 Original Article
Original Article

Affiliation:
1Research and Development, Hybrinomics Life Science and Diagnostics LLP, Bangalore 560077, Karnataka, India
Affiliation:
1Research and Development, Hybrinomics Life Science and Diagnostics LLP, Bangalore 560077, Karnataka, India
Email: prabmeg@hybrinomics.com
ORCID: https://orcid.org/0000-0003-3171-6899
Affiliation:
2Department of Bioinformatics, Bharathiar University, Coimbatore 641046, Tamil Nadu, India
ORCID: https://orcid.org/0000-0002-8548-8150
Explor Med. 2024;5:530–543 DOI: https://doi.org/10.37349/emed.2024.00237
Received: April 09, 2024 Accepted: June 25, 2024 Published: July 17, 2024
Academic Editor: Yun-Long Wu, Xiamen University, China
Aim: Present study was done to understand the dimerization of HER2/ERBB2 in normal and cancer cells using in-silico study.
Methods: Pathway analysis was done using Reactome. Structure of HER2/ERBB2 protein was obtained from PDB database, and using Schrödinger software protein structure was analysed and dimerization was done.
Results: In normal cells, HER2/ERBB2 is present at low levels and forms a stable complex with HSP90 (heat shock protein 90), CDC37 (cell division cycle 37), and ERBIN (an adaptor protein of the HER2/ERBB2 receptor). HER2/ERBB2 lacks a ligand-binding site, so it cannot bind ligands to activate HER2/ERBB2 signaling directly. Instead, it heterodimerizes with other EGFR family members, using their ligand-binding sites to activate cell proliferation signaling cascades. In cancer, overexpression of HER2/ERBB2 leads to ligand-independent activation of signaling through dimerization. During this process, HER2/ERBB2 dissociates from the HSP90 complex. Normally, HSP90 helps to correct misfolded and aggregated proteins, but it fails to correct mutated HER2/ERBB2 in cancer cells.
Conclusions: This discussion focuses on the structural changes that HER2/ERBB2 undergoes, particularly in the form of homodimers, under normal and cancerous conditions. This analysis highlights the mutated state of HER2/ERBB2 and the role of HSP90 in this context. Notably, a single-point mutation outside a protein’s active site can significantly alter its structure. This is a critical consideration in drug discovery, underscoring the need to evaluate the entire protein conformation during simulations.
HER2/ERBB2 (v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2), a member of the EGFR (epidermal growth factor receptor) family, comprises four principal domains: (1) an extracellular domain (ECD) that facilitates dimerization on the cell surface; (2) a transmembrane domain (TMD) situated within the lipid bilayer, crucial for receptor activation; (3) a juxtamembrane domain (JMD) in the cytoplasm, linking the TMD and kinase domain; and (4) a kinase domain (KD) in the cytoplasm, responsible for initiating signaling cascades through phosphorylation [1].
HER2/ERBB2 functions as a tyrosine-protein kinase receptor with enzymatic activity in its cytosolic domain. This kinase domain phosphorylates tyrosine residues on target proteins using phosphate groups from ATP hydrolysis. The ATP-binding site and catalytic center within the kinase domain are essential for this phosphorylation process, leading to the activation of cell proliferation signaling cascades [2].
Dimerization of HER2/ERBB2 occurs in the ECD, which is subdivided into four domains: D-I, D-II, D-III, and D-IV. D-I and D-III interact to block ligand binding, while D-II stabilizes protein-protein interactions during dimer formation. The function of D-IV remains less understood [3, 4].
Under normal cellular conditions, HER2/ERBB2 expression is low and stable, forming a complex with HSP90 (heat shock protein 90), CDC37 (cell division cycle 37), and ERBIN (an adaptor protein of the HER2/ERBB2 receptor). HER2/ERBB2 primarily forms heterodimers with other EGFR family members due to its lack of a ligand-binding site, thereby activating downstream signaling [5–7]. However, in cancer, HER2/ERBB2 is overexpressed, leading to homodimer formation and ligand-independent signaling activation. This occurs through dissociation from the HSP90, CDC37, and ERBIN complex [7–9].
Dimer formation results in trans-autophosphorylation within the kinase domain, where one kinase domain phosphorylates the tyrosine residues of its partner kinase domain, reciprocally enhancing activation of downstream signaling pathways [10, 11]. This process culminates in the growth factor-independent activation of the RAS/RAF/MAP kinase signaling cascade. Binding of SH1, a signaling adaptor, further recruits the GRB2:SOS1 complex, facilitating RAS guanine nucleotide exchange and subsequently activating the RAS/RAF/MAP kinase cascade (Figure 1) [12, 13]. This pathway has been extensively studied using the Reactome pathway database, providing a comprehensive understanding of HER2/ERBB2’s role in cellular signaling and its implications in oncogenesis [14].
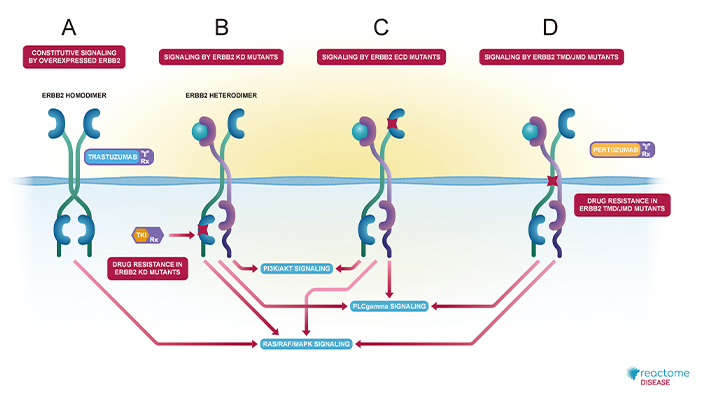
Diagrammatic representation of HER2/ERBB2 dimerizations and their respective activated signaling cascades from Reactome pathway database. (A) Constitutive signaling by overexpressed HER2/ERBB2, where it homodimerizes and activates RAS/RAF/MAPK signaling and also shown, trastuzumab as inhibitor; (B) signaling by HER2/ERBB2 KD mutants, where HER2/ERBB2 heterodimerizes and activates P13/Akt, PLCgamma and RAS/RAF/MAPK signaling and also indicates drug resistance; (C) signaling by HER2/ERBB2 ECD mutants, which heterodimerizes and activates P13/Akt, PLCgamma and RAS/RAF/MAPK signaling; and (D) signaling by HER2/ERBB2 TND/JMD mutants, where it heterodimerizes and activates PLCgamma and RAS/RAF/MAPK signaling and shown pertuzumab as inhibitor, but also drug resistance is shown. ECD: extracellular domain; JMD: juxtamembrane domain; KD: kinase domain; TMD: transmembrane domain
Note. Adapted from “Signaling by ERBB2 in Cancer” by Reactome; c2024 (https://reactome.org/content/detail/R-HSA-1227990). CC BY.
HSP90 is a chaperone that ensures protein homeostasis under stress by refolding misfolded and aggregated proteins, while CDC37, a co-chaperone, assists HSP90 in identifying client proteins [15].
In normal cells, HSP90 and CDC37 are secreted under stress conditions, but in cancer cells, where constant stress prevails, these proteins are persistently secreted, facilitating cell proliferation. HSP90 plays a critical role in regulating HER2/ERBB2 activity by binding to a specific loop in its kinase domain, stabilizing and preventing its dimerization, activation, and phosphorylation under normal conditions [7, 9].
In cancer, overexpression of HER2/ERBB2 due to gene amplification disrupts this chaperone binding, promoting HER2/ERBB2-mediated transactivation. HER2/ERBB2 is overexpressed in 15–20% of gastric adenocarcinoma (GC) patients, and while trastuzumab plus chemotherapy is the established first-line treatment, newer therapies like trastuzumab deruxtecan (T-Dxd) are under investigation, with T-Dxd receiving FDA approval in 2021 for advanced GC [16].
LIV-1, a transmembrane protein with zinc transporter and metalloproteinase activity, was initially identified as an estrogen-induced gene in breast cancer and later linked to epithelial-to-mesenchymal transition (EMT). Despite expression variations among cancer subtypes, LIV-1 is moderately to highly expressed in most breast cancers and is targeted by the antibody-drug conjugate (ADC) ladiratuzumab vedotin (SGN-LIV1A), currently in early-phase clinical trials showing promising results as monotherapy or in combination with other treatments [17].
Additionally, hyperglycemia during ipilimumab treatment exacerbates cardiotoxicity and reduces anticancer efficacy in breast cancer cells, a process mediated by the NLRP3 receptor. Combining ipilimumab with empagliflozin or lowering glucose levels mitigates these effects, enhancing treatment response and reducing cardiotoxicity. This is the first evidence that hyperglycemia worsens ipilimumab’s side effects and efficacy in MCF-7 and MDA-MB-231 cells [18].
The stability of HER2/ERBB2 is critically dependent on HSP90, primarily due to interactions within the kinase domain. This study focuses on the structural and sequential changes in HER2/ERBB2 between normal and cancer cells, emphasizing the mechanism of HER2/ERBB2 homodimerization and the role of HSP90. Understanding the dissociation of HSP90 from the HER2/ERBB2 complex during homodimerization in cancer cells, compared to its stable association in normal cells, is crucial for advancing targeted cancer therapies.
Using the Reactome pathway database (https://reactome.org/), the comprehensive cellular roles of HSP90 were analyzed, including its involvement in autophagy, cell cycle regulation, cellular responses to stimuli, disease processes (particularly cancer), immune system function, programmed cell death, and signal transduction [14]. Specifically, the role of HSP90 in HER2/ERBB2 homodimerization and its contribution to constitutive signaling by overexpressed HER2/ERBB2 was examined. This pathway involves the activation of the RAS/RAF/MAP kinase cascade through interactions with the SH1 domain and the GRB2:SOS1 complex.
X-ray crystal structures of HER2/ERBB2 with high resolution and validation were retrieved from the Protein Data Bank (PDB) (https://www.rcsb.org/). For normal cell HER2/ERBB2, PDB ID 2A91 was used, while for cancer cell ERBB2, PDB ID 3WLW was selected [19]. The homodimer interactions of HER2/ERBB2 in both normal and cancerous conditions were studied using Schrödinger software (Schrödinger Release 2023-4: BioLuminate, Schrödinger, LLC, New York, NY, 2023) [20–22].
In Schrödinger, both PDB structures were prepared and refined using the Protein Preparation Wizard, with parameters adjusted according to the specific requirements of the protein structures (Protein Preparation Wizard; Epik; Impact; Prime, Schrödinger, LLC, New York, NY, 2023) [23]. The homodimerization of HER2/ERBB2 structures was then performed using the protein-protein docking suite, and the docking results were analyzed based on the best PIPER scores [24–26].
FASTA files of both normal and cancerous ERBB2 were retrieved from the PDB and aligned using the Smith-Waterman algorithm via local alignment tools BLASTP (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and EMBOSS WATER (https://www.ebi.ac.uk/jdispatcher/psa/emboss_water). Sequential comparison was performed based on identity, similarity, and gap scores [27, 28].
Secondary structure prediction of both HER2/ERBB2 forms was conducted using PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/), which employs color patterns to differentiate between strands, helices, and coils based on the provided FASTA sequences [29].
Lastly, a comparative analysis of the 3D structures of normal and cancerous HER2/ERBB2, retrieved from PDB, was performed using UCSF Chimera software (https://www.rbvi.ucsf.edu/chimera). This involved superposing the structures to analyze differences and similarities in spatial conformation [30].
In aggrephagy, misfolded and aggregated proteins are targeted for degradation through ubiquitination, a process facilitated by HSP90. During this process, misfolded proteins are tagged with ubiquitin, allowing HSP90 to recognize and either refold or eliminate them. Subsequently, the HSP90 complex dissociates after addressing the misfolded proteins (see also Figure S1) [31].
In the cell cycle, HSP90 stabilizes newly synthesized proteins to ensure proper cellular function by forming a complex with FKBPL, which is responsible for protein stability, and CDKN1A, a cell cycle inhibitor. This complex also plays a crucial role in correcting DNA damage to prevent the formation of defective cells, and if the damage cannot be corrected, it leads to apoptosis, thereby conferring resistance to toxicity (see Figure S2) [32–34].
In response to external stimuli, HSF1 is normally present in an inactive state complexed with HSP90. External stresses such as heat, hypoxia, cellular senescence, or an increased amount of unfolded protein in the cell trigger the dissociation of this complex (see Figure S3). Following dissociation, HSF1 activates transcription factors involved in gene expression, which helps regulate stress conditions within the cell [35].
In the immune system, during viral infection, cytosolic viral RNA activates mitochondrial antiviral-signaling protein (MAVS), leading to the formation of the MAVS signalosome. Activated MAVS recruits TANK-binding kinase 1 (TBK1), interferon regulatory factor 3 (IRF3), and IRF7 to the mitochondria, resulting in the activation of IRF3/IRF7 and the subsequent production of type I interferons. Both TBK1 and IRF3 are associated with HSP90, which facilitates signal transduction from TBK1 to IRF3 in infected cells [36].
TOMM70, located on the outer mitochondrial membrane, mediates the translocation of mitochondrial protein precursors from the cytosol into the mitochondria. Molecular chaperone complexes of HSP90 and HSP70 deliver these precursor proteins to TOMM70 for import (see Figure S4). HSP90 forms a complex with TBK1 and IRF3 in the cytosol, delivering them to the MAVS signalosome on the mitochondria, thereby inducing the IRF3-mediated host antiviral response [37, 38].
In programmed cell death, HSP90 participates in regulated necrosis by forming a complex with CDC37. The pseudokinase mixed lineage kinase domain-like protein (MLKL), the terminal effector in the necroptosis pathway, is activated through phosphorylation by receptor-interacting protein kinase-3 (RIPK3). Once activated, MLKL translocates to cellular membranes, causing membrane destabilization and subsequent cell death. HSP90 and CDC37 together form a complex with MLKL, facilitating its activation (see Figure S5) [39–41].
Similarly, the activation of RIPK3, following the induction of necroptosis, requires the activity of the HSP90-CDC37 complex. This complex physically associates with RIPK3, playing a critical role in regulating necrosis (see Figure S5) [42].
In cancer cell signaling, the association or dissociation of HSP90 with members of the EGFR family (EGFR, ERBB1, ERBB2, ERBB3, and ERBB4) modulates the activation of growth and cell proliferation signaling cascades, including RAS/RAF/MAP kinase, Akt signaling, and DAG/IP3 signaling pathways. When HSP90 associates with these complexes (see Figure S6), it facilitates the loading of client kinases onto EGFR family members. Conversely, upon dissociation, it allows the EGFR members to dimerize (see Figure S7), leading to the activation of signaling cascades through a continuous series of tyrosine residue phosphorylations.
The structures of normal and cancer cell HER2/ERBB2 were retrieved from the PDB based on literature references, selecting entries with the highest resolution (< 3 Å) and optimal PDB validation reports for a detailed understanding of their X-ray crystal structures. For normal cell HER2/ERBB2, PDB ID 2A91 was used, while for cancer cell HER2/ERBB2, PDB ID 3WLW was chosen. The retrieved PDB structures of both forms of HER2/ERBB2 are depicted in Figure 2.
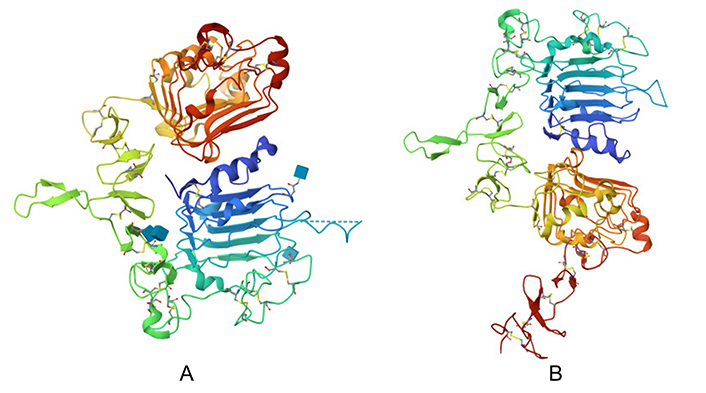
Protein structure of HER2/ERBB2. (A) PDB structure retrieved for normal cell HER2/ERBB2 (PDB ID 2A91); and (B) PDB structure retrieved for cancer cell HER2/ERBB2 (PDB ID 3WLW)
Note. Figure 2A adapted from the RCSB PDB (RCSB.org) of PDB ID 2A91 (Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, et al. The crystal structure of a truncated HER2/ERBB2 ectodomain reveals an active conformation, poised to interact with other HER2/ERBB2 receptors. Mol Cell. 2003;11:495–505). CC0. Figure 2B adapted from the RCSB PDB (RCSB.org) of PDB ID 3WLW (Hu S, Sun Y, Meng Y, Wang X, Yang W, Fu W, et al. Molecular architecture of the ErbB2 extracellular domain homodimer. Oncotarget. 2015;6:1695–706). CC0.
Upon homodimerizing the normal cell HER2/ERBB2 structures (PDB ID 2A91) using Schrödinger software, 12 dimerized structures were generated, none of which were stable. A PIPER energy score above –900 is considered indicative of a strong docking interaction, but no such scores were achieved, indicating weak interactions between the normal cell ERBB2 homodimers. The most stable structure identified was POSE2 (Figure 3A), with a PIPER pose energy of –133.7 and PIPER pose score of –71.382 as shown in Table 1. This homodimer exhibited only 12 interacting residues (Figure 3B), preventing a comprehensive interaction analysis in Schrödinger software due to the lack of solid interactions. Consequently, it was determined that normal HER2/ERBB2 cannot form a stable homodimer.
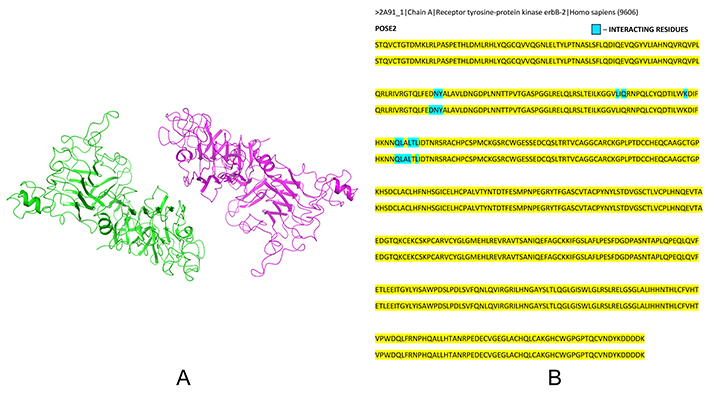
Dimerization of normal HER2/ERBB2. (A) Dimerized structure of normal cell HER2/ERBB2 (PDB ID 2A91) – POSE 2 (using Schrödinger Release 2023-4: BioLuminate, Schrödinger, LLC, New York, NY, 2023); and (B) record of interacting residues in normal cell HER2/ERBB2 dimer noted from Schrödinger software, where blue coloured residues indicate interacting residues
PIPER results of dimerized normal HER2/ERBB2 structure (PDB ID 2A91) showing unstable dimer values (using Schrödinger Release 2023-4: BioLuminate, Schrödinger, LLC, New York, NY, 2023)
| Title | PIPER pose energy | PIPER pose score | PIPER model number | PIPER cluster size |
|---|---|---|---|---|
| POSE1 | –93.038 | –74.6 | 272 | 260 |
| POSE2 | –133.7 | –71.382 | 21 | 228 |
| POSE3 | –55.632 | –106.158 | 975 | 145 |
| POSE4 | –55.871 | –10.35 | 961 | 68 |
| POSE5 | –55.752 | –12.125 | 966 | 63 |
| POSE6 | –62.301 | –54.411 | 792 | 59 |
| POSE7 | –62.404 | –27.389 | 790 | 52 |
| POSE8 | –60.198 | –66.592 | 847 | 42 |
| POSE9 | –59.333 | –32.963 | 870 | 40 |
| POSE10 | –70.686 | –28.635 | 603 | 37 |
| POSE11 | –59.265 | 0 | 871 | 3 |
| POSE12 | –56.614 | 0 | 934 | 3 |
Homodimerization of cancer cell HER2/ERBB2 (PDB ID 3WLW) resulted in 15 stable dimer structures, all exhibiting PIPER pose energy scores above –900, indicative of strong interactions (Table 2). The most stable interaction was observed in POSE1, with a PIPER pose energy of –1,238.94. The best stable POSE1 homodimer structure is depicted in Figure 4A. Further interaction analysis using Schrödinger software revealed a total of 165 strong to very strong interacting residues, as detailed in Figure 4B and Figure S8. These findings demonstrate that HER2/ERBB2 in cancer cells forms a stable homodimer with robust interactions, leading to the continuous activation of cell proliferation signaling cascades, thereby rapidly increasing cell population.
PIPER results of dimerized cancer HER2/ERBB2 structure (PDB ID 3WLW) showing stable dimer values (using Schrödinger Release 2023-4: BioLuminate, Schrödinger, LLC, New York, NY, 2023)
| Title | PIPER pose energy | PIPER pose score | PIPER model number | PIPER cluster size |
|---|---|---|---|---|
| POSE1 | –1,238.94 | –192.131 | 50 | 93 |
| POSE2 | –1,099.976 | –481.194 | 251 | 90 |
| POSE3 | –1,075.053 | 90.941 | 341 | 77 |
| POSE4 | –1,234.521 | 288.706 | 53 | 51 |
| POSE5 | –967.27 | –65.814 | 969 | 50 |
| POSE6 | –1,162.564 | –34.34 | 139 | 45 |
| POSE7 | –1,132.691 | –130.897 | 187 | 40 |
| POSE8 | –965.985 | –119.112 | 984 | 35 |
| POSE9 | –1,156.086 | 307.185 | 141 | 33 |
| POSE10 | –1,212.369 | –80.513 | 73 | 32 |
| POSE11 | –1,083.691 | –10.777 | 297 | 32 |
| POSE12 | –974.888 | 43.566 | 899 | 32 |
| POSE13 | –1,006.026 | –18.822 | 683 | 31 |
| POSE14 | –1,041.641 | 142.197 | 479 | 29 |
| POSE15 | –1,041.1 | 61.322 | 483 | 24 |
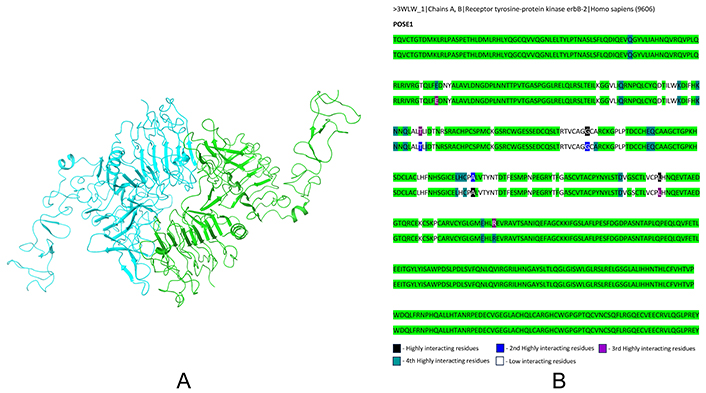
Dimerization of cancer HER2/ERBB2. (A) Dimerized structure of cancer cell HER2/ERBB2 (PDB ID 3WLW) – POSE1 (using Schrödinger Release 2023-4: BioLuminate, Schrödinger, LLC, New York, NY, 2023); and (B) record of interacting residues in cancer cell HER2/ERBB2 dimer structure obtained from Schrödinger software, where the pattern of colours represents strong to very strong interactions between the dimer
Sequential comparative analysis between normal and cancer HER2/ERBB2 using BLASTP and EMBOSS WATER revealed 99% and 99.6% identity, respectively, as shown in Figure 5. This high similarity indicates the presence of point mutations in the HER2/ERBB2 sequence in cancer cells. Secondary structure comparison using the PSIPRED tool showed shifts in loops, helices, and sheets in the cancer HER2/ERBB2 sequence compared to the normal HER2/ERBB2, as illustrated in Figure 6.

Sequential comparative analysis. (A) Using NCBI BLASTP showing 99% identity between normal and cancer cell HER2/ERBB2 sequence; and (B) using EMBOSS WATER, 99.6% identity found between normal and cancer cell HER2/ERBB2 sequence indicating point mutations in certain regions of the FASTA sequence

Secondary structure prediction of (A) normal cell HER2/ERBB2 (PDB ID 2A91); and (B) cancer cell HER2/ERBB2 (PDB ID 3WLW), using their FASTA sequence as input in PSIPRED tool
Finally, the 3D structures obtained from the PDB were compared by superposing them using Chimera software. Slight differences and shifts in protein folding were observed in the cancer cell HER2/ERBB2 structure compared to the normal cell HER2/ERBB2, as shown in Figure 7. These structural changes, caused by point mutations, result in the formation of strong homodimers in cancer cells, leading to the rapid activation of HER2/ERBB2 signaling and a significant increase in cell population.
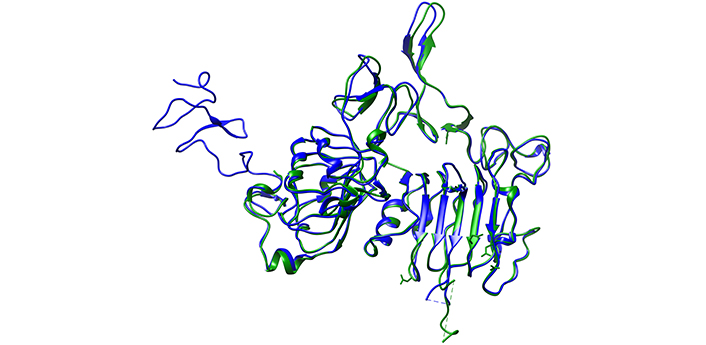
Comparative analysis by superposing both HER2/ERBB2 and visualizing their 3D structure using UCSF Chimera software
The comprehensive analysis delineates a stark contrast between the dimerization capabilities of HER2/ERBB2 in normal and cancerous cells. In normal cells, HER2/ERBB2 struggles to form stable homodimers and the identification of only 12 interacting residues, underscoring the protein’s inability to engage in substantial interactions necessary for stable dimer formation (Table 3). This finding is pivotal as it highlights the inherent stability issues within the normal HER2/ERBB2 homodimers, suggesting a natural regulatory mechanism that prevents the activation of cell proliferative signaling pathways. Conversely, in cancerous cells, HER2/ERBB2 demonstrates a remarkable ability to form stable homodimers and a robust network of 165 interacting residues (Table 3). This stark contrast underpins the pathogenic potency of cancerous HER2/ERBB2, as these stable homodimers relentlessly activate cell proliferation signaling, leading to uncontrolled cell growth. The near-identical sequence homology between normal and cancerous HER2/ERBB2, with single point mutations, yet profound differences in dimerization capabilities, underscores the critical role of structural nuances in protein function and disease progression. These findings beckon a deeper exploration of the structural and functional dynamics of HER2/ERBB2, offering potential avenues for therapeutic intervention aimed at destabilizing these pathogenic homodimers in cancerous cells, thereby mitigating the aggressive proliferation characteristic of cancer.
Summary of result analysis
| Feature | Normal cell HER2/ERBB2 | Cancer cell HER2/ERBB2 |
|---|---|---|
| Number of dimerized structures obtained | 12 | 15 |
| PIPER pose energy score (best docking score) | –133.7 (No score above –900) | –1,238.94 (Scores above –900) |
| Number of interacting residues | 12 | 165 |
| Stability of homodimers | No stable dimer found | Stable dimer structures obtained |
| Interaction analysis in Schrödinger | Unable to perform due to weak interactions | Conducted successfully, showing strong to very strong interactions |
| Sequence identity (BLASTP) | - | 99% similarity with normal HER2/ERBB2 |
| Sequence identity (EMBOSS WATER) | - | 99.6% similarity with normal HER2/ERBB2 |
| Secondary structure comparison (PSIPRED) | - | Shift in loops, helices, and sheets observed compared to normal HER2/ERBB2 |
| 3D structure comparison (Chimera) | - | Slight difference and shift in protein folding compared to normal HER2/ERBB2 |
Overall, the structural characterization of HER2/ERBB2 proteins underscores the pivotal role of dimerization and interaction strength in modulating the signaling cascades that drive cell proliferation. These findings offer valuable insights into the molecular mechanisms underlying HER2/ERBB2-mediated carcinogenesis, emphasizing how alterations in protein structure and interactions can lead to uncontrolled cell growth and cancer progression. This knowledge is crucial for developing targeted therapies for HER2/ERBB2-positive cancers, where interrupting or modulating these interactions could effectively inhibit tumor growth.
The analysis revealed that even single-point mutations in non-active regions of HER2/ERBB2 can induce significant alterations in the overall protein architecture. This phenomenon is critical because it demonstrates that mutations outside the active site can still have profound effects on protein function by altering its folding, stability, and interaction capabilities. Such structural changes can enhance the protein’s ability to form stable homodimers, as observed in cancer cells, leading to the continuous activation of proliferative signaling pathways.
These insights have substantial implications for drug discovery and development. Traditional molecular docking and dynamics simulations often focus predominantly on the active sites of target proteins, aiming to identify compounds that can effectively bind and inhibit these regions. However, the findings from this study highlight the necessity of considering the entire protein structure in drug design strategies. By accounting for potential conformational changes induced by mutations outside the active site, researchers can develop more effective therapeutic agents that target the protein’s overall architecture and interaction networks.
Moreover, this comprehensive approach can help identify allosteric sites and secondary binding pockets that could be exploited for therapeutic intervention. Allosteric modulators, which bind to regions other than the active site, can induce conformational changes that affect the protein’s activity and interactions. This strategy could provide a more nuanced and effective means of regulating HER2/ERBB2 function and mitigating its role in cancer.
In summary, the structural characterization of HER2/ERBB2 proteins not only enhances our understanding of HER2/ERBB2-mediated carcinogenesis but also informs the development of more sophisticated and effective targeted therapies. By considering the entire protein structure and the impact of mutations beyond the active site, researchers can better design drugs that address the complex molecular dynamics of HER2/ERBB2 and similar oncogenic proteins. This holistic approach holds promise for improving therapeutic outcomes in HER2/ERBB2-positive cancers and potentially other diseases driven by similar molecular mechanisms.
A limitation of this study is that we were able to analyze only a single protein structure and its related pathways. For a more comprehensive understanding, it is essential to explore additional protein structures and associated pathways. Furthermore, these findings need to be validated under in vivo conditions to confirm their relevance and applicability in a physiological context.
CDC37: cell division cycle 37
EGFR: epidermal growth factor receptor
HER2/ERBB2: v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2
HSP90: heat shock protein 90
IRF3: interferon regulatory factor 3
MAVS: mitochondrial antiviral-signaling protein
MLKL: mixed lineage kinase domain-like protein
PDB: Protein Data Bank
RIPK3: receptor-interacting protein kinase-3
TBK1: TANK-binding kinase 1
The supplementary figures for this article are available at: https://www.explorationpub.com/uploads/Article/file/1001237_sup_1.pdf.
We thank Hybrinomics Life Science & Diagnostics LLP, for their willingness to allow public access to their web services, particularly Schrödinger software.
JS: Methodology, Investigation, Writing—original draft. PM: Conceptualization, Methodology, Investigation, Supervision, Writing—review & editing. HV: Formal analysis, Writing—review & editing. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

