
Abstract
Aim:
To screen differentially expressed genes related to gastric cancer based on The Cancer Genome Atlas (TCGA) database and construct a gastric cancer diagnosis model by machine learning.
Methods:
Transcriptional data, genomic data, and clinical information of gastric cancer tissues and non-gastric cancer tissues were downloaded from the TCGA database, and differentially expressed genes of gastric cancer messenger RNA (mRNA) and long non-coding RNA (lncRNA) were screened out. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyzed the differentially expressed genes, and the protein-protein interaction (PPI) of differentially expressed genes was constructed. Core differentially expressed genes were screened by Cytoscape software’s molecular complex detection (MCODE) plug-in. The differential genes of lncRNA were analyzed by univariate Cox regression analysis and lasso regression for further dimension reduction to obtain the core genes. The core genes were screened by machine learning to construct the gastric cancer diagnosis model. The efficiency of the gastric cancer diagnosis model was verified externally by the Gene Expression Omnibus (GEO) database.
Results:
Finally, 10 genes including long intergenic non-protein coding RNA 1821 (LINC01821), AL138826.1, AC022164.1, adhesion G protein-coupled receptor D1-antisense RNA 1 (ADGRD1-AS1), cyclin B1 (CCNB1), kinesin family member 11 (KIF11), Aurora kinase B (AURKB), cyclin dependent kinase 1 (CDK1), nucleolar and spindle associated protein 1 (NUSAP1), and TTK protein kinase (TTK) were screened as gastric cancer diagnostic model genes. After efficiency analysis, it was found that the random forest algorithm model had the best comprehensive evaluation, with an accuracy of 92% and an area under the curve (AUC) of 0.9722, which was more suitable for building a gastric cancer diagnosis model. The GSE54129 data set was used to verify the gastric cancer diagnosis model with an AUC of 0.904, indicating that the gastric cancer diagnosis model had high accuracy.
Conclusions:
Machine learning can simplify the bioinformatics analysis process and improve efficiency. The core gene discovered in this study is expected to become a gene chip for the diagnosis of gastric cancer.
Keywords
Machine learning, gastric cancer, diagnostic model, bioinformaticsIntroduction
Gastric cancer is the fifth most common cancer globally and ranks third in the world in terms of cancer mortality [1], and is one of the most common malignant tumors in China [2]. Gastric cancer is characterized by high malignancy, susceptibility to distant metastasis, poor prognosis, and heavy disease burden [3, 4]. Currently, the diagnosis of gastric cancer still relies on upper gastrointestinal endoscopy [4–6]. The invasive examination increases the difficulty of early screening [7]. For gastric cancer diagnosis, the specificity and sensitivity of traditional serum markers, such as carcinoembryonic antigen (CEA), are low [8]. Therefore, finding more accurate predictive markers for molecular diagnosis of gastric cancer is crucial in screening, early diagnosis and treatment.
With the development of high-throughput technology, RNA sequencing (RNA-seq)-based diagnostic markers for gastric cancer have been widely studied. Non-coding RNA (ncRNA) affects the expression of oncogenes or oncogenes and is expected to be a molecular marker for the early diagnosis of gastric cancer [9, 10]. Long ncRNA (lncRNA) has been widely studied among ncRNAs in gastric cancer. LncRNA is an RNA transcript that has more than 200 nucleotides in length and is usually found in the nucleus [11]. LncRNA plays an important role in epigenetic regulation, cell cycle, genomic imprinting, chromatin modification, transcriptional interference, protein activation, etc. [12, 13]. It was found that lncRNA is dysregulated in gastric, liver, breast, and cervical cancers and other tumors [14, 15], suggesting that lncRNA may be a potential biological marker for early diagnosis, efficacy chemoresistance and other assessments.
The volume of sequencing data is too large to be adequately analyzed by traditional means. Machine learning is algorithms that use statistical data analysis to build models for making predictions about the outcome of unknown data. Compared with existing statistical methods, machine learning has higher evaluation accuracy and personalized prediction ability when big data is used to analyze medical problems [16]. The New England Journal of Medicine believes that machine learning will bring a significant breakthrough in medicine [17]. For example, machine learning can predict the structures and functions of proteins based on the arrangement of genetic factors. Therefore, this study obtained core genes and built a diagnostic model for gastric cancer based on bioinformatics analysis of The Cancer Genome Atlas (TCGA) database, and then verified the model by machine learning and further validated the accuracy of the model using an external dataset (GEO dataset, Gene Expression Omnibus–NCBI), to provide a new method for early diagnosis of gastric cancer.
Materials and methods
Data acquisition and processing
The transcriptomic data, genomic data, and clinical information on gastric cancer from the TCGA database were downloaded from the Genomic Data Commons (GDC) Data Portal (https://portal.gdc.cancer.gov/) on June 28, 2021, including 376 gastric cancer patients and 31 non-gastric cancer tissue samples. On July 8, 2021, this study downloaded transcriptomic and genomic data and clinical information of lung cancer patients from the TCGA database as a control group, including 543 lung cancer patients with cancer tissues and 51 normal tissue samples. The number of gastric cancer patients reached 376, and the number of non-gastric cancer patients reached 625. After that, the GEO public database (https://www.ncbi.nlm.nih.gov/geo) was searched with “gastric cancer” as the keyword, and the GSE54129 dataset was selected as the external validation dataset. The GSE54129 dataset is based on the GPL570 platform, which contains the gene expression information of 111 gastric cancer patients and 21 normal controls. The RNA-seq data expression matrix was merged with R language (version 4.0.4) to obtain the complete RNA-seq expression profile, and the count data were normalized and ID transformed. The messenger RNA (mRNA) and lncRNA were extracted separately to generate gene expression matrices.
mRNA data processing
Screening for differential genes
The “DESeq2” package was called in R language to screen the differential genes with the criteria of |log2 fold change (FC)| ≥ 3 and false discovery rate (FDR) < 0.05, and the “heatmap” package was used to plot the differential gene heat map and the “ggplot” package to plot the differential gene scatter map.
Functional enrichment analysis of differential genes and protein interaction network construction
Using R language, this research first performs gene ontology (GO) analysis [18] and gene function annotation of differentially expressed genes, including biological process (BP), cell composition (CC), and molecular function (MF). Subsequently, a Kyoto Encyclopedia of Genes and Genomes (KEGG) [19] analysis was performed to obtain signaling pathways that may have a role. This study used the search tool for the retrieval of interacting genes (STRING) (an online biological database that provides gene analysis and constructs networks of gene interactions at the protein level [20]). Then, the program downloaded and installed the molecular complex detection (MCODE) plug-in in Cytoscape 3.6.1 and imported the above the protein-protein interaction (PPI) data in tab-separated values (TSV) format with a degree cutoff = 2, Node score cutoff = 0.2, and κ-core = 0.2. The PPI network’s most densely associated regions were obtained using degree cutoff = 2, Node score cutoff = 0.2, κ-core = 2, max. depth = 100 as the screening criteria. The most densely associated regions in the PPI network were obtained, which are the core genes related to gastric cancer screening in this study [21].
LncRNA data processing
Screening for differential genes
The “DESeq2” package was called in R to screen the differential genes with the criteria of |log2 FC| ≥ 3 and FDR < 0.05. The differential gene heat map is plotted by the “heatmap” package. The differential gene volcano was shown through the “ggplot” package in R language.
Screening for key genes
Since lncRNAs are ncRNAs, the univariate Cox regression model was first used to investigate the relationship between lncRNA expression levels and overall patient survival for the screened differentially expressed lncRNAs, and the identification criterion was P < 0.05. To avoid overfitting of the model, lasso regression was used to process the data. Lasso regression is based on linear regression, and the addition of a penalty term in the model estimation can compress extremely small regression coefficients to 0, at the cost of some estimation bias to obtain higher model prediction accuracy and model generalization ability. This study performed lasso regression analysis by “glmnet” package in R language to further screen lncRNAs associated with survival prognosis by increasing the penalty strength and narrowing the regression coefficients.
Construction of the model
This study used three machine learning algorithms (MLAs) [random forest (RF) [22, 23], naive Bayesian classification (NBC) [24], and k-nearest neighbor (KNN) [25, 26]] to construct and compare diagnostic models for gastric cancer (see the supplementary file for details of the algorithm).
Model optimization and validation
This study used accuracy, sensitivity, specificity, and area under the curve (AUC) to assess the performance of the gastric cancer diagnostic model. Accuracy refers to the proportion of samples correctly predicted by the model to all samples, and sensitivity refers to the proportion of correct predictions where the true value is a positive case. The AUC is the area under the receiver operating characteristic (ROC) curve. The ROC curve shows the sensitivity and specificity of the model prediction, and the larger the value, the better the prediction. To further verify the model’s efficacy, data from the GEO dataset GSE54129 were applied to further validate the accuracy of the model.
Results
Acquisition of key mRNA genes for gastric cancer
Screening for gastric cancer differential genes
Expression data were downloaded from the TCGA database for 407 patients, including 376 gastric cancer tissues and 31 control tissues. By differential comparison, this study screened a total of 947 differential mRNAs, of which 419 were upregulated and 526 downregulated, and plotted a heat map and volcano map (Figure 1).
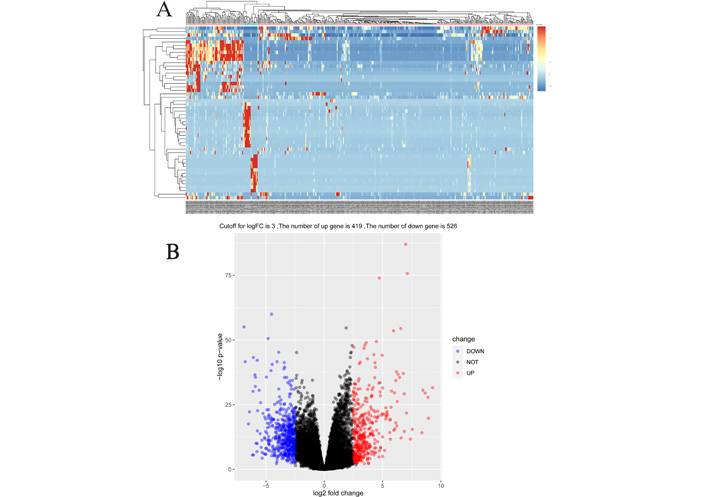
Heat map and volcano map of mRNA differential genes in gastric cancer. (A) Heat map of 947 mRNA differential genes in gastric cancer, and the top 50 most representative genes were selected to draw the heat map; (B) volcano map of 947 genes obtained at a cutoff value of 3, of which 419 were upregulated and 526 downregulated
Biological process analysis of differential genes in gastric cancer
GO enrichment analysis showed that gastric cancer upregulated differential genes (UDEGs) were mainly distributed in the extracellular region part, proteinaceous extracellular matrix (ECM), the ECM, and other tissues. The UDEGs were involved in biological processes such as cell adhesion, biological adhesion, response to wounding, and mainly had molecular functions such as ECM structural constituent and glycosaminoglycan binding. Gastric cancer downregulated differential genes (DDEGs) were mainly distributed in the apical part of cells, the extracellular region and other tissues, involved in biological processes such as digestion, lipid catabolic process and response to the metal ion. The DDEGs mainly had molecular functions such as steroid binding and coenzyme binding (Figure 2).
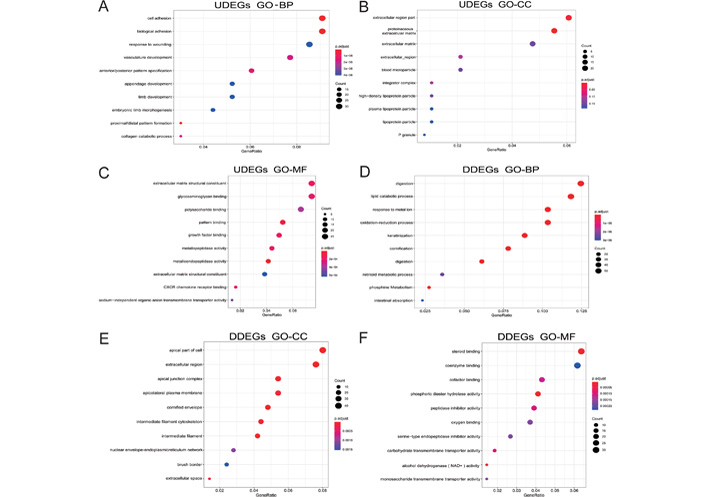
Biological process analysis of differential genes in gastric cancer. (A–C) The results of GO analysis of UDEGs in gastric cancer; (D–F) the results of GO analysis of DDEGs in gastric cancer. CXCR: CXC chemokines; NAD+: Dihydrouracil Dehydrogenase; p. adjust: adjust P-values for multiple comparisons; P granule: germ cell ribonucleoprotein granules
Analysis of the signaling pathways involved in differential genes of gastric cancer
KEGG enrichment analysis showed that gastric cancer UDEGs were highly expressed in signaling pathways such as focal adhesion, ECM-receptor interactions, and leukocyte transendothelial migration. In contrast, gastric cancer DDEGs were enriched in expression in pathways such as metabolism of xenobiotics by cytochrome P450, drug metabolism-cytochrome P450, and retinol metabolism (Figure 3).
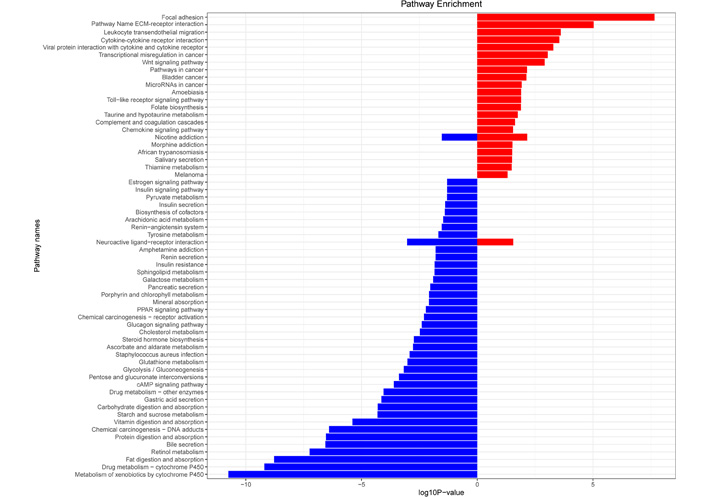
KEGG pathway analysis of differential genes in gastric cancer. Red represents UDEGs, and blue represents DDEGs. PPAR: peroxisome proliferator-activated receptor; cAMP: cyclic adenosine monophosphate
PPI network construction and core gene identification
Interactions between differential genes were predicted using the STRING database, and the information of 947 differential genes was imported into Cytoscape software for visualization study. Nine hundred and eighteen nodes and 1,209 edges were involved in the PPI network (Figure 4A). Ten gastric cancer-associated core differentially expressed genes were screened, and they were cyclin dependent kinase 1 (CDK1), non-SMC condensin I complex subunit G (NCAPG), cyclin B1 (CCNB1), kinesin family member 11 (KIF11), Aurora kinase B (AURKB), cell division cycle associated 8 (CDCA8), threonine kinase B (BUB1B), nucleolar and spindle associated protein 1 (NUSAP1), TTK protein kinase (TTK), and mitotic arrest deficient 2 like 1 (MAD2L1) according to the degree of node association from highest to lowest (Figure 4B).
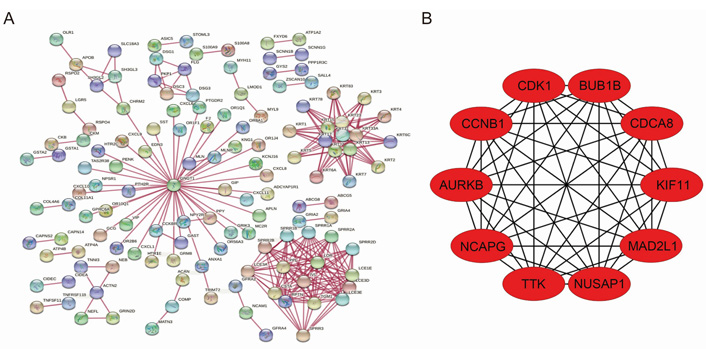
PPI network of differential genes and top 10 core genes in gastric cancer. (A) PPI map of differentially expressed genes in gastric cancer; (B) top 10 core genes in gastric cancer
Acquisition of key lncRNA genes in gastric cancer
The expression data of patients were downloaded from the TCGA database, including 31 control tissues and 376 cancer tissues, and 66 differentially expressed lncRNAs were screened using the “DESeq2” package, of which 29 lncRNAs were upregulated, and 37 lncRNAs were downregulated. Among the 66 differentially expressed lncRNAs, 19 lncRNAs were further analyzed by univariate Cox regression with P < 0.05. Subsequently, lasso regression was used to further downscale the model, and the results showed that the model error was minimized when the number of variables was 5, which corresponded to λ = 0. 075. The five screened lncRNAs were long intergenic non-protein coding RNA 1821 (LINC01821), AL138826.1, gastric cancer associated transcript 3 (GACAT3), AC022164.1, and adhesion G protein-coupled receptor D1-antisense RNA 1 (ADGRD1-AS1), which were used as gastric cancer lncRNA key genes (Figure 5).
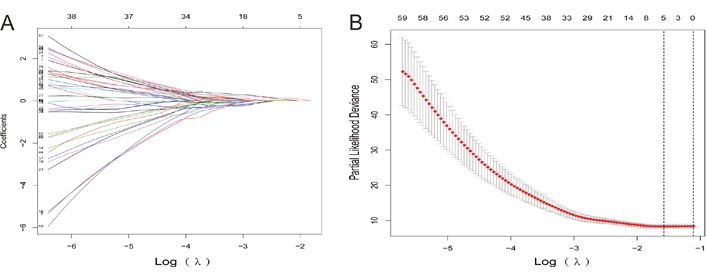
Lasso regression process. (A) Lasso diagram shows the dynamic process of screening variables; (B) the selection process of the cross-validation parameter Log(λ)
Construction of data tables for gastric cancer diagnosis model
The 10 key genes of mRNA and 5 key genes of lncRNA were used to construct a gastric cancer diagnosis model, with column names as gene names, row names as the number of each sample, and content as the expression of each gene in each sample, and the genetic data of 543 cases of lung cancer tissues and 51 cases of paired paracancerous normal tissues were added as non-gastric cancer patients introduced into the model for the validation of the gastric cancer prediction model. As a result, the number of gastric cancer patients reached 376 cases, and the number of non-gastric cancer patients reached 628 cases.
Model performance analysis
The above 15 genes were further screened by the “Feature Importance” algorithm to identify 10 key genes, namely LINC01821, AL138826.1, AC022164.1, ADGRD1-AS1, CCNB1, KIF11, AURKB, CDK1, NUSAP1, TTK. The 10 genes were modeled using the best feature subset, by three MLAs: RF, NBC, and KNN. As for the performance of RF, NBC, and KNN, algorithm boosting was measured with 6 main metrics, namely AUC, ROC, correctness, sensitivity, specificity, and precision. Among the three models, RF has the highest AUC and ROC of 0.9722, higher than the NBC of 0.9088 and the KNN algorithm of 0.8656. RF has the highest accuracy of 0.920 among all models, slightly higher than the NBC of 0.824 and the KNN algorithm of 0.797 (Figures 6 and 7). Therefore, the RF algorithm was finally chosen to construct the model.
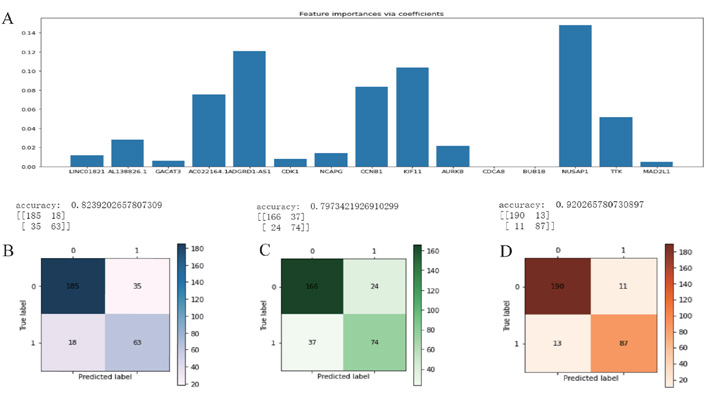
Feature selection and machine learning. (A) The initial screening of 15 genes by the “Feature Importance” algorithm and further screening of 10 key genes; (B) the NBC algorithm model; (C) the KNN algorithm model; (D) the RF algorithm model
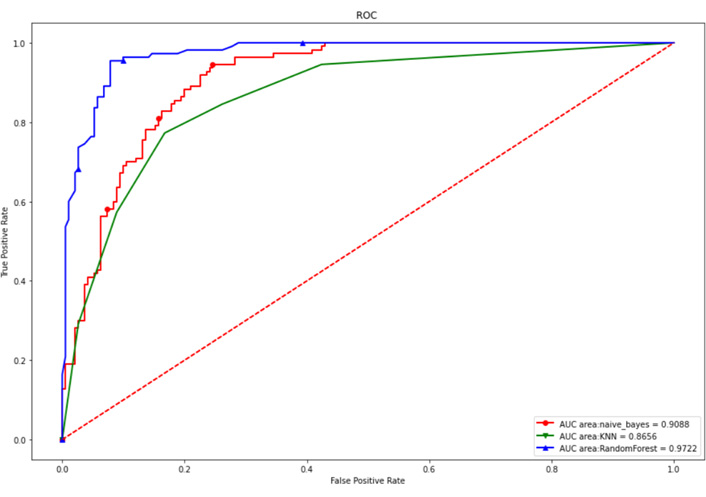
Internal test set ROC curves. Red dashed line is the reference line, red solid is the NBC algorithm model, green is the KNN algorithm model, and blue is the RF algorithm model
External validation of the gastric cancer diagnostic model
The GSE54129 dataset was selected as an external validation dataset in the GEO database. The GSE54129 dataset was based on the GPL570 platform, which contained the gene expression information of 111 gastric cancer patients and 21 non-gastric cancer patients. As is shown in Figure 8, the area of the ROC curve (AUC) for the validation sets was 0.9144. As a result, 0.9144 greater than 0.70 indicates that the model has good predictability.
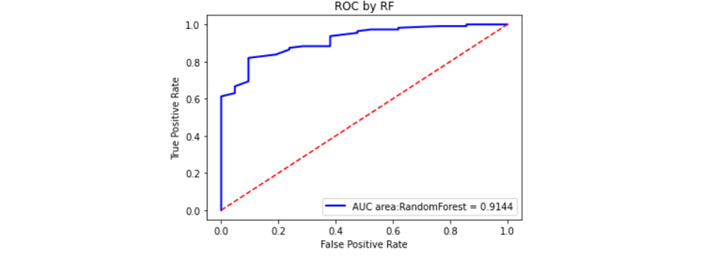
External validation set ROC curve. The red dashed line is the reference line, and the blue is the ROC curve
Discussion
This study downloaded the transcriptomic and genomic data of 372 gastric cancer patients and 625 paired non-gastric cancer patient samples. All patients’ clinical information was included in the study through the TCGA database. This study first analyzed the data quality using R software, and the results showed that the data quality was good, indicating that the data of this study were reliable. Subsequently, the mRNA and lncRNA differentially expressed gastric cancer genes were screened out, and the core differentially expressed genes were initially screened out after the related pathway analysis. By using the “Feature Importance” algorithm, 10 key genes related to gastric cancer were finally screened: LINC01821, AL138826.1, AC022164.1, ADGRD1-AS1, CCNB1, KIF11, AURKB, CDK1, NUSAP1, and TTK. It was found that the accuracy of the model built by the RF algorithm was 92% with an AUC of 0.897, which was more suitable for building a diagnostic model of gastric cancer. Then external validation was performed by data in the database, and the AUC of the model was found to be 0.9144 through validation, indicating that the gastric cancer diagnostic model has high accuracy and is expected to be an early diagnosis model for gastric cancer.
The research screened 10 key genes respectively from 947 differentially mRNAs and 66 differentially expressed lncRNAs. CCNB1 is one of the major members of the cell cycle protein B family and has an important role in the G2/M transition phase of the eukaryotic cells [27]. Yasuda et al. [28] showed that overexpression of CCNB1 mainly occurred in the early stages of gastric malignancies; Gao et al. [29] found that down-regulation of CCNB1 expression could help cordycepin-induced cell arrest at the G2/M phase, and high expression of CCNB1 could be one of the diagnostic genes for gastric cancer. KIF11 is a member of the kinesin family that affects the formation of spindle bipolarity [29, 30], causes chromosomal instability, leads to abnormal cell division and proliferation, promotes tumor formation, and plays a role in gastric cancer progression [31, 32]. AURKB is a member of the aurora kinase family, which plays a role in the assembly of the two-level spindle, maintains normal mitosis, and regulates stem cell self-renewal, reprogramming, and differentiation [33, 34]. In recent years, AURKB has been highly expressed in breast cancer, bladder cancer, gastric cancer, and other tumors [35–37]. Nie et al. [38] found that AURKB may promote gastric tumorigenesis through epigenetic activation of CCND1 expression. NUSAP1 is a microtubule-associated protein that plays an important role in cell division and chromosome segregation [39–41]. NUSAP1 can also be used as a molecular marker for prostate cancer, promoting cell proliferation and migration [42, 43]. Recently, it has been shown that NUSAP1 is highly expressed in gastric cancer cell lines and tissues and promotes malignant proliferation and invasion of gastric cancer cells [44]. Furthermore, the genes of LINC01821, AL138826.1, AC022164.1, ADGRD1-AS1, CDK1, and TTK in gastric cancer need to be further investigated.
Machine learning can summarize the patterns in a large amount of data information, explain the inner connection, and efficiently explore the value of data [45, 46]. The biggest advantage of this study is using an MLA, which reduces manual repetitive work, makes the efficiency of detection greatly improved, and accomplishes complex computational work that is impossible to be done manually by computer. It has been used to predict tumors by learning from gene expression data, such as Leng et al. [47] collected a large amount of data, including 474 lung adenocarcinoma samples and 491 lung squamous carcinoma samples, and learned 1,099 differentially expressed mRNA data by the extreme gradient boosting (XGBoost) algorithm to predict lung cancer subtypes, lung squamous cell carcinoma and lung adenocarcinoma. The XGBoost algorithm showed high predictive power in this study, outperforming the logistic regression algorithm and supporting vector machine algorithm for lung early diagnosis and treatment of squamous and lung adenocarcinoma. Yang et al. [48] selected the most important DNA methylation features as a model using RF, and the authors construct a support vector machine classifier for hepatocellular carcinoma diagnosis. Tian et al. [49] proposed that the normal gastric cell and its cancer counterpart can be distinguished by multiple cellular mechanical phenotypes (CMPs) based on MLA. More accurate prognostic biomarkers can be obtained through MLA, which provides a new method to verify the prognosis of cancer.
The advantages of the model created in this study: 1. the AUC of the model is greater than 0.9 in both the internal validation group and the external validation group, indicating that the model has high accuracy; 2. the model includes not only coding RNA but also ncRNA (lncRNA), which further improves the accuracy of the model and avoids the bias caused by a single gene; 3. the potential for clinical translation application: for those who are financially well-off and unwilling to receive invasive examinations, after obtaining the whole genome sequencing through blood samples, the core gene model can be used to assist in the diagnosis of gastric cancer, which is expected to avoid invasive examinations such as gastroscopic biopsies for the negative population. However, the model still has shortcomings: 1. since the original purpose of our study was to establish a model for diagnosis of gastric cancer, this study did not select data from gastric cancer tissues and paracancerous tissues for validation but used gastric mucosal tissues from non-gastric cancer patients as controls, considering that the sampling of the paracancerous tissue specimens is not standardized in the clinical practice. The definition of paracancerous tissues (length from cancerous tissues, etc.) is not uniform. This avoided the bias caused by sampling and increased our difficulty in finding external validation data sets, resulting in a sample size that was not ideal. In addition, in the external validation, the final selection of the GSE54129 dataset by the microarray resulted in the lack of 2 lncRNAs (AL138826.1 and AC022164.1) among the 10 core genes due to the insufficient number of lncRNA annotations, but this did not have a significant impact on this model, and the AUC of the model remained at 0.9144. 2. In the study, the number of available samples was limited. It would be better to collect more samples to strengthen the machine learning ability and improve the model’s accuracy. 3. The machine learning process used sample tissues of lung cancer as a control group because there were not enough normal sample tissues, which may also bias the study results. 4. Since the TCGA database did not provide us with the population source, there are differences between databases in terms of ethnicity, region, nationality, and disease characteristics, which pose a challenge to the generalizability of this finding. 5. This study has not been designed to validate the fresh specimens from clinical patients, and there is no answer to whether the finding can be applied to the Chinese population. The next step will be to improve the design of the study so that the future research can better answer the above questions.
Abbreviations
| ADGRD1-AS1: | adhesion G protein-coupled receptor D1-antisense RNA 1 |
| AUC: | area under the curve |
| AURKB: | Aurora kinase B |
| CCNB1: | cyclin B1 |
| CDK1: | cyclin dependent kinase 1 |
| DDEGs: | downregulated differential genes |
| ECM: | extracellular matrix |
| FC: | fold change |
| GEO: | Gene Expression Omnibus |
| GO: | gene ontology |
| KEGG: | Kyoto Encyclopedia of Genes and Genomes |
| KIF11: | kinesin family member 11 |
| KNN: | k-nearest neighbor |
| LINC01821: | long intergenic non-protein coding RNA 1821 |
| lncRNA: | long non-coding RNA |
| MLAs: | machine learning algorithms |
| mRNA: | messenger RNA |
| NBC: | naive Bayesian classification |
| ncRNA: | non-coding RNA |
| NUSAP1: | nucleolar and spindle associated protein 1 |
| PPI: | protein-protein interaction |
| RF: | random forest |
| RNA-seq: | RNA sequencing |
| ROC: | receiver operating characteristic |
| TCGA: | The Cancer Genome Atlas |
| TTK: | TTK protein kinase |
| UDEGs: | upregulated differential genes |
Supplementary materials
The supplementary material for this article is available at: https://www.explorationpub.com/uploads/Article/file/100194_sup_1.pdf.
Declarations
Acknowledgments
We acknowledge TCGA and GEO databases for providing their platforms and contributors for uploading their meaningful datasets.
Author contributions
FK and WY conceived and designed this study; ZY constructed the machine learning and IT support. FK, NL, PW, and SF designed the statistical analysis and analyzed the data. FK, NL, and WY interpreted the results. FK wrote the first draft of the manuscript. FK, PW, WY, and NL contributed to the writing of the manuscript. All authors have read and confirmed the data in the manuscript. They can take responsibility for the data’s integrity and the data analysis’s accuracy and read and approved the submitted version.
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
The transcriptomic data and clinical information of gastric cancer and lung cancer for this study can be found in the TCGA database and downloaded from the Genomic Data Commons (GDC) Data Portal (https://portal.gdc.cancer.gov/); the GSE54129 dataset for this study can be found in the GEO public database (https://www.ncbi.nlm.nih.gov/geo).
Funding
Not applicable.
Copyright
© The Author(s) 2022.