proteins in intrinsic apoptosis
BCL-2 family proteins on the mitochondrial surface are important regulators of apoptosis. When a cell encounters an apoptotic stimulus such as reactive oxygen species (ROS), a DNA damage, oncogene dysregulation, toxins, or growth factor inhibition, the pro-apoptotic proteins known as BH3-only activators, that include BCL-2-interacting mediator of cell death (BIM) [1], truncated BH3-interacting domain death agonist (tBID) [2], and p53-upregulated modulator of apoptosis (PUMA) [3], trigger mitochondrial dysfunction and caspase activation inducing apoptosis. The process involves oversaturating the BH3 region of pro-survival proteins such as BCL-2, BCL-W, BCL extra-large (BCL-XL), Bcl-2 homolog from fetal liver 1 (BFL1), and myeloid cell leukemia sequence 1 (MCL1) proteins [4]. When a cell is primed for apoptosis, the excess BH3 activator proteins can activate pro-apoptotic BCL-2-associated X protein (BAX) [5], and BCL-2 antagonist/killer (BAK) at the mitochondrial membrane [6]. Subsequently, the activated paired proteins will form pores to facilitate mitochondrial outer membrane permeabilization (MOMP) [7]. However, cells can undo MOMP by the later action of pro-survival proteins BCL-XL, BFL1, and MCL1 [4]. In cytosol, following MOMP, the apoptosis progresses towards a series of apoptogenic cascades of serine protease Omi (Htr2A), scaffolding-like apoptotic protease-activating factor 1 (APAF1), second mitochondria-derived activator of caspases (SMAC), cytochrome c, and caspases 9, 7, 3 (Figure 1). The intrinsic apoptosis pathway induces apoptosis by directly activating caspase-3 or by cleaving bid, resulting in mitochondrial dysfunction and subsequent release of cytochrome c and activation of caspase-9 and caspase-3. Caspase-3 promotes the characteristic features such as DNA fragmentation and cell death. However, the cascade can be inhibited by X-linked inhibitors of apoptosis protein (XIAP) [8–10]. Interestingly caspase-8 from extrinsic pathway can also activate the truncated tBID to add extra pressure for apoptotic fate in cells having plentiful XIAP protein. Such loop interaction suggests BCL-2 proteins as apoptotic amplifiers on cell types with limited death signal [11–13].
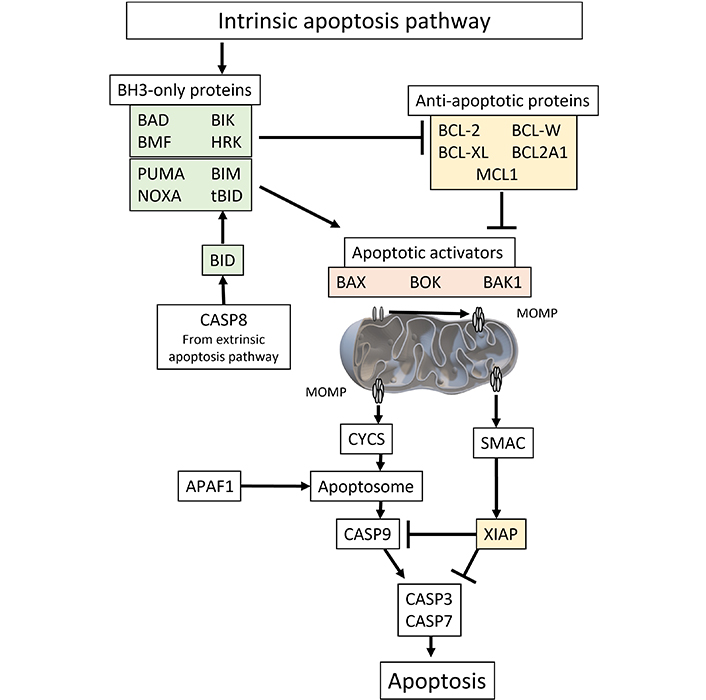
The molecular pathway of intrinsic apoptosis can be initiated by various internal or external stimuli, such as oxidative stress, DNA damage, alterations in the endoplasmic reticulum, changes in microtubules, or depletion of growth factors. Subsequently, pro-apoptotic effectors play a crucial role in causing MOMP and cell death. Among the effectors involved in the early stages of apoptosis are BAX and BAK. Afterwards, MOMP exposes the mitochondrial space, composed of a bilayer, to proapoptotic proteins originating from the cytosol, such as CYCS and SMAC. Following assembly of CYCS with APAFa, dATP, and pro-CAS9, the apoptosome is activated, leading to the activation of CASP9 and subsequent activation of caspases CASP3 and CASP7. These caspases are regulated by SMAC and may be either sequestered or degraded by members of the XIAP protein family, which act as anti-apoptotic proteins. BAD: BCL-2-associated agonists of cell death; BMF: Bcl-2-modifying factor; BIK: Bcl-2-interacting killer; HRK: activators of apoptosis Harakiri; PUMA: p53-upregulated modulator of apoptosis; BIM: BCL-2-interacting mediator of cell death; NOXA: known as phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1); tBID: truncated BH3-interacting domain death agonist; BCL-XL: BCL extra-large; BCL2A1: BCL-2-related protein A1; MCL1: myeloid cell leukemia sequence 1; CASP8: caspase-8; BAX: BCL-2-associated X protein; BOK: BCL-2-related ovarian killer; BAK1: BCL-2 antagonist/killer 1; MOMP: mitochondrial outer membrane permeabilization; CYCS: cytochrome c; SMAC: second mitochondria-derived activator of caspase; APAF1: apoptotic protease-activating factor 1; XIAP: X-linked inhibitors of apoptosis protein
In relation to BID activation, the cleavage of small tBID has been observed to enhance its binding affinity to the BCL-2 protein, as compared to the wild-type tBID. A study has substantiated this observation by demonstrating that tBID can undergo cleavage resulting in 11 kDa, 15 kDa, and 13 kDa fragments, particularly when BID possesses an additional cleavage site at Asp-98 [14–16]. Notably, the study highlighted the specific association of the 13 kDa and 11 kDa fragments with mitochondria. Furthermore, while tBID activates BAK, its activation does not coincide with that of BCL-XL activation [15–17]. Along with BH3-only activators, different BH3-weaker-acting proteins, so-called BH3-only sensitizers, can cooperate with BH3-only activators in inhibiting BH3 sequestration release of pro-survival proteins in order to activate MOMP permeabilization [4, 16]. Examples of BH3-only sensitizers are BCL-2-associated agonists of cell death (BAD), NOXA [known as phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1)], and activators of apoptosis Harakiri (HRK). The binding affinity of BH3-only proteins differs according to the BH3 domain constriction of its 9–13 amino acids [16].
Certain cancer cells such as human cervical (HeLa) and human osteosarcoma (U2OS) cells have the ability to undergo incomplete MOMP, also known as minority MOMP [17, 18]. This process can lead to the release of mitochondrial deoxyribonuclease (DNase), potentially causing DNA mutations [17]. However, these cells may become resistant to cancer immunotherapy treatments that specifically target caspases [19]. It is hypothesized that when caspase-deficient cells experience post-minority MOMP, an alternative pathway involving pro-inflammatory signaling, such as cyclic guanosine monophosphate-adenosine monophosphate (GMP-AMP) synthase-stimulator of interferon genes (cGAS-STING), may be activated [20]. This activation subsequently leads to the release of type-I interferons, which act as damage-associated molecular patterns (DAMPs), potentially enhancing the response to immunotherapy.
Controlling the balance: regulatory mechanisms of BCL-2 family proteins
BCL-2 family proteins are transcriptionally regulated via different factors such as p53, myelocytomatosis viral oncogene homolog (MYC), signal transducer and activator of transcription 3 (STAT3), NF-κB, and hypoxia-inducible factor 1 (HIF1). These factors can modulate BCL-2 gene expression according to cellular stress or damage status. p53, for example, increases expression of BAX, NOXA, and PUMA, but downregulates expression of BCL-2, in cell types dependent on BCL-2 priming, including hematopoietic stem cells (HSCs) (see next subsection); such priming proteins are age-dependent; expression of the BCL-2 proteins is downregulated over time [21, 22]. MYC activates expression of BAX, BID, and BIM, increasing sensitivity to apoptosis in dividing cells [23]. On the other hand, STAT3 and NF-κB promote expression of BCL-2 and BFL1, respectively, promoting cell survival. HIF1 can upregulate MCL1 transcription during hypoxic conditions [24, 25]. In addition, PTMs comprise an abundant regulatory mechanism for regulating mRNA expression of BCL-2 family proteins.
The functional redundancy of pro-apoptotic and anti-apoptotic BCL-2 proteins in embryonic development
BCL-2 family proteins play an important role in maintaining apoptosis; the hemostasis between pro-apoptotic and anti-apoptotic cells is crucial for health and preventing diseases. These observations are mainly validated using mouse models. Interrupting apoptotic homeostasis via disrupting protein-coding genes can cause irreversible damage to development and health, including hematogenesis.
In studies of mice, deletional BCL-2L1 or Mcl1 genes show abrogated neonate development and induce embryonic lethality at different stages, suggesting the importance of those proteins in early development [26, 27]. In a study using a BCL-XL-only deletion model, the mice exhibit developmental deficiencies at erythroid progenitor and central nervous system levels, indicating the specific survival role of BCL-XL in these tissues [27]. In studies using BCL-2-deficient mice, several embryonic organ defects included kidneys, central nervous system (CNS), lymphocytic cell depletion, and hair graying, thus indicating the importance of BCL-2 in these tissues [28–34].
In hematopoiesis, haploinsufficiency of protein-coding genes MCL1 or BCL-XL does not exhibit developmental deficiency; this may reflect the degree of redundancy in BCL-2 family proteins [35–39]. On the other hand, knockout studies using conditional deletion of MCL1 show tissue defects on hematopoietic stem cells at development and maintenance stages, including hematopoietic stem cells and progenitor cells [40], immature and mature lymphocytes [41–45], natural killer cells [46], neutrophils [47], basophils and mast cells [48], and plasma B cells [49, 50]. These emphasize further the critical role of MCL1 in the survival and development of blood and immune cells. Overlapping actions of different BCL-2 family proteins are seen in many tissues and cells and can protect against apoptosis. Conversely, disruption of certain functions can lead to severe health disorders in different tissues, as seen in studies showing multiple defects of the pro-apoptotic proteins BAX, BAK1, and BCL-2-related ovarian killer (BOK) [51, 52].
Regulation of erythropoiesis via cell signaling and BCL-XL activation
Maturation of red blood progenitors necessitates a multi-step process (Figure 2), including ATA-1 activation, continuous Epo receptor (EpoR) signaling, and apoptosis inhibition by activating anti-apoptotic BCL-XL protein. Following EpoR activation, the JAK2-STAT5 complex activates BCL-XL expression to expanding and maturing red cells [53, 54]. EpoR has been postulated to accelerate the red cell maturation process via shortening G1 of the cell cycle [53]. In the erythroid niche, mature erythroblasts can interact with maturing erythroblasts using Fas ligands (FasL). Fas receptors of maturing erythroblast are then triggered and followed by the proteolytic action of caspases, which could breakdown erythroid transcription factors such as GATA binding protein 1 (GATA1). As a result, BCL-XL activation is dismissed, resulting in apoptosis [55]. Independent of the p53 apoptotic pathway, leukemia/lymphoma-related factor (LRF) is a transcription factor of POZ-Kruppel family. LRF is also directly activated by the antiapoptotic role of GATA1 and has been shown to have a role at late-stage erythroblast. BIM, as BCL-2 family protein, acts upon LRF activation [54]. It is also shown that anti-apoptotic protein, BCL-XL, may be overexpressed during LRF activation via binding protein Src-associated in mitosis 68 kDa protein (SAM68) [56].
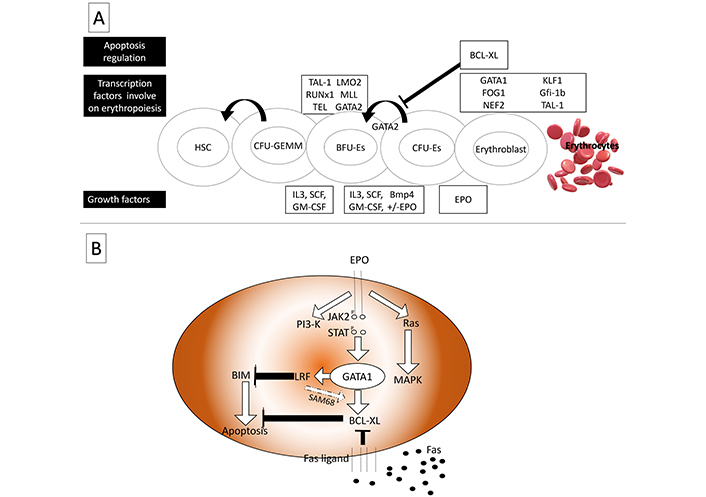
Transcription factors involved in erythropoiesis. (A) As a feedback inhibitor for GATA2 anti-apoptotic protein, BCL-XL is involved in regulating BFU-erythrocytes’ expanding; (B) in presence of EPO signaling, BIM is deactivated by LRF, LRF also activates BCL-XL by SAM68. In contrast, Fas signaling deactivates BCL-XL to control erythropoiesis in a rate-limiting fashion. HSC: hematopoietic stem cell; CEU-GEMM: colony-forming unit-granulocyte erythrocyte monocyte megakaryocyte; BFU-Es: burst-forming unit-erythroid; CFU-Es: colony-forming unit-erythroid; TAL-1: T-cell acute lymphoblastic leukemia 1; LMO2: LIM domain only 2; RUNx1: runt-related transcription factor 1; MLL: mixed lineage leukemia; TEL: translocation ETS leukemia; GATA2: GATA-binding factor 2; BCL-XL: BCL extra-large; FOG1: friend of GATA; NEF2: nuclear factor erythroid-derived 2; KLF1: Krüppel-like factor 1 (erythroid); Gfi-1b:growth-factor independent 1b; IL3: interleukin-3; SCF: stem cell factor; GM-CSF: granulocyte-macrophage colony-stimulating factor; Bmp4: bone morphogenetic protein 4; EPO: erythropoietin; PI3-K: phosphoinositide 3-kinase; JAK2: Janus kinase 2; STAT: signal transducer and activator of transcription; MAPK: mitogen-activated protein kinase; BIM: BCL-2-interacting mediator of cell death; LRF: leukemia/lymphoma-related factor; SAM68: Src-associated in mitosis 68 kDa protein
Death receptors, another apoptotic signal, are cell-surface receptors which belong to tumor necrosis factor receptor superfamily (TNFRSF), and are characterized by death domain such as tumor necrosis factor receptor 1 (TNFR1), TNF-related apoptosis-inducing ligand receptors (TRAIL-R), and CD95/Fas [57, 58]. The intra-cytoplasmic tail of death domain anchors protein-protein interactions with effectors such as Fas-associated death domain protein (FADD). When the ligand activates, the death receptors then activate intrinsic apoptosis followed by release of caspases and activating pro-apoptotic proteins to form death-inducing signaling complex (DISC). DISC signaling, however, does not overlap with mitochondrial death cascade. Some proteins can act as survival signal such as TNFR1 as a negative feedback loop [57]. Apoptosis activation is not limited to TP53 protein. It also involves other TP53 family proteins such as TP63 and TP73. Both proteins can bind specifically and activate some TP53-modulated death genes such as Fas, BAX, BCL-2 binding component 3 (BBC3) (also known as PUMA), TP53I3 (PIG3), caspase-1, and p53 apoptosis effector related to PMP-22 (PERP) [59].
Many p53-targeted proteins have been extensively studied, especially those belonging to TNF superfamily such as the FasL/CD95L death receptor Fas (CD95), TNFRSF10A (DR4), TNFRSF10B (DR5), TNFRSF10C (DcR1), and TNFRSF10D (DcR2). During extrinsic stimuli, apoptosis is initiated via TRAIL-R and Fas response to downstream signaling of pro-apoptotic pathway in mitochondria [60]. Furthermore, recent studies on TP53 transcription targets identify novel targets such as TMEM219 death receptor which is activated via insulin-like growth factor-binding protein 3 (IGFPB3) ligand [61, 62]. Little is known about IGFPB3 in relation to leukemia, but Ikezoe et al. [63] showed that the retinoid effect of IGFPB3 was enhanced in acute myeloid leukemia (AML) cell culture, after internalization of IGFPB3 constructed plasmid [63, 64]. That is, such inhibitory mechanisms could be a useful therapeutic application.
TP53, as apoptotic protein, can migrate into mitochondria and induce apoptosis via BCL-2 family proteins, including MOMP regulators, such as BAX, BID, PMAIP1 (NOXA), BBC3 (PUMA), and probably BCL-2 interacting protein 3 like (BNIP3L), apoptosis inducing factor mitochondria associated 2 (AIFM2), six-transmembrane epithelial antigen of prostate (STEAP3), TP53 regulated inhibitor of apoptosis (TRIAP1) and TP53 regulated inhibitor of apoptosis 1 (TP53AIP1). Therefore, TP53 works as fine-tuner of mitochondrial permeability and/or cytochrome c secretion [65, 66]. In addition, several genes are ubiquitous in pyroptosis (apoptosis due to inflammatory response), and such as pathways are also TP53-regulated including cytosolic caspase activators such as APAF1, p53-induced protein with a death domain 1 (PIDD1), and NOD-like receptor family CARD domain-containing protein 4 (NLRC4) [59].
PTMs modify affinities and ubiquitousness of BCL-2 family proteins
PTMs use phosphorylation and ubiquitin-proteasome to tune the expression level of BCL-2 family proteins, therefore modifying protein-protein interactions in stressful conditions such as protein instability, protein abundance, and protein binding affinity [67, 68]. That is, the interplay between pro-apoptotic or pro-survival functions of BCL-2 family proteins are contained for the sake of cell survival [69].
Protein cleavage is the primary activation process for pro-apoptotic proteins. During apoptosis, death receptors become activated via the DISC. It is initiated via adaptor proteins such as FADD, tumor necrosis factor receptor type 1-associated death domain (TRADD), and FLICE-inhibitory protein (FLIP) [70]. In the cytosol, active FADD guides activated caspase-8 to cleave wild-types BID (p22), resulting in tBID. The tBID (p15) is further integrated into the mitochondrial surface, where tBID (p11, p13) fragments enter the mitochondria for cytochrome c activation during intrinsic apoptosis [71, 72]. PTMs are involved via N-myristylation at Gly-60 of 15 kDa tBID fragment, which integrates membrane to downstream signaling interactions [73]. Caspase and granzyme B (GB) are then cleaved by binding to tBID at Asp-59 and Asp-75, respectively [71]. The role of GB is also extended by inducing microcentric ROS, which in turn digest anti-apoptotic factors such as pro-survival factors and aid in DNA fragmentation and lysosomal lysis [74].
BCL-2, as an anti-apoptotic protein, plays a major role in lymphocyte survival during rigorous T-cell differentiation and double positive (DP) thymocyte process [75]. BCL-2 protein initiates interconnection to other BCL-2-family regulators. A recent study shows that BCL-2 is potentially regulated via epigenetics, such as Robnr gene as a long non-coding RNA molecule [75], using CRISPER-Cas system to suppress Robnr; BCL-2 protein downregulation thus potentiating cell death [76]. Robnr and other lncRNAs could be potential drug targets to downregulate BCL-2 gene in cancers such as T-cell leukemia and lymphoma. Studies into the regulation of BCL-2 proteins and MOMP have led to the development of BH3-mimetics as drugs targeted towards apoptotic pathways. These novel molecules can compete with BH3 sensitizers to potentially activate apoptosis. Ongoing research may give better insight into the mechanisms of PTM and cell survival.
PTM of TP53 protein provides the stabilization required for apoptotic activation. This is attained through TP53 phosphorylation at serine residues S15 and S20. Some pro-apoptotic proteins, such as TP53AIP1, require further TP53 phosphorylation at S46, which is regulated via TP53 pro-apoptotic protein TP53INP1. Other proteins, such as ASPP coupled p53 [either PPP1R13B (ASPP1) or TP53BP2 (ASPP2)], require TP53 as co-modulator for completely activating BAX, Fas, BBC3 (PUMA), and TP53I3 (PIG3) [77]. This may reflect the important role of PTMs in fine-tuning TP53’s regulatory roles.
BCL-2 family protein dysregulation in blood neoplasms
BCL-2 gene expression and blood neoplasms
BCL-2 is believed to be a consensus cancer gene. It has been documented in many cancers such as T-cell acute lymphoblastic leukemia (TAL) and lymphoma. BCL-2 is an apoptosis antagonist (pro-survival). Unlike oncogenic proteins, BCL-2 activation leads to sequestering of apoptotic cascades of BCL-2 family proteins, predominantly at mitochondria. It is overly expressed in different lymphoid neoplasms and reflects a poor prognosis and chemotherapy resistance [78].
In Forbes et al. study [79], whole-exome sequencing (WES) was done on lymphoma patients. Results reveal a high mutation rate of BCL-2 mutations in patients, some with multiple hits. These mutations occur within spanning regions c.11–c.200 and c.320–c.600 [79, 80]. In addition, increased novel mutations in BCL-2 gene are largely related to long exposure to BH3 mimetic agents such as venetoclax, resulting in low affinity and therapy refractoriness [81]. Many novel mutations, including Gly101Val and Phe104Ile, have been detected in venetoclax-treated blood malignancies [82, 83]. Venetoclax resistance is seen more often as more patients are treated with venetoclax. However, treatment trials using combinational regimens could circumvent the issue to some extent [84, 85].
In terms of single nucleotide polymorphisms (SNPs) analysis, the evolution of single-cell analyses and chromatin accessibility studies has enabled the characterization of variants of trait-cell type and their precise functions [86]. An exciting example is BCL-2 rs17758695 SNP, which can downregulate hematopoiesis. rs17758695 is an intron nucleotide in the BCL-2 gene region, which acts as anti-enhancer at monocyte-erythroid progenitor (MEP). It decreases BCL-2 expression, which is seen as a count reduction of eosinophil monocytes and erythrocytes [87]. Knowing such a trait can pave the way for exploring germline variants in controlling stem cell proliferation.
In lymphomas and leukemias, the persistent proliferation of cancer cells may hinder apoptosis via promoting the expression of BCL-2 protein. Methylation studies show that chronic lymphocytic leukemia (CLL) progression is correlated with the hypomethylated BCL-2 gene at its promoter site; this may involve elevating the expression of BCL-2 gene [88]. In addition, genomic translocation t(14;18) (BCL-2; IgH) and 13q deletion concurrently affect BCL-2 expression [89, 90]. Fortunately, polymerase chain reaction (PCR) is an easy and affordable molecular approach to detect chromosomal indels to monitor cancer progression.
Globinopathies and BCL11A gene regulation
After birth, the newborn fetus can produce hemoglobin-forming red blood cells. These red cells contain fetal hemoglobin (HbF), which is progressively replaced by hemoglobin A (HbA). At the globin level, γ-globin switches to β-globin by activating Krüppel-like factor 1 (KLF1), BCL11A, and MYB with other transcription factors and co-repressors. BCL11A acts as a γ-globin repressor when activated by KLF1 [91]. KLF1 directly activates BCL11A to switch expression from γ-globin to β-globin. It is also suggested that KLF1 indirectly inhibits γ-globin expression by activating BCL11A [92, 93].
In β-thalassemia, the protein level of HbF is relatively higher than normal because of activated binding sites of Hbγ gene at three binding sites for Hbγ gene, which include BCL11A suppressor complexes on Hbγ coding region, alpha-delta (Aγ-δ) intragenic region, and GGCCGG motif in the HbG proximal promoter. These regions are also seen in the milder form of hereditary persistence fetal hemoglobin (HPFH) [94]. In addition, certain SNPs on BCL11A could assist HbF high-level production with better prognosis for HPFH. Gene editing therapy using BCL11A is clinically plausible for treating hemoglobinopathies [95].
Tyrosine kinases and BCL-2 engagement in myeloproliferative neoplasms: mechanisms and precision targeted therapies
To evade apoptosis, cancer cells use chromosomal translocations to indirectly activate anti-apoptotic genes such as BCL-XL. In chronic myeloid leukemia (CML), for example, the breakpoint at Philadelphia chromosome (Ph) chromosome produces breakpoint cluster region-Abelson tyrosine kinase 1 (BCR-ABL1) fusion protein which is a major pathogenic determinant. As a result, the leukemic cells exploit ABL1, a tumor-influencing tyrosine kinase, to be continuously activated by coiled-coil BCR domain. The fused protein p210 (210 kDa molecular weight) is the common and moderately aggressive phenotype in CML when compared to the p190 smaller phenotype; CML cells, therefore, can evade apoptosis via different mechanisms including activation of Ras pathway for boosting proliferation, diminishing adhesion effects of cell-cell inhibition interactions with bone marrow (BM) stromal cells, and upregulating anti-apoptotic BCL-XL protein following STAT5 pathway activation. Different in vivo studies using BCR-ABL gene of transgenic mouse show a surge in myeloid progenitors that mimics the survival mechanisms in CML [96, 97].
Tyrosine kinase inhibitors (TKIs) such as imatinib, nilotinib, and dasatinib have established their efficacy as treatments for Ph+ CML, targeting BCR-ABL kinases [97]. Despite their success in Ph+ CML, TKIs have exhibited unsatisfactory outcomes in advanced CML, particularly in patients with blast-phase CML (CML-BP) (Ph+) [98]. Recent investigations have delved into the potential of combining TKIs with venetoclax, an anti-BCL-2 protein agent, specifically in heavily pretreated advanced CML-BP Ph+ patients. Results indicate that the synergy of venetoclax and TKIs yielded a 60% overall response rate (ORR) and a median overall survival (OS) of 2 months in CML-BP Ph+, while the OS reached 10.9 months in CML-BP. Another study on TK-naïve and newly diagnosed CML patients reported a median OS of 9–12 months [99, 100]. Furthermore, preliminary findings using TKIs with venetoclax have shown promising responses among CML-BP (Ph+) patients, even among those resistant to TKIs [101, 102].
In AML, mutations involving the tyrosine kinase domain (TKD) and internal tandem duplication (ITD) in the FLT3 gene are prevalent in newly diagnosed cases, and these mutations are associated with reduced OS and elevated relapse rates [103, 104]. A recently published phase I/II study demonstrated the potential of a dual combination therapy involving azacytidine and a second-generation FLT3 inhibitor, quizartinib. This approach displayed remarkable responses in a cohort of older or unfit individuals with newly diagnosed FLT3 mutated AML. The study yielded 87% complete remission or complete remission with incomplete count recovery (CR/CRi) rate and a median OS of 19 months. This outcome highlights the concept of synergistic lethality between the two agents [105]. However, the LACEWING phase III trial, which investigated azacitidine with or without gilteritinib, an FLT3 inhibitor, was prematurely terminated during interim analysis due to a lack of discernible benefit [106].
Multiple preclinical investigations have examined the potential for synergistic lethality between FLT3 inhibitors and venetoclax [107–109]. The combination of giltertinib with venetoclax has demonstrated promising efficacy, with a composite CR (CRc) rate exceeding 75%. Notably, approximately 60% of responsive patients showed a FLT3 molecular response (< 10–2) that persisted, even among those previously treated with FLT3 inhibitors [110]. Moreover, a recent study explored the effectiveness of a triplet agent approach involving hypomethylating agents (HMA), FLT3 inhibitors, and venetoclax in a subgroup of older or unfit patients newly diagnosed with FLT3 mutated AML. This investigation revealed a remarkable 93% CR/CRi rate, 70% CR rate, and 83% minimal residual disease (MRD) negativity using both FLT3-PCR and flow cytometry analysis [103].
BCL-2 family proteins and development of autoimmune diseases
In addition to contributing in the pathogenesis of blood cancer, BCL-2 family proteins have also emerged as key players in autoimmune blood cell disorders, for example, idiopathic thrombocytopenic purpura (ITP). ITP is a type of bleeding disorder due to low platelet numbers; many studies shows that ITP is related to an imbalance in regulation of BCL-2 family proteins, possibly leading to immature platelets clearance [111–113]. In Leytin’s in vivo study [114], anti-GPIIb administration led to antibody-mediated platelet apoptosis via exposing phosphatidylserine onto platelet surface, caspase activation, and thrombocytopenia. This suggests that BCL-2 family proteins may play a role in accelerated platelet clearance in ITP.
Glucocorticoids are the first-line treatment for ITD, suppressing the immune response in favor of increasing platelet counts. Many patients, however, experience intolerable side effects, necessitating dose reductions or the use of alternative treatments [115]. Anti-BCL-2 agents, such as venetoclax, have been linked to emergent autoimmune cytopenias (AICs) and ITP in the treatment of CLL. These reported conditions are simple to handle and do not cause interruption of the treatment schedule [116]. However, immunomodulation of anti-BCL-2 proteins in platelets may improve platelet clearance of refractory autoimmune hemolytic anemia [117].
Other autoimmune blood disorders are also implicated by the BCL-2 family proteins; these include autoimmune hemolytic anaemia (AIHA) and autoimmune neutropenia (AIN). In a similar manner to ITD, these disorders are characterized by faulty labeling of affected cells via immune response, leading to acceleration of destruction and anemia in AIHA and neutropenia and susceptibility to infection in AIN. Regulation of BCL-2 family proteins has been observed to be affected in both disorders and is also seen as an effect modulated by anti-BCL-2 agents [117, 118].
Venetoclax as an anti-neoplastic drug
There has been a shift in the treatment approach for B-cell lymphoproliferative disorders, moving away from traditional chemotherapy to novel small-molecule inhibitors. One particular target of interest is the (BCL-2), which plays a crucial role in modulating apoptotic cell death pathway. Venetoclax belongs to class of chemicals called as pyrrolopyridines and it was the first BH3 mimetic (BCL-2 inhibitor) which was approved by the Food and Drug Administration (FDA), USA, in 2016. Venetoclax, as an anti-cancer drug, has shown effectiveness in various subtypes of lymphoid malignancies (more effectively in patients with 17p deletion), including patients with relapsed CLL with an ORR for these patients reaching nearly 80% [119, 120]. Figure 3 provides the chemical structure of venetoclax.
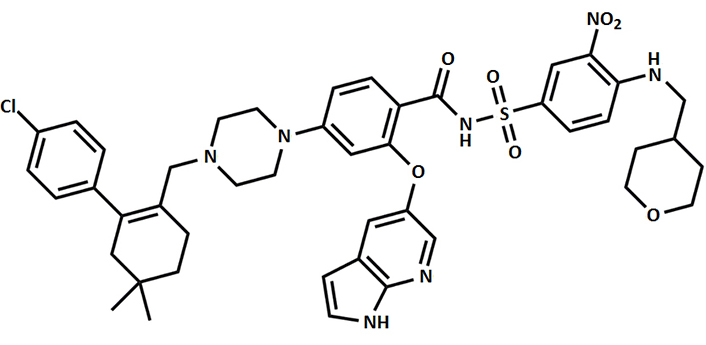
Modulating apoptosis equilibrium through synergistic venetoclax combinations in blood cancers
The evasion of apoptosis is one of the hallmarks of cancer, enabling pre-cancerous cells to survive and progress into malignancy. Thus, an imbalance in the pace of DNA repair and damage in aged HSCs may be associated with blood cancer in older age. BCL-2 family proteins are strongly associated with blood cancer. An in vivo study by Ke and colleagues [51] showed that mature lymphocytes accumulated in mice as a result of BAX and BAK1 double knockouts, suggesting that lymphocytes, as well as neutrophils and platelets, are mainly controlled by intrinsic apoptosis despite other apoptotic pathways. During lymphocyte development, auto-reactive lymphocytes are typically eliminated through intrinsic apoptosis. However, in cancer therapy, this may also be associated with higher thrombocyte apoptosis, leading to thrombocytopenia as a side effect when using BH3 mimetics against the BCL-XL protein.
The BCL-2 gene is selectively targeted by ABT-199 (venetoclax), which has been approved for treating progressive cases of CLL. Venetoclax achieves an improved response rate either as a single or combined agent [121, 122]. Recent studies have shown that the synergistic effect of using a cyclin-dependent kinase (CDK) inhibitor with venetoclax can achieve a better response rate in AML patients. Alvocidib, a CDK inhibitor, can promote apoptosis alongside venetoclax, thus modulating the cell-death equilibrium through abrogating MCL-1 and upregulating BIM, NOXA, and other pro-apoptotic proteins. In vivo and ex vivo studies show conditional synergy, as the positive effects require no overexpression of BCL-XL or BIM. This also suggests that the major role of intrinsic apoptosis for successful synergism is mainly via CDK9 inhibition [85, 123].
However, some patients experience cancer rebound due to venetoclax resistance, as reported in CLL and diffuse large B-cell lymphoma (DLBCL) cases. This is mainly due to BCL-2 unresponsiveness caused by gene mutations at both the driver and evolving mutation stages. For example, somatic mutations on Gly101Val, Phe104Ile, Gly33Arg, Ala4Thr, Arg6Lys, c.2G>A, and Arg12Trp are commonly detected in patients. These mutations may potentially affect the function of the BCL-2 protein by altering its ability to interact with other proteins, inhibiting apoptosis, and promoting cancer cell survival [82, 83, 124, 125]. This necessitates the development of venetoclax analogues and the use of synergistic combinations. It also highlights the importance of regularly screening the BH3 profile during leukemia remission to measure the response rate of BH3 mimics.
Patients receiving venetoclax often experience cytopenias, including neutropenia [126]. This is because the BCL-2 protein, which supports the survival of granulocyte-macrophage precursor cells, is affected by venetoclax. Neutropenia, in particular, is commonly seen. Neutrophils are crucial in the innate immune system as they directly attack invading bacteria or fungus and release cytokines to recruit inflammatory responses at the infection site [127]. Hence, patients with quantitative or qualitative deficits in neutrophils are at a higher risk of bacterial and fungal infections. The severity and duration of neutropenia directly impact the likelihood of infection. A recent study examining 350 patients taking venetoclax revealed that dose reductions or interruptions were most frequently due to neutropenia [128]. The study also found that 15% of patients with severe neutropenia (grade III/IV) experienced serious infections, although death resulting from infectious complications was uncommon.
Therapy regimens containing venetoclax have entered clinical trials for different hematological malignancies, such as AML and myelodysplastic syndrome. The mechanisms of venetoclax resistance have been investigated, including the role of p53 in affecting apoptosis pathways or altering energy metabolism [129–132]. In myelodysplastic syndrome, high-risk cases have shown a therapeutic response to venetoclax monotherapy, suggesting the potential therapeutic effectiveness of venetoclax in other blood-related cancers [133, 134].
Laboratory investigation of hematological cancers based on BCL-2 family proteins
BH3 profiling is a useful tool to indirectly measure BCL-2 antiapoptotic activities in cancer. This technique uses synthetic peptides of the BH3 domain of BCL-2 pro-apoptotic proteins to competitively bind to BCL-2 anti-apoptotic proteins on cancer cells, thus assessing the MOMP activities as indicators of cellular dependence on certain anti-apoptotic BCL-2 family proteins [135]. Examples are BCL-2, BCL-XL, and MCL1 dependence in CLL, AML, and multiple myeloma (MM), respectively [136–138]. BH3 profiling is also a useful approach for evaluating the efficacy of treatment for blood cancer. According to a study by Pei and colleagues [139], venetoclax-resistant AML is associated with cancer subclones that express the anti-apoptotic MCL1 protein at high levels; these findings are relevant to monocytic-type AML. A general understanding of BCL-2 dependencies may aid in planning therapy using BH3 mimetics and other anti-apoptotic inhibitors. It is important to understand the mechanism of action of BCL-2 family inhibitors, to gain maximum treatment utilization. Molecular dynamics were tested using simulations of interactions between BCL-2 family proteins and their binding targets [140, 141]. As with BH3 profiling, these data may enhance the treatment decision through optimizing treatment efficacy.
BH3 profiling has been used in assessing the combination treatment using anti-apoptotic BCL-2 inhibitors. In a study on precursor B-cell acute lymphoblastic leukemia, Seyfried et al. [142] suggest the potential to predict efficacy of BCL-2 inhibitor venetoclax (ABT-199) to induce apoptosis and provide heterogenous treatment efficacy [142, 143]. These observations may provide an optimistic result of combining anti-apoptotic inhibitors in hematological malignancies. In this respect, BH3 profiling employing modern technology offers a high-throughput assay providing a prompt and meaningful solution in screening a large number of BCL-2 family inhibitors [143]. These make it possible to identify novel therapies that specifically target anti-apoptotic proteins and thus become potential treatment choices [142–144].
In addition to BH3 profiling, BCL-2 expression profiling is employed in DLBCL to define DLBCL types based on BCL-2 family proteins within B cell maturation at the germinal center. Immunohistochemical labelling differentiates DLBCL germline center subtypes which include CD10/BCL-2-positive, BCL6-positive, activated B cell-like (ABC), and unclassifiable DLBCL [145]. The MUM1 mutation correlates with interferon regulatory factor 4 (IRF4)/BCL6 expression in high-grade lymphoma of BCL6 types [146]. It is further distinguished by immunoglobulins (IG) and IRF gene translocation and the lack of an abnormal BCL-2 gene [147].
Conclusions
In conclusion, this review raises a number of intriguing questions for future research, such as the potential clinical use of apoptosis-modulating agents and the feasibility of combination strategies that can simultaneously enhance apoptosis and offset compensatory pathways of regulated cell death (RCD). More basic research is needed to examine and develop agents that specifically augment apoptotic signaling without interfering with other critical processes that rely on apoptosis regulators, such as cell differentiation, multiplication, and the inflammatory response. Future research could identify the most effective strategies for inducing apoptosis in various disorders, taking into account their significant pathophysiological aspects. Topics related to improving therapeutic response should be researched, such as developing strategies for combining treatments that target apoptosis and capping other compensatory RCD pathways. One critical aspect is determining whether such interventions inhibit apoptosis and whether they have an effect on other processes that rely on apoptotic regulators. This assessment provides a thorough understanding of such interventions and aids in the development of an effective and safe treatment plan. Overall, addressing these research questions will broaden our understanding of apoptotic cell death and lead to the development of novel therapeutic strategies with improved specificity and efficacy in hematological and non-hematological malignancies.
Abbreviations
| AML: | acute myeloid leukemia |
| BAK: | BCL-2 antagonist/killer |
| BAX: | BCL-2-associated X protein |
| BBC3: | BCL-2 binding component 3 |
| BCL-2: | B cell lymphoma-2 |
| BCL-XL: | BCL extra-large |
| BCR-ABL1: | breakpoint cluster region-Abelson tyrosine kinase 1 |
| BFL1: | Bcl-2 homolog from fetal liver 1 |
| BIM: | BCL-2-interacting mediator of cell death |
| CDK: | cyclin-dependent kinase |
| CLL: | chronic lymphocytic leukemia |
| CML: | chronic myeloid leukemia |
| CML-BP: | blast-phase chronic myeloid leukemia |
| CR: | complete remission |
| DISC: | death-inducing signaling complex |
| DLBCL: | diffuse large B-cell lymphoma |
| FADD: | Fas-associated death domain protein |
| HbF: | fetal hemoglobin |
| IGFPB3: | insulin-like growth factor-binding protein 3 |
| ITD: | internal tandem duplication |
| ITP: | idiopathic thrombocytopenic purpura |
| KLF1: | Krüppel-like factor 1 |
| LRF: | lymphoma-related factor |
| MCL1: | myeloid cell leukemia sequence 1 |
| MOMP: | mitochondrial outer membrane permeabilization |
| NOXA: | known as phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1) |
| OS: | overall survival |
| Ph: | Philadelphia chromosome |
| PTMs: | post-translational modifications |
| PUMA: | p53-upregulated modulator of apoptosis |
| SNPs: | single nucleotide polymorphisms |
| STAT3: | signal transducer and activator of transcription 3 |
| tBID: | truncated BH3-interacting domain death agonist |
| TKIs: | tyrosine kinase inhibitors |
| TNFRSF: | tumor necrosis factor receptor superfamily |
Declarations
Author contributions
FT: Conceptualization, Writing—original draft, Writing—review & editing.
Conflicts of interest
The author declares no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2024.