 Mini Review
Mini Review

Affiliation:
1Department of Cardiology, Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital, Fudan University, Shanghai 200032, China
2National Clinical Research Center for Interventional Medicine, Shanghai 200032, China
3State Key Laboratory of Cardiovascular Diseases, Zhongshan Hospital, Fudan University, Shanghai 200032, China
4NHC Key Laboratory of Ischemic Heart Diseases, Shanghai 200032, China
5Key Laboratory of Viral Heart Diseases, Chinese Academy of Medical Sciences, Shanghai 200032, China
ORCID: https://orcid.org/0009-0004-5783-5065
Affiliation:
1Department of Cardiology, Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital, Fudan University, Shanghai 200032, China
2National Clinical Research Center for Interventional Medicine, Shanghai 200032, China
3State Key Laboratory of Cardiovascular Diseases, Zhongshan Hospital, Fudan University, Shanghai 200032, China
4NHC Key Laboratory of Ischemic Heart Diseases, Shanghai 200032, China
5Key Laboratory of Viral Heart Diseases, Chinese Academy of Medical Sciences, Shanghai 200032, China
Email: jren_aldh2@outlook.com
ORCID: https://orcid.org/0000-0002-0275-0783
Explor Med. 2024;5:684–693 DOI: https://doi.org/10.37349/emed.2024.00249
Received: June 21, 2024 Accepted: August 05, 2024 Published: October 25, 2024
Academic Editor: Esma R. Isenovic, University of Belgrade, Serbia
Diabetic vascular disease, as one of the common complications in diabetic patients, threatens the quality of life and health of patients. Autophagy maintains cellular homeostasis and survival as an important intracellular self-repair mechanism. In recent years, with the gradual deepening of autophagy research, more and more studies have found that vascular cells such as endothelial cells, smooth muscle cells, and inflammatory cells are closely associated with various autophagy disorders. Different autophagy regulatory mechanisms can lead to different or similar cellular outcomes, and there are complex crosstalk mechanisms in the process. Therefore, we will summarize the latest research progresses with regards to the role of autophagy in diabetic vascular disease, focusing on the complex regulatory mechanisms of mitophagy, epigenetic modifications, apoptosis, inflammation, and autophagy in the development of diabetic vascular disease, aiming to provide effective therapeutic targets for diabetic vascular injury.
Diabetes mellitus, a metabolic anomaly characterized by hyperglycemia, has become a significant global public health concern. It is estimated that 693 million adults will be afflicted with diabetes by 2045 [1]. Diabetes triggers the development of a wide variety of complications including diabetic cardiomyopathy and diabetic vascular anomalies. Diabetic vascular diseases are common in patients with diabetes and can be divided into two categories: diabetic macroangiopathy (peripheral arterial disease, cardiovascular disease, and cerebrovascular disease) and diabetic microangiopathy (diabetic nephropathy, diabetic retinopathy, and diabetic cardiomyopathy). These anomalies represent the primary causes of morbidity and mortality in diabetic patients [2]. Diabetic vascular complications remain a major threat to human health, although the underlying pathogenesis remains poorly understood [3].
Autophagy is a complex process of self-degradation through the formation of autophagosomes that randomly or selectively wrap damaged organelles, abnormally aggregated proteins, or other cellular components, followed by the formation of autophagosome-lysosome complexes that degrade the phagocytosed molecules [4]. As a self-protection mechanism of organisms, autophagy is essential for maintaining homeostasis and regulating cell survival and death [5, 6]. Autophagy can maintain the homeostasis of most cells originating from blood vessels, including endothelial cells, smooth muscle cells, and inflammatory cells [7]. However, defective autophagy can cause disease and hasten its progression under certain pathological conditions [8, 9]. Autophagy is a double-edged sword that plays a dual role in diabetes-induced vascular injury, and how it works warrants further investigation [10, 11]. Based on this, this review will present the latest findings on the molecular regulation of diabetic vascular diseases by autophagy.
Autophagy selectively recognizes, isolates, and degrades specific targets through autophagosomes. This process is commonly known as the selective autophagy [12]. Mitophagy, represents the mitochondria-selective autophagy, which can selectively eliminate damaged mitochondria, and maintain the dynamic balance of mitochondria and intracellular environmental homeostasis, involving the regulation of multiple molecular mechanisms and signaling pathways, such as BCL2 interacting protein 3 (BNIP3)-mediated mitophagy, FUN14 domain-containing protein 1 (FUNDC1)-mediated mitophagy, and the PTEN-induced kinase 1 (PINK1)/Parkin RBR E3 ubiquitin-protein ligase (PARKIN) pathway, which are closely linked to the development of vascular injury in diabetes [6, 13, 14].
Diabetic patients generally possess elevated circulating lactate levels. In vitro experiments have demonstrated that lactate induces oxidative stress, and apoptosis of vascular smooth muscle cells (VSMCs), to accelerate VSMCs calcification by inhibiting BNIP3-mediated mitophagy [15]. Induction of mitophagy by brain-derived neurotrophic factor (BDNF) has been demonstrated to exert a protective role on brain microvascular endothelial cells (BMECs) under hyperglycemia conditions. BDNF binding to the tropomyosin receptor kinase B (TrkB) induces downstream hypoxia-inducible factor-1α/BNIP3-mediated mitophagy and protects BMECs from hyperglycemia-induced oxidative stress and apoptosis [16]. In retinal vascular endothelial cells from diabetes mice, activity of the Hedgehog pathway was reduced, and the dissociation between Beclin1 and BCL-2 was induced by upregulation of BNIP3, leading to autophagy activation. In contrast, metformin downregulated autophagy through activation of the Hedgehog pathway, dependent upon the downregulation of BNIP3, resulting in a protective effect on vascular endothelial cells [17].
Empagliflozin, as an antidiabetic drug, activates FUNDC1-mediated mitophagy by upregulating the AMP-activated protein kinase catalytic subunit alpha 1 (AMPKα1)/Unc-51-like autophagy activating kinase 1 (ULK1) pathway during cardiac microvascular ischemia/reperfusion injury, resulting in attenuated mitochondrial apoptosis, stabilization of mitochondrial membrane potential, and improvement of cardiac microvascular endothelial cells (CMECs) function and microvascular structure [18]. In a hyperglycemic state, the knockdown of phosphoglycerate mutase family member 5 (PGAM5) upregulates prohibitin2 expression and ameliorates diabetic myocardial dysfunction by upregulating mitophagy-related genes (PARKIN; BNIP3; and FUNDC1) and suppressing the expression of mitochondrial fission-related genes [Dynamin-related protein 1 (Drp1), mitochondrial fission protein 1 (FIS1), and mitochondrial fission factor (MFF)] [19]. The latest work has indicated that levels of FUNDC1 are overtly elevated in the hearts of diabetic patients compared to those without diabetes. Under hyperglycemia, cardiomyocyte-specific knockdown of FUNDC1 inhibits the formation of mitochondria-associated endoplasmic reticulum membranes induced by diabetes, reduces mitochondrial Ca2+ flux, and improves mitochondrial function and cardiac function [20].
Diabetic retinopathy has a complex etiology and a slow progression, in which endothelial cell injury is the key factor in hyperglycemia damage. In endothelial cell injury, crosstalk is evident for endothelial cells with inflammatory cells and pericytes [21]. The process of mitophagy, an endogenous system that protects primary human retinal endothelial cells from the adverse effects of high glucose, is one of the mechanisms contributing to the slow onset of diabetic retinopathy [22, 23]. Nevertheless, prolonged exposure to high glucose microenvironments has been demonstrated to affect mitochondrial metabolism, which leads to endothelial dysfunction. High glucose increases mitochondrial fission and mitochondrial damage through activation of the protein kinase C δ (PKCδ)/Drp1 signaling, which subsequently inhibits formation of LC3B-II and p62 degradation of p62, thereby inhibiting mitophagy by downregulating PINK1/PARKIN signaling pathway. This process ultimately leads to retinal microvascular damage in diabetic retinopathy rats and retinal microvascular endothelial cells (RMECs) dysfunction [24]. G-protein-coupled bile acid receptor (TGR5), as one of the major bile acid receptors, reduces mitochondrial fission by inhibiting Ca2+/PKCδ/Drp1 signaling and enhances mitophagy by upregulating PINK1/PARKIN signaling pathway, attenuating RMECs dysfunction and alleviating the progression of diabetic retinopathy angiopathy [25]. The herbal medicine scutellarin enhances mitophagy and protects vascular endothelial cells from hyperglycemia-induced injury by upregulating the PINK1/PARKIN signaling pathway [26]. In contrast, db/db mice had significantly increased Beclin1, PARKIN, and PINK1 expression in the thoracic aorta and activated mitophagy. Treatment of db/db mice with salvianolic acid B for 6 weeks significantly reduced the expression of Beclin1, PARKIN, and PINK1 in thoracic aorta, which ameliorated vascular endothelial dysfunction in diabetic mice [27].
Epigenetic modifications are heritable changes that regulate gene expression without changing the DNA sequence, such as DNA modifications, histone modifications, and non-coding RNAs (ncRNAs), which play a role in autophagy regulation in the development of diabetic vascular disease [28].
Histone acetylation, one of the widely studied epigenetic modifications, is co-regulated by histone deacetylase (HDAC) and histone acetyltransferase, which affects gene expression and plays an important role in the pathological process of diabetic vascular diseases [29]. Levels of caveolin-1, histone H3 lysine 9, and histone H3 lysine 56 were greatly increased in the vascular wall of patients with type 2 diabetes mellitus (T2DM), while levels of microtubule-associated protein 1 light chain 3 beta (LC3B) and Sirt6 were downregulated. Specific overexpression of Sirt6 in aortic endothelial cells of diabetic ApoE–/– mice downregulated the level of acetylated caveolin-1, induced autophagy, increased the expression of aortic LC3B, and delayed the progression of hyperglycemia-induced atherosclerosis. Activation of Sirt6 serves as a novel therapeutic strategy for diabetic atherosclerosis [30]. Overtly elevated renal function-related indexes (serum creatinine, and uric acid) and significant renal tissue fibrosis were found in db/db mice. Rutin, as a widely available bioflavonoid, displays biological effects of hypoglycemic, anti-inflammatory, and antioxidation [31]. Rutin treatment improved renal function in db/db mice and inhibited HDAC1 by suppressing the phosphatidylinositol 3-kinase (PI3K)/protein kinase (Akt)/mammalian target of rapamycin (mTOR) signaling pathway, thereby restoring autophagy and attenuating renal endothelial-to-mesenchymal transition [32]. Oxidative stress induced decreased HDAC6 expression in human umbilical vein endothelial cells (HUVECs), leading to increased autophagy and impaired endothelial migration and adhesion functions. While GLP-1 is a common hypoglycemic agent with anti-autophagic property, activation of GLP-1 was found to decrease EKR1/2 phosphorylation and upregulate HDAC6 expression, leading to suppressed oxidative stress-induced autophagy in endothelial cells and endothelial dysfunction [33].
ncRNAs, including microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs), have been identified to play an essential role in vascular injury and autophagy in diabetic patients (Table 1). miRNAs are widespread single-stranded ncRNAs with a length of approximately 20–25 nucleotides [34]. Increased expression of miR-32 in macrophages and their extracellular vesicles, stimulated by hyperglycemia, inhibits autophagy in SMCs and promotes arterial calcification in T2DM mice through the myocyte enhancer factor 2D/cGMP/PKG signaling pathway [35]. Downregulation of miR-199a-3p and miR-130a in peripheral blood is associated with developing diabetic vasculopathy in T2DM patients. Compared to the healthy population, there was a downregulation of miR-199a-3p expression observed in the peripheral blood of T2DM patients. miR-199a-3p overexpression could activate the PI3K/AKT/nuclear transcription factor-κB (NF-κB) signaling pathway, thereby inhibiting the apoptosis of HUVECs induced by high glucose and promoting the autophagy, proliferation, and migration of HUVECs [36]. miR-130a was downregulated in the peripheral blood of patients with T2DM. Overexpression of miR-130a promotes Bcl-2 expression and thus improves endothelial progenitor cells (EPCs) apoptosis by regulating runt-related transcription factor 3 upregulation, which subsequently reduces Beclin1 expression and the number of autophagosomes and promotes EPCs survival [37].
The role of ncRNAs in the regulation of autophagy in diabetic vascular diseases
| ncRNAs | Expression | Autophagy related genes | Autophagy | Result | Refs |
|---|---|---|---|---|---|
| miR-32 | Up | ATG5, Beclin1, p62 | Inhibition | Promoting arterial calcification in T2DM mice | [35] |
| miR-199a-3p | Down | LC3B-II | Inhibition | Decreasing proliferation and migration of HUVECs | [36] |
| miR-130a | Down | Beclin1 | Promotion | Inducing inflammation and apoptosis of EPCs | [37] |
| circMKLN1 | Up | LC3B-II, Beclin1, p62 | Promotion | Inducing retinal microvascular leakage in diabetic mice | [38] |
| circADAM9 | Up | LC3-II/LC3-I, p62, ATG7 | Promotion | Inducing proliferation and migration of EPCs | [39] |
| lncRNA TUG1 | Down | LC3-II/LC3-I | Inhibition | Decreasing EPCs migration, invasion, and tube formation | [41] |
| lncRNA LYPLAL1-DT | Down | SIRT1, LC3-II/LC3-I | Promotion | Triggering inflammation and apoptosis | [42] |
| lncRNA THRIL | Up | ATG4D, LC3-II/LC3-I, Beclin1 | Promotion | Inducing proliferation and migration of RMECs | [43] |
ATG4D: autophagy-related gene 4D; EPCs: endothelial progenitor cells; HUVECs: human umbilical vein endothelial cells; LC3B: microtubule-associated protein 1 light chain 3 beta; T2DM: type 2 diabetes mellitus; RMECs: retina microvascular endothelial cells
circRNAs are ncRNAs with a covalent closed-loop structure. Novel pathways that regulate autophagy were found to be involved in the regulation of microvascular dysfunction in diabetic mice, and retinal microvascular leakage in diabetic mice could be ameliorated by inhibiting the expression of circMKLN1. In vitro research has demonstrated that circMKLN1 adsorbed miR-26a-5p as a molecular sponge to target Rab11a and promote elevated autophagy levels in RMECs under high glucose stimulus [38]. In addition, circADAM9 acts as a miRNA sponge for mir-20a-5p and interacts with mir-20a-5p to upregulate levels of PTEN and ATG7, inhibit the phosphorylation of AKT and mTOR, and induce autophagy and apoptosis in high glucose-induced EPCs [39]. lncRNAs are ncRNAs with a length of more than 200 nucleotide sequences. Recently several lncRNAs were identified to play a role in the development of vascular injury in diabetes [40]. The migration, invasion, and tubulogenesis of EPCs were downregulated under the stimulation of high glucose. In contrast, overexpression of lncRNA taurine upregulated gene 1 (TUG1) ameliorated angiogenic dysfunction caused by high glucose by upregulating miR-29c-3p to activate platelet-derived growth factor-BB (PDGF-BB)/Wnt pathway and upregulate LC3 protein expression to foster autophagy [41]. lncRNA LYPLAL1-DT was overtly downregulated in leukocytes from T2DM patients with macrovascular complications in comparison with healthy controls. lncRNA LYPLAL1-DT attenuates endothelial cell apoptosis through downregulation of miR-204-5p, upregulation of SIRT1, and inhibition of autophagy [42]. Under the challenge of high glucose, elevated expression of lncRNA THRIL inhibited miR-125b-5p and enhanced autophagy-related gene 4D (ATG4D) expression, increased LC3-II/LC3-I ratio, and Beclin1 protein levels, leading to facilitated autophagy and RMECs proliferation and migration. To this end, lncRNA THRIL could be a potential target for the treatment of diabetes retinopathy [43]. These findings suggest an essential role for ncRNAs in the regulation of vascular injury in diabetes. Therefore, targeting ncRNAs to regulate the functional state of autophagy should provide new treatment options for this devastating complication in diabetes.
Apoptosis, a type of programmed cell death, serves as a key pathological feature in diabetic vascular injury. While autophagy is an important regulatory mechanism, the role of interaction between autophagy and apoptosis in diabetic vascular injury remains to be explored [44] (Figure 1).
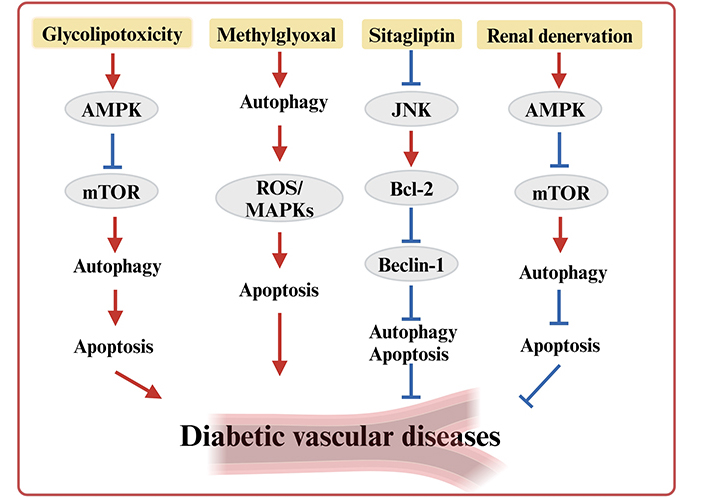
Interaction between autophagy and apoptosis in diabetic vascular diseases. AMPK: AMP-activated protein kinase; JNK: c-Jun N-terminal kinase; mTOR: mammalian target of rapamycin; MAPK: mitogen-activated protein kinase; ROS: reactive oxygen species
Methylglyoxal (MGO) serves as a precursor of advanced glycation end products, and the accumulation of MGO is associated with the pathogenesis of vascular injury in diabetes [45]. Recent studies have shown that MGO triggers DNA damage in HUVECs, which activates p53, inhibits downstream mTORC1, and induces autophagy [46]. After activation of autophagy induced by MGO, it promotes endothelial cell apoptosis by activating reactive oxygen species (ROS)/MAPKs signaling cascade and inhibits angiogenesis by activating the mTOR/Akt pathway, leading to aortic endothelial cells dysfunction [47]. Sitagliptin is not only a hypoglycemic agent. It also has a protective role in the vascular endothelium. Sitagliptin reduces the expression of aortic autophagy in diabetic rats by inhibiting the expression of LC3-II and Beclin1 and increasing the expression of Bcl-2 to prevent hyperglycemia-induced apoptosis and attenuate hyperglycemia-induced damage to vascular endothelial cells [48]. Glycolipotoxicity activates autophagy via increasing AMPK phosphorylation and preventing mTOR phosphorylation, resulting in a significant amount of autophagosomes, which induces apoptosis and impairs endothelial cells’ function [49]. On the contrary, renal denervation (RDN) as a new treatment for hypertension, RDN improves endothelial dysfunction in T2DM rats by activating AMPK phosphorylation, inhibiting mTOR phosphorylation, and inducing autophagy [50]. Mammalian sterile 20-like kinase 1 (Mst1) acts as a regulator of apoptosis and autophagy, and Mst1 knockdown improved cardiac microvascular structure and cardiac contractile function in diabetic mice. In the hyperglycemic microenvironment, Mst1 knockdown upregulated Sirt1 expression, increased LC3 expression, and enhanced autophagy, thereby reducing apoptosis in CMECs [51].
The inflammatory response is a common physiological and pathological event within the body. Inflammation-related genes are related to the pathogenesis of diabetic vascular diseases and autophagy function, among which nucleotide-binding oligomerization domain-like receptors protein 3 (NLRP3) inflammasome gained much attention in recent years. Quercetin ameliorates hyperglycemia-induced retinal angiogenesis by inhibiting NLRP3 inflammasome and downstream factors (IL-1β, IL-18) and autophagy signaling [52]. In contrast, kakonein activates autophagy in an AMPK-dependent way, significantly increases LC3-II expression, and decreases p62 expression, which reduces NLRP3 inflammasome activation and restores hyperglycemia-induced disruption of vascular endothelial junctions [53]. High glucose stimulates increased levels of ROS and inflammatory factors (TNF-α, IL-1β, and IL-6) in RMECs. Stachydrine promotes autophagy and alleviates high glucose-induced inflammation by activating the AMPK/SIRT1 pathway [54]. In diabetic mice, autophagy attenuates aortic calcification by reducing matrix vesicle body-mediated IL-1β release [55]. Specific knockdown of mTORC1 in vascular endothelial cells of diabetic mice with peripheral arterial disease activated autophagy by enhancing LC3-II level and downregulating p62 protein, resulting in alleviation of mitochondrial ROS production and levels of endothelial inflammation (TNF-α, IL-6, and IL-1β), ultimately promoting improved endothelial integrity, angiogenesis, and protection against hindlimb ischemia in diabetic mice [56].
In summary, the interactions among apoptosis, inflammation, and autophagy may participate in multiple pathways and molecular targets in diabetic vascular diseases, while their complex relationships remain to be elucidated by further studies.
Taken together, accumulating evidence has demonstrated the complexity of autophagy in diabetic vascular injury likely through a rather intricated interplay among epigenetic modification, apoptosis, and inflammation. Given the discrepant roles of autophagy in different tissues, further studies are needed to reveal whether the complex role of autophagy is related to specific cell types. The thorough understanding of the regulatory mechanisms of autophagy in diabetic vascular injury may help to develop safe and effective therapeutic targets.
EPCs: endothelial progenitor cells
HUVECs: human umbilical vein endothelial cells
MGO: methylglyoxal
NLRP3: nucleotide-binding oligomerization domain-like receptors protein 3
RMECs: retinal microvascular endothelial cells
ROS: reactive oxygen species
T2DM: type 2 diabetes mellitus
YA: Writing—original draft, Writing—review & editing. JR: Writing—review & editing.
The authors declare that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
No original data were involved in this work.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

