
Abstract
The renin-angiotensin system (RAS) is a key regulator of blood pressure and electrolyte homeostasis. Besides its importance as regulator of the cardiovascular function, the RAS has also been associated to the modulation of higher brain functions, including cognition, memory, depression and anxiety. For many years, angiotensin II (Ang II) has been considered the major bioactive component of the RAS. However, the existence of many other biologically active RAS components has currently been recognized, with similar, opposite, or distinct effects to those exerted by Ang II. Today, it is considered that the RAS is primarily constituted by two opposite arms. The pressor arm is composed by Ang II and the Ang II type 1 (AT1) receptor (AT1R), which mediates the vasoconstrictor, proliferative, hypertensive, oxidative and pro-inflammatory effects of the RAS. The depressor arm is mainly composed by Ang-(1-7), its Mas receptor (MasR) which mediates the depressor, vasodilatory, antiproliferative, antioxidant and anti-inflammatory effects of Ang-(1-7) and the AT2 receptor (AT2R), which opposes to the effects mediated by AT1R activation. Central Ang-(1-7) is implicated in the control of the cardiovascular function, thus participating in the regulation of blood pressure. Ang-(1-7) also exerts neuroprotective actions through MasR activation by opposing to the harmful effects of the Ang II/AT1R axis. This review is focused on the expression and regulation of the Ang-(1-7)/MasR axis in the brain, its main neuroprotective effects and the evidence regarding its involvement in the pathophysiology of several diseases at cardiovascular and neurological level.
Keywords
Angiotensin-(1-7), Mas receptor, brain, neuroprotectionThe renin-angiotensin system
The renin-angiotensin system (RAS) constitutes one of the most important systems in the control of the cardiovascular function. The RAS is a key regulator of blood pressure and electrolyte homeostasis. RAS cascade begins with the secretion of renin from the juxtaglomerular cells into the circulation, which triggers the systemic production of a pressor peptide, angiotensin (Ang) II [1]. The hepatocytes synthesize and release angiotensinogen (AGT) into the circulation, which contains at its N-terminal end the decapeptide Ang I that is released by the interaction of AGT with circulating renin [2]. The angiotensin converting enzyme (ACE) is a dipeptidyl carboxypeptidase that catalyzes the hydrolysis of Ang I into Ang II [3]. For many years, Ang II has been considered the main bioactive component of the RAS. However, the existence of many other biologically active RAS components has currently been recognized, with similar, opposite, or distinct effects compared to Ang II. Generation of the different components of the RAS is presented in Figure 1. Ang-(1-12) is a 12 amino acid peptide present in plasma, heart, kidney and other tissues and derived from AGT from a renin-independent cleavage, as a precursor for Ang II formation independent from renin action. Like Ang II, Ang-(1-12) exerts a systemic pressor response [4]. Ang III is a biologically active peptide with similar actions to those of Ang II, formed from Ang II by the action of aminopeptidase A [5]. Ang A is an octapeptide derived from Ang II with an amino acid sequence that differs from Ang II only in the first amino acid: alanine instead of aspartic [6]. Like Ang II, Ang-(1-12), Ang III and Ang A cause vasoconstriction and smooth muscle cell proliferation by activation of Ang II type 1 receptor (AT1R) [7–9]. At the peripheral level, Ang III induces natriuresis and diuresis by acting on Ang II type 2 receptor (AT2R) [10].
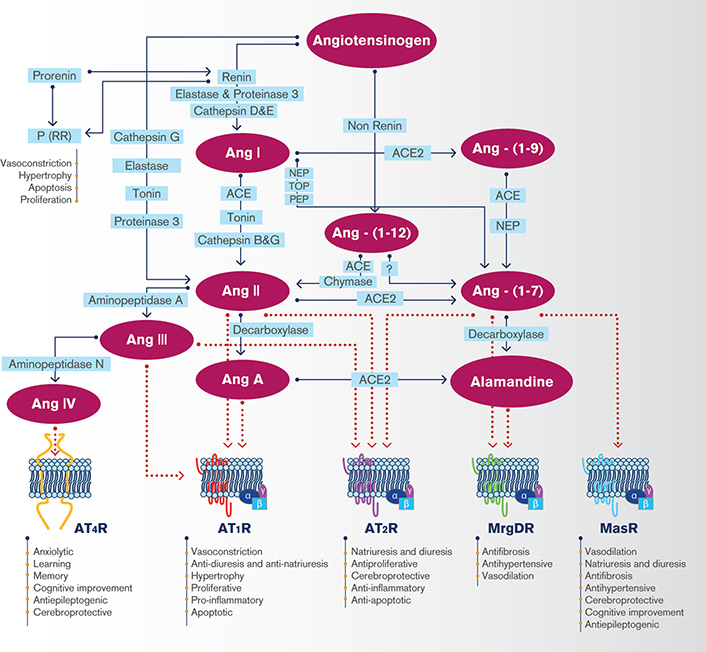
The brain renin-angiotensin system. AT4R: angiotensin type 4 receptor; MasR: Mas receptor; MrgDR: Mas-related G protein-coupled receptor member D; NEP: neutral endopeptidase (neprilysin); PEP: prolyl endopeptidase; (P)RR: prorenin receptor; TOP: thimet oligopeptidase
Other peptides like Ang IV, Ang-(1-7), Ang-(1-9) and alamandine exert protective effects opposite to those displayed by Ang II. Ang IV is synthetized from Ang III by the action of aminopeptidase N and can be generated from Ang II by the activity of aminopeptidase D [11]. Ang-(1-7) is a peptide formed by seven amino acids that lacks the phenylalanine residue at position 8 of the Ang II sequence. Ang-(1-7) can be formed from Ang I through the enzymatic activity of neutral endopeptidase (NEP), prolylendopeptidase, and thimet oligopeptidase, from Ang-(1-9) by ACE or NEP or from Ang II by an enzyme homologous to ACE named ACE2 [12]. ACE2 also acts on Ang I, although with an enzymatic efficiency 400 times lower, generating Ang-(1-9), which can be cleaved to Ang-(1-7) by ACE or NEP [13]. Both Ang-(1-7) and Ang II can, in turn, be generated from AGT through a renin-independent pathway, from Ang-(1-12) [12]. Alamandine is a heptapeptide that can be originated from Ang-(1-7) by decarboxylation at the N-terminal aspartic residue through an unknown yet enzymatic pathway or from Ang A by the action of ACE2 [14]. ACE2 is a multifaceted enzyme that negatively regulates systemic and local RAS function. Besides the generation of Ang-(1-7), ACE2 limits the production of Ang II and can act on other vasoactive peptides with important functions in the regulation of blood pressure, such as apelin or dynorphin [15]. ACE2 also participates in the signaling mediated by integrins in human hearts [16] and functions as the key receptor of severe acute respiratory syndrome (SARS) coronavirus [17]. Thus, ACE2 is implicated in the regulation of lung, cerebral, cardiovascular and renal function. Recently, ACE2 has gain a lot of attention, since this enzyme is the functional receptor for SARS-CoV-2, the etiological agent of coronavirus disease 2019 (COVID-19) [18].
The actual view is to consider the RAS as a system constituted by two opposite arms. The pressor arm is composed by Ang II and the AT1R, which mediates the vasoconstrictor, proliferative, hypertensive, oxidative, pro-inflammatory and profibrotic effects of the RAS [19]. The depressor arm of the RAS is mainly composed by Ang-(1-7), its receptor Mas (MasR) which acts as a mediator of the depressor, vasodilatory, antiproliferative, antioxidant, anti-inflammatory and antifibrotic effects of Ang-(1-7) and the AT2R, which opposes the effects mediated by AT1R activation [20]. The first evidence showing that Ang-(1-7) is the physiological agonist of MasR was based on the loss of intracellular signaling and effects of Ang-(1-7) and the reduced Ang-(1-7) binding to kidney sections in mice with genetic deletion of MasR [21]. Binding studies in Mas-transfected CHO cells demonstrated high affinity and specific binding of Ang-(1-7), which was not modified by an AT1R or AT2R antagonist, thus excluding Ang-(1-7) binding to these receptors [21]. It was recently reported that MasR is not an Ang-(1-7) receptor because Ang-(1-7) binding, which was performed with Ang-(1-7) labelled with a mixture of 125I: 127I (1:19), was not specific in tissue membrane preparations [22]. However, since the authors did not evaluate competitive binding in the presence of A-779 (MasR antagonist) [22], Ang-(1-7) specific binding to MasR cannot be excluded under these experimental conditions. In contrast, unpublished results from our lab (manuscript under preparation) showed that the specific 125I-Ang-(1-7) binding to - macrophages from a human monocytic leukemia cell line (THP-1) was specific with a Kd value around 250 nmol/L, demonstrating that at least in these cells MasR is a specific receptor for Ang-(1-7).
There is substantial evidence showing the existence of tissue RAS at renal, cardiac, adipose, arterial wall and cerebral level, among others, which are independently regulated from the systemic RAS [23]. According to Sigmund et al. [23], data supporting the existence of a RAS system in the brain can be classified into two categories. The first category is represented by evidence showing that all components of the RAS are synthesized de novo in the brain. All RAS essential constituents have been detected in the brain, including the substrates and enzymes necessary for the synthesis and metabolism of the different Angs as well as their specific receptors [24]. The second category is represented by evidence demonstrating the physiological functions of the central RAS. The brain RAS is a key regulator of blood pressure and electrolyte homeostasis and exerts a critical role in the development of the nervous system and cognitive processes [12, 19, 25]. The brain RAS is also involved in the pathogenesis of neurogenic hypertension and in the development of neurological disorders such as Alzheimer’s disease (AD), stroke, anxiety, depression and emotional stress [25–29].
Regarding the protector axis of the RAS, all its components have been detected in almost all cells of the brain (see the following section). Although there is extensive evidence showing the presence and functionality of central RAS in almost every species, it has been proposed that independent Ang generation in the brain is unlikely. Uijl et al. [30] could not detect renin levels that supported brain local synthesis. Van Thiel et al. [31] showed that Ang I was undetectable in brains of spontaneously hypertensive rats (SHRs), suggesting that there is no local Ang I production in the brain. The authors concluded that brain Ang II represents Ang II taken up from blood rather than locally generated Ang II. Circulating Ang II may gain access following situations where the blood-brain barrier (BBB) is disrupted, such as hypertension [32]. The biochemical and functional complexity of the RAS at central level, as well as the fact that the same detection methods applicable to the peripheral circulating system may not be applicable to the RAS in the brain may have contributed to originate this controversy.
The present review is focused on the expression of the brain Ang-(1-7)/MasR axis, its main cerebral effects and the evidence regarding its involvement in the pathophysiology of several diseases.
The RAS system in the brain
Unlike other tissues, the access of the peripheral RAS components to many of the brain regions is limited by the BBB. The existence of a BBB prevents, under physiological conditions, that circulating components of the RAS, including renin and Ang II, reach most of the brain areas, except for the circumventricular organs [33]. The RAS is present in different cell types within the brain, such as neurons, microglia, astrocytes and oligodendrocytes. The existence of an intracellular RAS with intracrine or autocrine functions has been widely described [34, 35]. Neurons present an intracellular RAS, mainly involving the mitochondria, nucleus and neurosecretory vesicles [35, 36].
Currently, only a limited array of methodologies is available to quantify Ang peptides at physiological concentrations [37]. For years, enzyme-linked immunosorbent assay (ELISA) and radioimmunoassay (RIA) have been the most used biochemical methods to quantify endogenous peptides [38]. One concern that arise when using these methodologies is antibody specificity, since Ang peptides in the RAS only differ by one or two amino acids, which can lead to cross-reactivity. This limitation in antibody specificity has raised the need to validate each antibody [39]. Over the last years, mass spectrometry (MS) has arisen as an important alternative for the development of assays designed to specifically identify Ang peptides [40]. More recently, a new technology was developed by coupling a laboratory-built capillary electrophoresis nanoelectrospray ionization platform to a high-resolution mass spectrometer, allowing the quantification of Ang peptides with high sensitivity [37].
The quantification of Ang-(1-7) and Ang II is challenging since endogenous levels of these peptides are quite low (concentrations in the fmol per gram or fmol per milliliter range) [38]. There is large discrepancy in the levels of Ang II and Ang-(1-7) reported in the literature. The differences found in Ang levels may be due to lack of proper standardization of the techniques used to obtain and process the samples, to extract and/or to measure Ang peptides in different tissues and in circulation. Most studies have reported human and animal plasma concentrations of Ang-(1-7) in the pmol/L range [41–44]. Using RIA-HPLC or HPLC-MS, Ang II and Ang-(1-7) were identified and quantified in the brain, with values in the fmol/g range [38, 42, 45–47]. Transgenic mice with elevated brain Ang II in the brain develop hypertension, highlighting the pathophysiological relevance of this local production of Ang II [48]. It has been proposed that Ang II may access to the brain under conditions where the BBB is partially disrupted, such as hypertension [30, 32]. Supporting this, Van Thiel et al. [31] showed that Ang I was undetectable by MS in brains of SHRs, suggesting that there is no local Ang I production in the brain. In this sense, the source of brain Ang II could be represented by circulating Ang II that binds to brain Ang receptors in conditions where the BBB integrity is compromised, such as deoxycorticosterone acetate (DOCA)-salt hypertension. It has been shown that DOCA-salt suppressed plasma and brain Ang II in parallel, while spironolactone simultaneously increased Ang II and lowered blood pressure, indicating that DOCA-salt hypertension is not mediated by brain RAS activation [49]. Contrary to this hypothesis, we have shown that Ang II levels were increased in brainstem neurons from SHRs compared with those from WKY animals. As this work was done using primary culture of neurons from brainstem, Ang II source could not be represented by circulating Ang II, since there was no blood [36].
The majority of Ang II central effects are mediated by the activation of AT1R [35]. AT1R is expressed in areas of the brain implicated in blood pressure control including the rostroventrolateral medulla (RVLM), caudoventrolateral medulla (CVLM), the nucleus of the solitary tract (NTS), the paraventricular nucleus (PVN) and the supraoptic nucleus (SON) of the hypothalamus [50]. AT1R hyperactivation exerts important actions in the promotion of vasoconstriction within the brain, exacerbating cognitive impairment, cell death and inflammation [34]. For further reading on Ang II and Ang receptors please refer to the reviews by Forrester et al. [19], Abiodun and Ola [26], Miller and Arnold [51], Cosarderelioglu et al. [29] and Ren et al. [32], since this review is focused on the expression and regulation of the Ang-(1-7)/MasR axis in the brain.
Renin and AGT
It has been shown that both renin and AGT are expressed in many regions of the brain [52]. Gene expression and enzymatic activity of renin in the brain has been reported [23]. One of the limitations that dampened the advance in understanding how the brain RAS is regulated is the fact that the expression levels of RAS genes, especially renin, are extremely low [52]. However, several strategies have been developed to overcome this obstacle, like large transgenes to increase renin expression levels, the knocked of Cre-recombinase into the renin locus and the use of reporter genes [23]. Using a double-reporter transgenic mouse model, Lavoie et al. [52] observed that renin and AGT are expressed in close proximity in the rostral ventrolateral medulla. Conditional gene knockout using Cre-recombinase has shown that renin mRNA transcription in the brain occurs at an alternative promoter compared to renin transcription in renal juxtaglomerular cells [53]. The brain-specific isoform of renin (called renin-b) differs from the classical isoform expressed in the kidney (called renin-a). Renin-b encodes a non-secreted form of prorenin. It has been proposed that there is a tight selective transcriptional regulation of renin-b and renin-a in the brain [53]. In neurons, renin is secreted as prorenin [35]. Brain prorenin binds with higher affinity to prorenin receptors (PRRs) compared to renin [54]. PRR stimulation by prorenin induces AGT cleavage and Ang II formation. In the central nervous system, PRRs are expressed mainly in neurons and to a lesser extent in astrocytes. In the regulatory nuclei of cardiovascular function of the brain stem and hypothalamus, PRRs are exclusively expressed in neurons, at the plasma membrane and cytosol level, and not in glial cells [55]. Renin-b has been postulated as a negative regulator of the RAS since selective deletion of this isoform is associated to hypertension and increased sympathetic nerve activity due to brain RAS activation [56]. It has also been shown that PRR is a key regulator of blood pressure that contributes to most of the Ang II generated in the brain [57, 58]. These results firmly support both Ang de novo synthesis and function in the brain. On the contrary, there is evidence suggesting that brain renin represents renin from blood, since brain tissue contains trapped blood when is homogenized [31]. Using a methodology based on the assessment of aliskiren-sensitive Ang I-generating activity (AGA) in homogenates from different brain regions, Van Thiel et al. [31], concluded that contamination with trapped blood would be the source of renin activity in mice brain tissue. The loss of AGA after buffer perfusion was considered as evidence that brain renin came from contamination of trapped blood in the brain. Thus, a controversy still exists regarding renin generation in the brain.
AGT is constitutively expressed in both glia cells and neurons [59]. The astrocytes have been identified as the main producers of brain AGT, although it has been shown that neurons also express this precursor [60]. Furthermore, it has been reported that AGT is secreted by pure rat neuronal cell cultures [61]. Evidence coming from functional studies firmly support AGT de novo synthesis in the brain [23]. Contamination with trapped blood seems less likely in this case, since several studies have identified brain AGT levels that do not run in parallel with those of plasma AGT [62]. Transgenic mice expressing an antisense targeting glial AGT of endogenous origin showed a 90% reduction in brain AGT protein expression, without changes in plasma levels of the precursor protein [63].
ACE2/Ang-(1-7)/MasR axis
ACE2 catalyzes Ang II degradation into Ang-(1-7) and has been identified within the brain, mostly in regions related to the control of the cardiovascular function [64]. ACE2 has been detected in the cytoplasm of neurons, but not in glial cells [64] and its gene expression has been shown to be present in primary cultures of rat cerebellum and brainstem astrocytes [65]. Furthermore, ACE2 is highly expressed at the mitochondrial membrane of neurons [66].
A large-scale immunohistochemical analysis of ACE2 expression levels using antibodies validated according to criteria established by the International Working Group for Antibody Validation showed that human brain ACE2 protein expression was below detection limits [67]. The results disagree with those recently obtained by Chen et al. [68], who found that ACE2 mRNA expression is relatively high in human neurons from the middle temporal gyrus and posterior cingulate cortex.
Neuronal ACE2/Ang-(1-7)/MasR axis exerts antioxidant and anti-inflammatory effects [66]. In this way, MasR signaling facilitates cognition and cell survival. ACE2 overexpression in astrocytes results in improved cognition through both antioxidant and anti-inflammatory mechanisms [69]. ACE2 deficiency had a pro-oxidant effect in the brain of mice and was associated to autonomic dysfunction in comparison to control animals [70]. Conversely, brain ACE2 activation has been associated to significant beneficial effects on cardiovascular function, cognitive processes, brain damage and development of the nervous system. These effects were mediated by the Ang-(1-7)/MasR axis since they were abolished by the MasR antagonist, A-779 [71].
Besides ACE2, other enzymes are involved in Ang-(1-7) synthesis and depend on the cerebral area. For instance, NEP is the major enzyme accountable for the generation of Ang-(1-7) in the rat hippocampus [72]. Increased levels of NEP were identified in the hippocampus of epileptic rats, in accord with the rise in Ang-(1-7) levels that had been shown in the hippocampus all through the acute and silent periods in a model of epilepsy induced by pilocarpine in rats [73].
Ang-(1-7) has been identified in brain areas related to blood pressure regulation and electrolyte balance [74]. Ang-(1-7) immunostaining has been detected mostly in hypothalamic PVN, SON, and suprachiasmatic nuclei, bed nucleus of the stria terminalis, substantia innominate, median eminence, and neurohypophysis [74–76]. Ang-(1-7) has also been identified in neurons located at the hypothalamus, brainstem, and PVN from rats [77, 78].
The receptor for Ang-(1-7), the MasR, has been detected in the hippocampus, amygdala, cortex, hypothalamus and brain stem [27]. Within the hypothalamus and brainstem, MasR is preferentially located in areas related to the control of cardiovascular function such as NTS, CVLM and RVLM, and in the hypothalamic PVN and SON [27]. In adult rat cerebral cortex, MasR was predominantly expressed in neurons and microglia, both in the cytoplasmatic and nuclear fractions. MasR was not identified in astroglia [27, 78]. In addition, MasR expression was increased in brainstem and hypothalamic neurons from SHRs compared to normotensive Wistar-Kyoto rats [36, 79]. Although a study by Burghi et al. [39] raised concerns regarding specificity of commercial antibodies against MasR, we successfully proved the specificity of the MasR antibody used in our own experiments. Antibody specificity was evaluated by measuring MasR expression by Western-blot in non-transfected and MasR-transfected HEK293T cells [36]. These findings reinforce the need to evaluate antibodies specificity, not only for each application and experimental model, but also regarding the expression levels of the protein target.
Ang-(1-7)/MasR actions in the brain
Central Ang-(1-7) acts at sites involved in the control of the cardiovascular function, mainly in regions associated to tonic and reflex regulation of blood pressure, like the hypothalamic nuclei and the dorsomedial and ventrolateral medulla. Central overexpression of Ang-(1-7) has been shown to induce predominantly parasympathetic cardiovascular effects, while blockade of the RAS depressor axis by administration of A-779, a specific MasR antagonist, predominantly promotes sympathetic responses [80]. Therefore, the Ang-(1-7)/MasR axis has a fundamental role in autonomic modulation [80].
Injection of Ang-(1-7) in the NTS, CVLM, and PVN or in the anterior hypothalamic area induces a reduction in arterial pressure [12]. The hypotensive effect of Ang-(1-7) in the hypothalamus and in the CVLM is dependent on the generation of nitric oxide (NO) [81, 82]. At this localization, cardiovascular effects of Ang-(1-7) are blocked by A-779, evidencing that brain Ang-(1-7) effects are associated to MasR activation [83]. On the contrary, the injection of Ang-(1-7) in the RVLM induces a rise in arterial pressure and this effect is mediated by the generation of superoxide anion [84].
Intracerebroventricular (ICV) Ang-(1-7) infusion for 4 days induced enhancement of the baroreflex control of the sympathetic activity at renal level [85]. This effect was similarly seen in transgenic mice with human ACE2 overexpression in the brain [86]. Central administration of Ang-(1-7) induces a reduction of blood pressure in hypertensive rats. Long-term infusion (14–28 days) of Ang-(1-7) induced a reduction in the increased arterial pressure in DOCA-salt rats [87], TGR(mREN2)L-27 hypertensive rats [88] and Ang II induced hypertensive rats [89]. Accordingly, increased ACE2 selective overexpression in the RVLM or PVN reduced high arterial pressure in SHR [90] and Ang II induced-hypertensive rats [86], respectively.
The activation of MasR by Ang-(1-7) at central level is not only related to the regulation of the cardiovascular function. It has been shown that Ang-(1-7), through MasR, stimulates synaptic plasticity in rat hippocampus [91] and lateral amygdala, participating in the processes of learning and memory [92]. Ang-(1-7)/MasR axis stimulation is associated to neuroprotection. Ang-(1-7) through MasR stimulation elicits phospholipase A2 activation, arachidonic acid release and NO generation [12]. The Ang-(1-7)/MasR axis is also linked to stimulation of phosphatidylinositol 3-kinase/Akt, mitogen-activated protein kinase and protein kinase A (PKA) pathways [92]. Increased levels of NO, arachidonic acid and activation of the signaling pathways elicited by Ang-(1-7) promote neuronal survival [36]. The activation of the protective axis of the RAS has beneficial effects through improvements in the cerebral oxidative state and inflammation through the inhibition of brain nuclear factor-κB (NF-κB) [93]. The neuroprotector effects of Ang-(1-7)/MasR in cerebral ischemia are associated to a pro-angiogenic effect [69]. Ang-(1-7) also increases cerebral blood flow, which contributes to its neuroprotective effects [69]. The main mechanisms and signaling pathways implicated in the neuroprotection proposed for ACE2/Ang-(1-7)/MasR are presented in Figure 2.
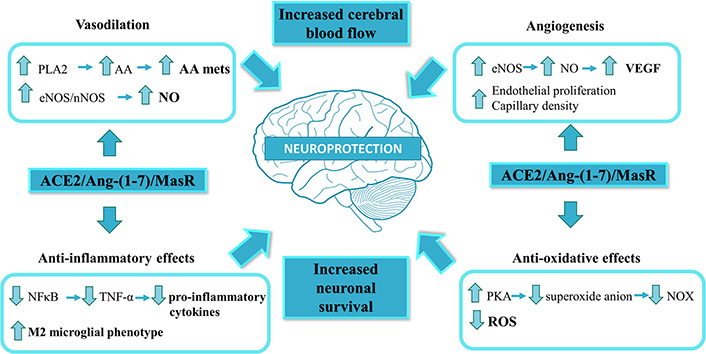
Main signaling pathways and mechanisms implicated in the ACE2/Ang-(1-7)/MasR axis-induced neuroprotection. The ACE2/Ang-(1-7)/MasR axis is known as the protective arm of the renin-angiotensin system. Ang-(1-7) through MasR stimulation elicits phospholipase A2 (PLA2) activation, arachidonic acid (AA) release and NO generation. Increased levels of NO, AA and the activation of PKA pathway promote neuronal survival. The production of NO and AA can cause vasodilation, while activation of endothelial nitric oxide synthase (eNOS), in addition to being involved in vasodilation, is associated to angiogenesis. The protective axis of the RAS has beneficial effects through improvements in the cerebral oxidative state and inflammation through the inhibition of NF-κB. Ang-(1-7) through MasR participates in the reduction of superoxide anion in the nucleus and mitochondria which in turn allows the inhibition of NOX, thus allowing greater neuronal survival and decreased oxidative stress. The ACE2/Ang-(1-7)/MasR axis is also involved in the change of macrophage population to an anti-inflammatory phenotype. AA mets: AA metabolites; nNOS: neuronal nitric oxide synthase; ROS: reactive oxygen species; VEGF: vascular endothelial growth factor
The existence of a close interaction between the RAS and other brain systems like dopaminergic, cholinergic and adrenergic systems has also been shown [34]. By interacting with other neurotransmitters, Ang-(1-7) might be involved in numerous brain processes, such as hormone synthesis, motor function and cognition. It has been shown that Ang-(1-7) influences the release of norepinephrine (NE), gamma aminobutyric acid (GABA), glutamate, substance P and dopamine at different regions of the brain, affecting in this way the effects elicited by these neurotransmitters [94–98].
Ang-(1-7) central effects on the regulation of cardiovascular function are related to variations in synaptic concentrations of NE. Ang-(1-7), counteracting Ang II effects, exhibits a sympathoinhibitory action [98]. Ang-(1-7) promotes a reduction in synaptic NE levels in neurons of the hypothalamus and brainstem of normotensive and SHRs. This effect is the result of a reduction in the release and synthesis of NE and a rise in its uptake by neurons [99]. Ang-(1-7) inhibition of NE release is mediated through NO generation by cyclic guanosine monophosphate/protein kinase G stimulation [100]. Besides inhibiting the release of NE, Ang-(1-7) reduces the protein expression of tyrosine hydroxylase, the enzyme that catalyzes the rate limiting step in the synthesis of catecholamines, by stimulating its degradation via ubiquitin-proteasome in hypothalamic neurons of normotensive and SHRs [78]. Ang-(1-7) also acts as a long-term stimulus to increase NE neuronal uptake. This effect is exerted by inducing the increase in the transcription and protein content of the NE transporter [79], which regulates NE levels in the synaptic cleft [101]. Ang-(1-7) stimulation of neuronal NE uptake is mediated by a mechanism dependent on Akt and extracellular signal-regulated kinase 1/2 (ERK1/2) activation [78]. It has been suggested that Ang-(1-7) could participate in the central motor control because it regulates the secretion of dopamine and GABA in the dorsal striatum [79]. Dopaminergic neurons exhibit a downregulation of the Ang-(1-7)/MasR axis with aging which could promote the aging-related vulnerability to neurodegeneration [66]. Brain Ang-(1-7) has also been linked to emotional stress and anxiety modulation [102]. MasR activation might be involved in these Ang-(1-7) induced actions, since knock out mice for MasR exhibited increased anxiety behavior [103]. On the other hand, central administration of Ang-(1-7) attenuated anxiety in rats with low brain AGT [104].
Central Ang-(1-7) effects are summarized in Table 1.
Central effects of Ang-(1-7)/MasR
| Ang-(1-7) induced effect | References |
|---|---|
| Decrease in blood pressure mediated by NO | Feng et al., 2010 [105] |
| Decrease in NE release | Gironacci et al., 2004 [99] |
| Decrease in NE synthesis | Lopez Verrilli et al., 2009[78] |
| NE transporter upregulation | Lopez Verrilli et al., 2012[79] |
| Angiogenesis | Goldstein et al., 2016 [106] |
| Decrease in BBB permeability | Wu et al., 2015 [107] |
| Protection of neurons and oligodendrocytes | Rabie et al., 2018 [108] |
| Decrease in infarct size | Mecca et al., 2011 [109] |
| Improvement in neurological deficits induced by stroke | Jiang et al., 2013 [28] |
| Promotion of cell survival and healthy synapse formation | Costa-Besada et al., 2018 [66] |
| Decrease in superoxide production in nucleus and mitochondria of neurons of the substancia nigra | Costa-Besada et al., 2018 [66] |
| Increase in vasopressin release | Qadri et al., 1998 [110] |
| Increase of BDNF production | Rabie et al., 2018 [108] |
| Increased neuronal survival due to reduction of oxidative stress mediated by PKA | Mo et al., 2019 [111] |
| Reduction in microglial activation and astrogliosis | Janatpour et al., 2019 [112] |
| Reduction in inflammation by inhibition of the MAPKs pathway | Moore et al., 2013 [113] |
| Increase of the M2 microglial anti-inflammatory phenotype in microglia | Liu et al, 2016 [114] |
| NOS upregulation | Cerrato et al., 2012 [82] |
| Reduction in oxidative stress | Jiang et al., 2013 [28]Costa-Besada et al., 2018 [66] |
| Stimulation of prostaglandin release in endothelial cells | Jaiswal et al., 1992 [115] |
| Stimulation of neuronal activity in hypothalamus and medulla | Fontes et al.,1994 [116] |
| Increased sensitivity to baroreflex | Campagnole-Santos et al., 1992 [117] |
| Increased IL-10 and decreased TNFα and IL1β | Sriramula et al., 2011[86] |
| Increase of arachidonic acid release | Cerniello et al., 2020 [36] |
| MAPKs and PKB/Akt activation | Rabie et al., 2018 [108] |
BDNF: brain derived neurotrophic factor; IL: interleukin; MAPK: mitogen-activated protein kinase; MCP-1: Monocyte chemoattractant protein-1; NOS: nitric oxide synthase; PKB: protein kinase B; TNFα: Tumor necrosis factor alpha
Ang-(1-7)/MasR axis and cardiovascular diseases
As previously mentioned, the Ang-(1-7)/MasR axis of the RAS has been found in various areas of the brain associated with the cardiovascular function like NTS, CVLM, RVLM and PVN of the hypothalamus [12]. In addition, MasR has been detected in autonomic preganglionic neurons, ganglia and terminals of the nerves [118], involved in neuromodulatory actions that can directly influence blood pressure control [102]. Chronically central Ang-(1-7) infusion decreases blood pressure in animal models of hypertension [119]. Ang-(1-7) ICV administration for 14 days reduced arterial pressure in transgenic hypertensive rats (mRen2)27 [88]. Ang-(1-7) also stabilized the baroreflex control of heart rate, reestablished cardiac autonomic balance, prevented cardiac hypertrophy, and reduced the altered ratio of Ang II/Ang-(1-7) in the heart [88]. Most of those actions were mediated by MasR, given the fact that they were abolished by its antagonist, A-779. Ang-(1-7) chronic ICV administration for 14 days also reduced the high blood pressure levels in DOCA-salt rats [87]. Central administration of Ang-(1-7) lowered arterial pressure levels, improved baroreflex bradycardia sensitivity indexes, restored the balance of cardiac autonomic tone and returned the levels of left ventricular mRNA expression of collagen type I to normal ones [87]. The augmentation of Ang-(1-7) levels in the brain chronically also induced beneficial cardiometabolic effects in rats fed a high fructose diet [120]. ICV administration of Ang-(1-7) for 4 weeks decreased cardiac sympathetic tone, stabilized blood pressure levels and restored baroreflex sensitivity. Ang-(1-7) also improved metabolic parameters like glucose and insulin levels, and improved glucose tolerance in rats with fructose overload. Ang-(1-7) chronic administration also diminished mRNA expression of neuronal nitric oxide synthase in hypothalamus and dorsomedial medulla [120]. ICV infusion of the MasR selective antagonist A-779 in female rats with aldosterone/NaCl induced hypertension aggravated the rise in blood pressure evidencing the considerable protective effects of Ang-(1-7) synthesized endogenously in situations of high blood pressure [89].
Ang-(1-7) administration into the basolateral amygdala in normotensive rats decreased the tachycardia and prevented the rise of arterial pressure provoked by acute emotional stress [121]. These actions were totally blocked by A-779, demonstrating the participation of MasR [121]. In contrast, Ang-(1-7) administration in the RVLM increased sympathetic nerve activity at renal level and the arterial pressure in two-kidney, one clip (2K1C)-operated rats [122]. Ang-(1-7) was also associated to enhanced cardiac sympathetic afferent reflex and sympathetic output in these rats by activation of MasR [122]. Ang-(1-7) administration in RVLM also increased blood pressure in animals with stress-induced hypertension, without differences in blood pressure levels in those animals in relation to their respective normotensive controls [123].
It must be pointed out that amounts of administered Ang-(1-7) exceed, by orders of magnitude, the expected concentrations of this peptide in tissues or in the blood. One reason to use a high concentration of Ang-(1-7) in pre-clinical studies is the short half-life of this peptide [124], as a way to assure that they reach the receptors in amount enough to measure a response. Using MS, we previously showed that Ang-(1-7) is degraded in hypothalamic and brainstem neurons from rats [78].
Ang-(1-7) effects in animal models of cardiovascular disease are summarized in Table 2.
Ang-(1-7) effects at central level in animal models of cardiovascular disease
| Animal model | Main finding | References |
|---|---|---|
| Transgenic hypertensive rats (mRen2)27 | ICV administration of Ang-(1-7) for 14 days:- decreased systolic and diastolic blood pressure- stabilized the baroreflex control of heart rate, reestablished cardiac autonomic balance, reversed cardiac hypertrophy, and reduced the altered ratio of cardiac Ang II/Ang-(1-7)- attenuated the rise in atrial natriuretic peptide, brain natriuretic peptide, collagen I, fibronectin and TGF-β cardiac expression | Kangussu et al., 2015 [88] |
| DOCA-salt hypertensive rats | ICV administration of Ang-(1-7) for 14 days:- decreased mean blood pressure- enhanced baroreflex bradycardia sensitivity indexes, improved the balance of cardiac autonomic tone and stabilized left ventricular mRNA expression of collagen type I | Guimaraes et al., 2012 [89] |
| Rats with fructose-induced metabolic syndrome | ICV administration of Ang-(1-7) for 4 weeks:- stabilized blood pressure levels- decreased cardiac sympathetic tone baroreflex sensitivity, reduced glucose and insulin levels and enhanced glucose tolerance | Guimaraes et al., 2014 [120] |
| Female rats with aldosterone/NaCl induced hypertension | - ICV infusion of the MasR selective antagonist A-779 exacerbated the increase in blood pressure | Xue et al., 2013 [89] |
| Two-kidney, one clip (2K1C)-operated rats with renovascular hypertension | - Ang-(1-7) injection into RVLM increased the renal sympathetic nerve activity and arterial pressure | Li et al., 2015 [122] |
| Rats with stress-induced hypertension | - Ang-(1-7) microinjection into RVLM increased blood pressure | Du et al., 2013 [123] |
TGF-β: Transforming growth factor beta
Ang-(1-7)/MasR axis in cerebral diseases
Stroke
It has been shown that Ang-(1-7) has a neuroprotective effect against stroke [93, 125]. Ang-(1-7) beneficial effects have been demonstrated in different animal models of stroke, such as endothelin1-induced middle cerebral artery occlusion (MCAO) [109], permanent MCAO [93, 126] and stroke-prone SHRs [127] (Table 3).
Ang-(1-7) in animal models of stroke
| Type of stroke induced | Animal model | Main finding | References |
|---|---|---|---|
| Ischemic stroke | Endothelin1-induced middle cerebral artery occlusion (MCAO) | - Ang-(1-7) central administration reduced the brain infarct size and neurological alteration 72 h after the insult.- Beneficial actions of Ang-(1-7) were mediated by MasR. | Mecca et al., 2011 [109] |
| Permanent middle cerebral artery occlusion (pMCAO) | - Intracerebral infusion of Ang-(1-7) decreased infarct volume and improved neurological alteration after 24 h following the insult.- Ang-(1-7) had an antioxidant effect, inhibited NF-κB activity and decreased pro-inflammatory cytokines and COX-2 in the peri-infarct areas.- Beneficial actions of Ang-(1-7) were mediated by MasR. | Jiang et al., 2012 [93] | |
| - Intracerebral infusion of Ang-(1-7) for 4 weeks reduced infarct size and neurological alteration after pMCAO.- Ang-(1-7) increased cerebral capillary density via stimulation of endothelial cell proliferation and eNOS activation.- Beneficial effects of Ang-(1-7) were mediated by MasR. | Jiang et al., 2014 [126] | ||
| - Ang-(1-7) infusion exerted a direct neuroprotective action during l ischemic injury in the brain by reducing cell swelling and death. These effects were mediated by decreased NOX expression with the consequent reduction of ROS production.- The beneficial effect of Ang-(1-7)/MasR stimulation on cerebral ischemic injury showed a greater protective effect in older animals.- Neuroprotective effects of Ang-(1-7) were mediated by MasR. | Zheng et al., 2014 [128] | ||
| Hemorrhagic stroke | Stroke-prone SHRs (spSHRs) | - ICV Ang-(1-7) infusion into spSHRs fed a high-sodium (4%) diet for 6 weeks decreased the number of hemorrhages in the striatum, enhanced neurological status, reduced the striatal microglia and increased survival.- Ang-(1-7) effects were partly prevented by ICV infusion of the MasR blocker, A-779. | Regenhardt et al., 2014 [127] |
| Intracerebral hemorrhage | - ICV infusion of Ang-(1-7) enhanced neurological function compared with rats that received control solution. | Del Bigio et al., 1996 [129] |
COX-2: cyclooxygenase 2; eNOS: endothelial nitric oxide synthase; NOX: NADPH oxidase; ROS: Reactive oxygen species
In models of ischemic stroke, Ang-(1-7) infusion decreased the area of infarct and the neurological alterations after the insult [93, 109, 126]. Ang-(1-7) protective effects in ischemic stroke are related to anti-inflammatory and anti-oxidative effects. In a stroke model of endothelin1-induced MCAO, Ang-(1-7) inhibited NF-κB activity and reduced the release of pro-inflammatory cytokines, such as interleukin (IL)-1b and IL-6 [93, 109]. Ang-(1-7) also showed an antioxidant effect and reduced cyclooxygenase-2 and inducible nitric oxide synthase gene expression in the peri-infarct regions of rats with permanent MCAO [93, 126]. These beneficial effects of Ang-(1-7) were blocked by pre-treatment with the MasR selective antagonist A-779, evidencing MasR participation. The beneficial effects of Ang-(1-7) in stroke are conserved and even increased in aged animals [128]. Ang-(1-7)-induced brain angiogenesis prevented the decrease of brain blood flow in a model of permanent MCAO, leading to improvements in stroke outcomes [126].
It has also been shown that Ang-(1-7) exerts neuroprotective actions through MasR activation by opposing to the detrimental effects of the Ang II/AT1R axis. Accordingly, high levels of Ang II have been detected in the brain during stroke [130]. Increased Ang II has been linked to vascular contraction, damage of the endothelium, neuronal apoptosis, inflammation, and increased levels of oxidative stress. All these effects are counteracted by Ang-(1-7)/MasR activation [69].
The protective effects of the Ang-(1-7)/MasR axis have also been shown in models of hemorrhagic stroke, including spSHR under a high-salt diet [127] and in rats with collagenase-induced intracranial hemorrhage [129]. In spSHR rats, central administration of Ang-(1-7) reduced the number of hemorrhages, increased the lifespan and had beneficial effects regarding the neurological status, attenuating cerebral inflammation [127]. In rats with collagenase-induced intracranial hemorrhage, Ang-(1-7) infusion improved neurological function compared with rats that received control saline solution [129].
All these findings highlight the importance of brain Ang-(1-7)/MasR axis in stroke prevention. In this sense, many strategies are being designed and evaluated to target the Ang-(1-7)/MasR axis in stroke. One suggested approach includes the use of an orally active Ang-(1-7) formulation, designed to protect Ang-(1-7) from breakdown in the stomach and small intestine. Oral administration of this formulation has shown antithrombotic effects, however more studies are needed to determine its benefits in the treatment of ischemic stroke [131]. Systemic administration of Ang-(1-7)-secreting stem cells in the following hours after a stroke has also been proposed [69]. This strategy is based on evidence showing that targeted delivery of ACE2 via endothelial progenitor cells reduced infarct volume and improved neurological deficits in a AGT transgenic (R+A+) mouse model [132].
AD
AD is a neurodegenerative disease characterized by memory loss following progressive impairment of cognition and personality alterations [133]. The formation of extracellular amyloid plaques composed of amyloid-β (Aβ) and the presence of intracellular neurofibrillary tangles formed by hyperphosphorylated tau protein constitute the two main pathological hallmarks involved in AD pathophysiology [134]. Besides these two mechanisms, the development of neuroinflammation exerts a fundamental role in the pathogenesis of the disease [135].
The brain RAS is implicated in the pathophysiology of AD through ACE/Ang II/AT1R hyperactivation, which leads to increased production of brain Aβ levels, tau phosphorylation, oxidative stress and pro-inflammatory processes [136]. The existence of increased expression of Ang II has been detected in postmortem brain tissue in the midfrontal cortex from AD patients [137]. The mechanisms proposed for Ang II induced accumulation and deposition of Aβ peptides in AD include increased amyloid precursor protein (APP) mRNA expression, β-secretase activity, and presenilin expression [136, 138]. It has also been proved that central Ang II increases the levels of hyperphosphorylated tau via glycogen synthase kinase 3β and other tau kinases [139].
Accumulating evidence indicates that dysregulation of the brain ACE2/Ang-(1-7)/MasR axis is also involved in the pathogenesis of AD. ACE2 activity was decreased about 50% in postmortem human brain tissue from AD patients [140]. ACE2 reduction was strongly associated to increased Aβ and phosphorylated tau levels. In addition, ACE2 enzyme activity correlated positively with albumin, a marker of BBB integrity, in cerebrospinal fluid from AD patients, suggesting RAS participation in relation to capillary damage in AD [141]. In Tg2576 mice, a recognized transgenic APP mouse model, augmentation of brain ACE2 activity inhibited cognitive impairment in young asymptomatic mice and improved cognitive function in aged mice [142].
Midfrontal cortex Ang II levels were significantly enhanced whereas Ang-(1-7) levels were unchanged, increasing Ang II/Ang-(1-7) ratio, in AD patients in comparison to age-matched controls [140]. In addition, Ang-(1-7) plasma levels from AD patients were lowered in relation to cognitive functioning compared with matched controls [141]. In accordance, Ang-(1-7) levels were significantly diminished in cerebral cortex and hippocampus in a mouse model of sporadic AD throughout disease evolution. Ang-(1-7) at central level negatively correlated with the levels of hyperphosphorylated tau [143]. Table 4 summarizes centrally ACE2/Ang-(1-7)/MasR expression in AD.
ACE2/Ang-(1-7)/MasR axis in AD
| Type of evidence (clinical or experimental) | Main finding | References |
|---|---|---|
| Human (postmortem human brain tissue from AD patients) | - ACE2 activity was decreased by about 50%- ACE2 reduction strongly associated to increased Aβ and phosphorylated tau levels | Kehoe et al., 2016 [140] |
| Human (cerebrospinal fluid from AD patients) | - ACE2 increased in association with aging in the control group but not in AD- ACE2 enzyme activity correlated positively with albumin, a marker of blood-brain barrier integrity | Kehoe et al., 2019 [141] |
| Human (postmortem human brain tissue from AD patients) | - Increased Ang II/Ang-(1-7) ratio in AD in comparison to age-matched controls | Kehoe et al., 2016 [140] |
| Human (plasma from AD patients) | - Decreased Ang-(1-7) plasma levels in AD compared with matched controls- Ang-(1-7) plasma levels were positively correlated to cognitive performance | Jiang et al., 2016 [143] |
| Animal (mouse model of sporadic AD) | - Reduced Ang-(1-7) levels in cerebral cortex and hippocampus during disease progression- Ang-(1-7) levels had a negative correlation with hyperphosphorylated tau | Jiang et al., 2016 [144] |
| Animal (established transgenic APP mouse model, Tg2576 mice) | - Enhancement of brain ACE2 activity lowered hippocampal Aβ and improved cognition- ACE2 enhancement restored hippocampal MasR levels | Evans et al., 2019 [142] |
All these findings support the fundamental role of the protective axis of the RAS in the pathophysiology of AD. ACE2/Ang-(1-7)/MasR axis opposes to the actions of the pressor axis of the RAS, with beneficial effects on cognitive processes such as memory and learning, which may be considered as a novel therapeutic target for potential treatment of AD.
Anxiety disorders
Brain Ang-(1-7)/MasR axis has strongly been associated to anxiety modulation [102]. Many studies have proven that Ang-(1-7) can mitigate the physiological responses to emotional stress at central level [121, 145]. The psychotropic activity of Ang-(1-7) was one of the first biological actions described for this compound [146]. Ang-(1-7) ICV administration increased the time spent and number of entries in the open arms of the elevated plus maze, enhanced the head-dipping behavior in open arms and decreased stretching in closed arms. All these effects were linked with an anxiolytic action of Ang-(1-7) [146]. It has been shown that Ang-(1-7) anxiolytic effects are related to components of the limbic system, comprising the amygdala, hippocampus and hypothalamus [102]. Ang-(1-7) administration to the basolateral amygdala decreased the pressor response in a model of stress induced by air jet [121]. Central Ang-(1-7) injection prevented the anxiety symptoms in two different transgenic models. Transgenic rats with low brain AGT demonstrated anxiety-like behavior in association to a depression-like state. Ang-(1-7) attenuated the anxiety and depressive-like behavior in this animal model [104]. Ang-(1-7) also exerted an anxiolytic action in transgenic hypertensive (mRen2)27 rats. Pre-treatment with A-779, prevented the change of anxiety- and depression-like behavior exerted by Ang-(1-7), evidencing MasR participation [147]. These results are in accordance with a previous study showing that knock out Mas–/– mice exhibited increased anxiety behavior [148]. On the other hand, male mice with ACE2 overexpression investigated the open arms of the elevated plus maze a higher number of times in comparison to wild-type mice, suggesting an anxiolytic effect related to ACE2 activity [149]. Administration of a MasR antagonist prevented the anxiolytic phenotype, evidencing one more time the participation of MasR in the central actions of Ang-(1-7). A centrally delivered ACE2 activator to C57BL/6 mice also prevented the symptoms of anxiety, reinforcing the evidence regarding an anxiolytic effect related to the activity of this enzyme [140]. Ang-(1-7) effects on modulation of stress behavior and anxiety are summarized in Table 5.
ACE2/Ang-(1-7)/MasR axis on stress behavior and anxiety
| Central area | Animal model | Main effects | Reference |
|---|---|---|---|
| Cerebral ventricles | Ang-(1-7) acute injection to rats | - First evidence of an anxiolytic action of Ang-(1-7) | Holy et al., 1992 [146] |
| TGR(ASrAOGEN)680R rats (rats with low brain AGT, expressing an antisense RNA against AGT in glial cells) | - Ang-(1-7) acute injection prevented the anxiety- and depressive-like behavior in transgenic rats | Kangussu et al., 2013 [104] | |
| Conscious male Wistar rats with induced tachycardia by acute stress (air jet 10 l/min) | - Ang-(1-7) acute injection decreased by ~45% the tachycardia induced by emotional stress | Lima et al., 2013 [83] | |
| Transgenic hypertensive (mRen2)27 rats | - Ang-(1-7) infusion promoted anxiolytic effects- Beneficial effects of Ang-(1-7) were mediated by MasR | Almeida Santos et al. 2016 [147] | |
| C57BL/6 mice with enhanced ACE2 activity by central delivery of diminazene aceturate (ACE2 activator) | - ACE2 activation reduced anxiety-like behavior- Beneficial effects of ACE2 activation were mediated by MasR | Wang et al. 2016 [149] | |
| Amygdala | Adult male Wistar rats | - Ang-(1-7) acute injection induced anxiolytic effects- Ang-(1-7) increased GPx specific activity and decreased the main peroxidation marker MDA- The majority of the behavioral parameters correlated with markers of oxidative stress | Bild et al., 2013 [145] |
| Wistar rats submitted to stress trials | - Ang-(1-7) prevented the tachycardia and the pressor response induced by acute stress- Beneficial effects of Ang-(1-7) were mediated by MasR | Oscar et al., 2015 [121] | |
| Hippocampus | Mas–/– mice | - Mas-deficient animals showed increased anxiety | Walther et al., 1998 [148] |
GPx: glutathione peroxidase; MDA: malondialdehyde
Many mechanisms have been suggested to explain the contribution of the ACE2/MasR/Ang-(1-7) axis to the mitigation of the biological response to emotional stress [102]. Some of these mechanisms include the modulation of synaptic plasticity at different brain regions that participate in the stress response such as amygdala and hippocampus [92, 93], the attenuation of the sympathetic outflow by inhibition of the release of NE in the hypothalamus [78], the reduction of the increased levels of hormones that modulate stress such as Ang II and corticosterone as well as the restoration of serotonin and dopamine levels in brain areas implicated in the stress response [150].
Hypertensive patients with comorbid depression showed reduced depressive symptoms after treatment with the ACE inhibitor captopril [151], which has been related to increased Ang-(1-7) levels [152]. Recombinant human ACE2 could be proposed as a novel therapeutic target in the regulation of stress and anxiety behavior, since it has been proved that its cardioprotective effects are mediated by Ang-(1-7) [153]. The stimulation of the ACE2/Ang-(1-7)/MasR axis has been associated to many benefits in modulation of emotional stress and anxiety, however, more studies are needed to confirm these findings at clinical level.
MasR regulation
G protein-coupled receptor (GPCR) can trigger signaling pathways at dynamic nanodomains localized both on the cell membrane and on membranes of intracellular compartments. Lateral diffusion and trafficking influence the place where GPCRs are located within the nanodomains, thus modulating in this way the timing and place of GPCR signaling [154, 155]. Trafficking of GPCR provides distinct signaling platforms critical for specifying receptor function. Upon agonist stimulation most GPCRs are internalized into early endosomes, from where they can activate others signaling pathways, leading in this way to biological responses different from those induced from the plasma membrane. Thus, GPCRs can have differential signaling consequences depending on their subcellular localization [156]. Once internalized, GPCR may be recycled back to the cellular membrane, be directed to lysosomes for degradation or be translocated to other organelles, like the nucleus or mitochondria [155, 157]. MasR belongs to GPCR family [12]. After being stimulated with Ang-(1-7), MasR is internalized into early endosome. MasR induces Akt and ERK1/2 activation from early endosomes, and then it returns to the plasma membrane via slow recycling vesicles in MasR-expressing HEK293T cells [158]. This way of internalization was also detected in brainstem neurons from normotensive Wistar-Kyoto rats [36]. However, in brainstem neurons from SHR, MasR shows a differential trafficking: the proportion of MasRs that were internalized into early endosomes was increased and the proportion of MasRs recycled back to the plasma membrane was reduced in SHR compared to WKY brainstem neurons. Surprisingly, in SHR neurons but not in WKY neurons, Ang-(1-7) promoted MasR translocation to the nucleus [36]. Up to now there is no evidence about the biological response induced by MasR translocation to the nucleus of neurons from SHR.
Most of GPCRs display changes in its functionality due to interaction with other receptors [159]. MasR has been shown to heteromerize with AT1R, reducing the synthesis of inositol phosphates and utilization of intracellular calcium stimulated by Ang II [160]. MasR-AT2R heteromerization induced CX3CR1 mRNA expression in cultured astrocytes from mice [161] and an increase in NO generation as well as diuretic and natriuretic responses in obese Zucker rat kidney [162]. In addition, MasR-B2R interaction was associated to a delay in sequestration of MasR from the cell membrane and to an increased affinity ligand binding for MasR [163]. Thus, biological responses mediated by MasR stimulation result from MasR trafficking as well as from interaction with other receptors.
Clinical evidence
The existence of RAS alterations in patients with neurodegenerative and neuropsychiatric diseases such as AD, PD and schizophrenia (SCZ) has been proved. Studies in humans showed that Ang-(1-7) plasma levels from AD patients were lowered in relation to cognitive function compared with matched controls [143]. Accordingly, a recent study showed that ACE activity was elevated in cerebrospinal fluid (CSF) of AD patients [144]. PD patients exhibited lower plasma levels of Ang-(1-7) compared to controls, in association with increased severity of depressive symptoms [164]. ACE activity was also increased in PD patients [165]. In fact, ACE gene may be associated to genetic susceptibility to PD, mainly in older individuals [166]. In patients with SCZ, an association between increased ACE activity in plasma and cognitive deficits was also suggested [167].
Clinical evidence shows that the blockade of the pressor arm of the RAS with an ACE inhibitor (ACEi) or an Ang receptor blocker (ARB) is associated with improvement in cognitive impairment. Elderly patients treated with centrally active ACEi such as captopril exhibited a reduced rate of cognitive decline [168]. ARBs such as losartan and valsartan also improved cognitive dysfunction and dementia [169]. The Perindopril Protection Against Recurrent Stroke Study (PROGRESS) was a randomized, double-blind and placebo-controlled clinical trial which showed that treatment with the ACEi perindopril reduced the risk of post-stroke dementia [170]. In the Heart Outcomes Prevention Evaluation (HOPE) study, ramipril reduced the cognitive decline associated with stroke [171]. In the Antihypertensives and Vascular, Endothelial, and Cognitive Function (AVEC) study, the ARB candesartan was shown to be superior to hydroclorotiazide and lisinopril in preserving the executive function [172]. ARBs reduced the incidence and progression of AD and dementia when compared to ACEIs in patients with cardiovascular disease older than 65 years [173]. Altogether this clinical evidence demonstrates that blockade of the pressor arm of the RAS, regardless of the drug class, have benefits on reducing the cognitive impairment associated to stroke and dementia.
Neuroprotective effects of ARBs and ACEi reported in animal models of PD have not yet been translated into clinical trials. A large cohort study showed that ACEi users had a lower risk of PD, suggesting a potential reduction of PD risk associated to these antihypertensive agents [174]. Treatment with telmisartan significantly decreased SCZ symptoms together with plasma levels of the pro-inflammatory marker IL-6, showing promising results regarding RAS modulation in SCZ treatment [175]. Further clinical studies in PD and SCZ are warranted to prove a net benefit regarding RAS blockade in these patients.
Conclusions
Besides being a central regulator of blood pressure and hydrosaline homeostasis, brain RAS also participates in the development of the nervous system and cognitive processes, including cognition, memory and learning. Stimulation of ACE2/Ang (1-7)/MasR axis at central level has been linked to neuroprotective effects, modulation of cognition, induction of angiogenesis and cerebral vasodilation. All these actions oppose to the harmful effects of the Ang II/AT1R axis. The existence of alterations in the brain Ang-(1-7)/MasR axis has been involved in the pathophysiology of neurogenic hypertension and in the development of neurological disorders such as AD, stroke, anxiety, depression and emotional stress. In this context, the depressor axis of the RAS could be thought as a potential target in the treatment of cardiovascular and neurological disorders. Central ACE2 activation and other strategies directed to increase central Ang-(1-7) levels may be considered as possible tools to treat neurodegenerative diseases in the brain. Future studies are needed to fully understand the role of central ACE2/Ang-(1-7)/MasR axis in neuroprotection.
Abbreviations
| Aβ: | amyloid-β |
| ACE: | angiotensin converting enzyme |
| AD: | Alzheimer’s disease |
| AGT: | angiotensinogen |
| Ang: | angiotensin |
| APP: | amyloid precursor protein |
| AT1: | Angiotensin II type 1 |
| AT1R: | Angiotensin II type 1 receptor |
| AT2R: | Angiotensin II type 2 receptor |
| BBB: | blood-brain barrier |
| CVLM: | caudoventrolateral medulla |
| DOCA: | deoxycorticosterone acetate |
| GPCR: | G-protein-coupled receptor |
| ICV: | intracerebroventricular |
| IL: | interleukin |
| MasR: | Mas receptor |
| MCAO: | middle cerebral artery occlusion |
| MS: | mass spectrometry |
| NE: | norepinephrine |
| NEP: | neutral-endopeptidase |
| NF-κB: | nuclear factor-κB |
| NO: | nitric oxide |
| PKA: | protein kinase A |
| SON: | supraoptic nucleus |
| NTS: | nucleus of the solitary tract |
| PRRs: | prorenin receptors |
| PVN: | paraventricular nucleus |
| RAS: | renin-angiotensin system |
| RVLM: | rostroventrolateral medulla |
| SHRs: | spontaneously hypertensive rats |
| spSHRs: | stroke-prone spontaneously hypertensive rats |
Declarations
Author contributions
NLRM wrote the first draft of the manuscript; AMP wrote sections of the manuscript; MMG conceived the idea, wrote sections of the manuscript and critically reviewed the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2021.