Review
Review

Affiliation:
Independent researcher, Elkins Park, PA 19027, USA
Email: burtabrams@hotmail.com
ORCID: https://orcid.org/0000-0002-0237-233X
Explor Musculoskeletal Dis. 2023;1:106–120 DOI: https://doi.org/10.37349/emd.2023.00015
Received: May 23, 2023 Accepted: July 28, 2023 Published: August 31, 2023
Academic Editor: Fernando Pérez-Ruiz, Cruces University Hospital, Spain
Hyperuricemia is known to be a necessary and causal condition for gout, but much more prevalent than gout. Medicine has standardized treatments for gout, but has no such determination for asymptomatic hyperuricemia. Nevertheless, people with hyperuricemia, gouty or not, too often continue to be at risk for shortened lifespans from life-threatening comorbidities, all of which are known to be consequences of obstructive sleep apnea (OSA), which is shown herein to cause most hyperuricemia. This review also presents the wide variety of OSA consequences, many of which are irreversible and life-threatening, as the rationale for treating all hyperuricemia (gouty and asymptomatic) by diagnostic testing and effective treatment for OSA as soon as hyperuricemia is detected. It advocates frequent ultrasonic screening for aggregated urate crystals. Multiple epidemiological studies have found OSA to be significantly more prevalent in those people with gout diagnosed with OSA than it is in those never diagnosed with it. A clinical study shows an even higher prevalence of OSA in people with gout. The pathophysiology of hypoxia from OSA explains how it would lead to both the overproduction and the underexcretion of uric acid, leading to hyperuricemia and the precipitation of monosodium urate crystals which cause a gout flare. Resolving OSA has been shown to prevent or even reverse life-threatening diseases that are recognized comorbidities of hyperuricemia and gout, and can prevent further gout flares. In order to extend the length and quality of life of people with gout or hyperuricemia, when either first manifests a patient sleep study is recommended, followed by effective OSA treatment as warranted.
The modern conception of gout is that it is a systemic disease for which hyperuricemia is a necessary condition. Treatment for gout is standard; treatment for asymptomatic hyperuricemia is not.
The extreme pain of a gout flare or diagnosis of hyperuricemia should be viewed as an alarm signal that something in the body is seriously amiss. It is up to the treating doctor to determine the underlying cause [1], leading to its resolution. Only prescribing non-steroidal anti-inflammatory drugs (NSAIDs), corticosteroids, or colchicine just muffles the alarm; only prescribing urate lowering therapy (ULT) just disables the alarm. These drugs, along with diet modification to reduce purines and therapy to improve kidney function, are beneficial for reducing the pain intensity, duration, and frequency of gout flares by inhibiting hyperuricemia. Even so, long-term health risks may remain from hyperuricemia’s underlying cause, too often undiagnosed, and a threat for premature death [2–4] from the life-threatening comorbidities of hyperuricemia and gout.
One aspect of gout which too often is overlooked in guidelines and practice is that most gout flares originate during sleep. The sleep connection has been known at least since Dr. Thomas Sydenham, himself a gout sufferer, wrote about it in 1683. Dr. Sydenham’s observation was confirmed in a 2015 study [5], finding that the risk of a gout flare is 2.4 times higher at night and early morning. It is a very important clue to the pathogenesis of hyperuricemia and gout.
In most cases, a gout flare or hyperuricemia diagnosis would be a warning of chronic intermittent hypoxia from obstructive sleep apnea (OSA) [6, 7], which likely is why most gout flares originate during sleep. This review compiles epidemiologic, pathophysiologic, and clinical evidence for a compelling hypothesis that most hyperuricemia and gout and their comorbidities [8] result from OSA, with gout occurring in those individuals who are genetically predisposed to gout [9]. Consequently, resolving the OSA can prevent any additional gout flares as well as reduce the risk of development of the life-threatening comorbidities of gout and hyperuricemia. Further clinical studies, as discussed in the conclusions section, are needed for corroboration.
The hypothesis of this review emphasizes the following take-home points for practitioners with an interest in hyperuricemia and gout:
In most cases, OSA is the principal cause of asymptomatic hyperuricemia and gout, starting early in OSA’s development.
Pathophysiological reasons for OSA causing hyperuricemia cease after the OSA has been resolved.
All known comorbidities of hyperuricemia and gout are known to be consequences of OSA, including those that are life-threatening.
Both gout and unresolved OSA significantly increase the risk of premature death.
Treating gout with urate lowering pharmaceuticals has only controversial benefit for preventing gout’s life-threatening comorbidities and for reducing its mortality.
Resolving OSA can prevent further gout flares; if resolved early enough it also reduces the risk of developing the life-threatening consequences of OSA, some of which are known comorbidities of gout and hyperuricemia, and even reverses some of them later.
A number of epidemiologic studies, synopsized in Table 1, have been reported that show gout to be significantly more prevalent in people diagnosed with OSA than it is in people never diagnosed with OSA [10–15]. Most of these studies access a health database to form a risk ratio of the number of gout patients who have been diagnosed with OSA relative to those with no OSA diagnosis, ranging from 1.75:1 up to 3.7:1. They do not take into account the fact that a very large percentage of people with OSA have never been diagnosed with it [16], so they would have been misplaced in the no OSA diagnosis category instead of the diagnosed OSA category. If these studies had taken that into account, the estimated risk ratios of OSA with gout could have been much higher.
Synopses of epidemological studies connecting gout with OSA
| References | Description of study | Number of participants | Results | Risk ratio |
|---|---|---|---|---|
| Roddy et al. [10], 2013 | Data from UK Clinical Practice Research Datalink. Comparison of gout incidence rate in cohort diagnosed with OSA vs. cohort (matched by age, sex, practice) never diagnosed with OSA. 5.8 years median follow-up. | 15,878 Diagnosed with OSA. 63,296 Never diagnosed with OSA* | OSA: 4.9% with gout. Incidence rate 7.83/KPyr No OSA: 2.6% with gout. Incidence rate 4.03/KPyr | 1.9:1 |
| Zhang et al. [11], 2015 | Data from UK general practice database. Comparison of gout prevalence in those diagnosed with OSA vs. those never diagnosed with OSA. | 1,689 Diagnosed with OSA. 6,756 Never diagnosed with OSA* | OSA: 0.7% No OSA: 0.3% | 2.3:1 |
| Singh and Cleveland [12], 2018 | Internet questioning about sleep disorders of people with physician-diagnosed gout who visited a gout education website. | 454 | 320 With gout. 77 With diagnosed OSA | N/A |
| Blagojevic-Bucknall et al. [13], 2019 | Data from 5% US Medicare beneficiary sample 2006–2012. Selected entries with new diagnosis of OSA. | 1.74 Million, follow-up of 10,448,472 person-years* | Incidence rates with: OSA: 14.3/KPyr No OSA: 3.9/KPyr | 3.7:1 |
| Singh [14], 2019 | Data from UK Health Improvement Network. Comparison of gout incidence rate with first SA diagnosis vs. cohort (matched by age, sex, BMI) never diagnosed with SA. | 9,865 With first SA diagnosis. 43,598 Never diagnosed with SA* | Incidence rates with: SA: 8.4/KPyr No SA: 4.8/KPyr | 1.75:1 |
| Kanbay et al. [15], 2014 | All participants tested for OSA, and questioned about history of cardiovascular disease. | 72 Controls (AHI < 5); 47 mild (5 < AHI < 15); 75 moderate OSA (15 < AHI < 30); 192 severe (AHI > 30) | SUA data show monotonic increase with AHI in those who had a cardiovascular event | N/A |
* Not all subjects were tested for OSA so the numbers are likely to underrepresent the true number of subjects with OSA. SA: sleep apnea; BMI: body mass index; AHI: apnea-hypopnea index; KPyr: kiloperson-years; N/A: not applicable; SUA: serum uric acid
A clinical study conducted by rheumatologists reported that 89% (a much higher 8:1 risk ratio) of 54 preselected people with gout were diagnosed by polysomnography with OSA [17], a percentage which is about as high as the sensitivity for OSA of one night of polysomnography [18]. This result, indicating a high risk of OSA with gout, demonstrates the limitations of the epidemiologic studies involving OSA.
There are no similar studies for asymptomatic hyperuricemia. Nevertheless, multiple medical journal papers have reported significantly elevated morbidity and mortality in hyperuricemic individuals [8, 19–23].
The epidemiology and clinical studies just described demonstrate the strong association of gout with OSA. The following description of the pathophysiology describes the mechanisms by which OSA would cause hyperuricemia and gout.
The principal mechanisms for hyperuricemia from OSA’s episodic hypoxemia comprise concurrent episodic overproduction of uric acid (UA) along with its concurrent episodic and long-term underexcretion. OSA’s chronic intermittent episodes of hypoxemia lead to concurrent catabolic episodes in every oxygen-starved cell of the body in which adenosine triphosphate degrades, leading to nucleotide turnover which culminates irreversibly in the generation of excess UA to be fed into the blood [24]. These episodes are accompanied by episodes of hypercapnia with reduced serum pH [25], which would reduce the solubility of SUA, as well as an increase in serum lactate [26], which leads to urate transporter 1 (URAT1) reducing renal reabsorption of UA thereby slowing excretion of SUA. OSA also leads to gradual deterioration of the glomerular filtration rate (GFR) [27] so that excretion of SUA is slowed even further.
Thus, with OSA there are repeated abrupt increases in the overproduction of UA fed into the blood along with its abruptly reduced solubility, plus kidney underexcretion. These would be perfect storm conditions for hyperuricemia to lead to the precipitation and deposition of monosodium urate (MSU) crystals, which cause a gout flare when they are deposited in a joint of an individual with the genetic predisposition for gout.
The pathophysiology just described is depicted in Figure 1, with the addition of other known contributing factors elevating SUA (diuretic medication, imbibing alcohol, fructose digestion, animal purine digestion) plus the chain of immune system reactions to nascent MSU crystals that lead to a gout flare in an individual genetically so predisposed [tumor necrosis factor-α (TNF-α), NLRP-3 (nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain inflammasome), caspase-1, interleukin-1β (IL-1β)]. The direct path from OSA to MSU crystal precipitation is the path of UA overproduction from OSA. Even with the use of ULT inhibiting overproduction by the cellular generation of UA, OSA on some nights may be severe enough to override ULT’s suppressive effect, acting as the tipping point for SUA to precipitate as nascent MSU crystals.
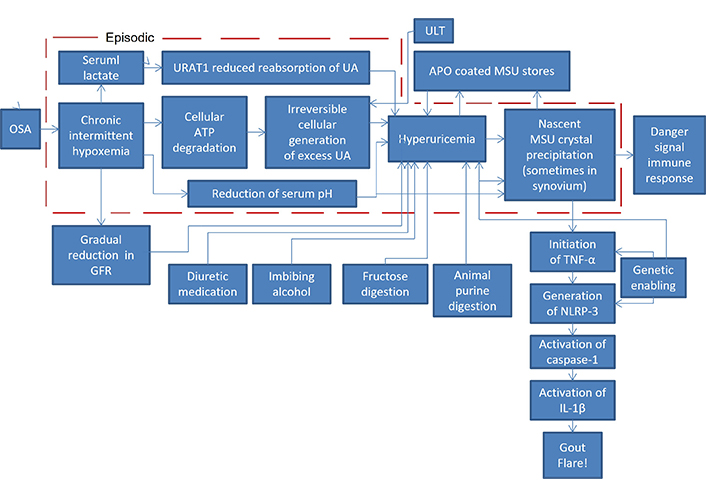
Pathophysiology diagram of OSA leading to a gout flare. OSA: obstructive sleep apnea; GFR: glomerular filtration rate; ATP: adenosine triphosphate; UA: uric acid; URAT1: urate transporter 1; APO: apolipoprotein; MSU: monosodium urate; TNF: tumor necrosis factor; IL: interleukin; NLRP-3: nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain inflammasome
Other mechanisms are operative as well. An important hypoxic pathway is driven by activation of the nuclear transcription factor, hypoxia‑inducible factor‑1α (HIF‑1α), the level of which has been found to be elevated in the plasma of patients with OSA [28], proposed as a biomarker to triage OSA severity [29]. In turn, HIF-1α is known to activate a survival pathway known as the polyol-fructokinase-xanthine oxidase pathway that generates UA [30–32]. This pathway tends to protect against hypoxia by reducing mitochondrial function while stimulating glycolysis, thereby reducing oxygen needs [33, 34]. However, chronic activation can lead to worsening features of metabolc syndrome and chronic inflammation.
Moreover, in chondrocytes hypoxia induces glucose transporter type 1 (GLUT1) and HIF-1α expression and a glycolytic shift that favors the Warburg effect, which causes the accumulation of lactate and the increased acidity of the extracellular microenvironment [35]. The acidification of the microenvironment causes the release of calcium ions which has been shown to nucleate the crystallization of MSU. This, along with the release of IL-1β, attracts resident macrophages that are activated by the MSU crystals leading downstream to a gout flare [36, 37]. A similar metabolic mechanism has been shown to exist in MSU-stimulated macrophages [37].
Once formed, the MSU crystals almost never dissolve on their own. Instead, over a period of one to two weeks the immune system enshrouds each of them with an APO [38, 39] so they no longer are sensed by other aspects of the immune system, and then the gout flare subsides. The shrouded MSU remains as urate stores throughout the body, usually considered to be benign. If ULT is used to reduce hyperuricemia, these MSU crystals begin to dissolve, replenishing the SUA concentration so that it remains in equilibrium for months until the MSU fully dissolves.
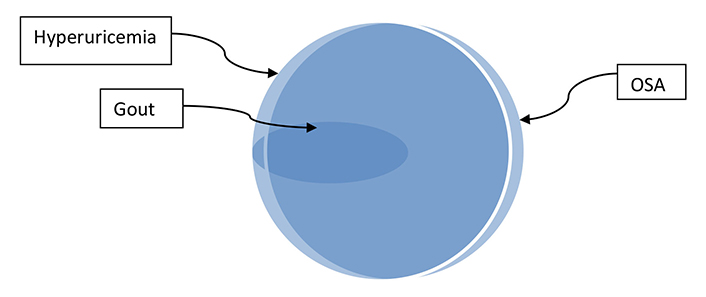
As a result of this pathophysiology, almost everyone with OSA would be expected to have hyperuricemia with MSU deposits, an expectation supported by the estimates that about 25% of US adults have OSA [40] and about 25% have hyperuricemia, both asymptomatic and with gout [41]. But only a small percentage (about 14% in the US, per the Supplementary material) of those with OSA, presumably only the ones who are genetically so predisposed, will develop gout when nascent MSU crystals are deposited in a joint. The Venn diagram of Figure 2 illustrates the envisioned overlaps of hyperuricemia and gout with OSA.
Furthermore, the meaningfulness of a blood test for SUA taken during waking hours is questionable in an individual with OSA. After awakening and normal breathing is restored, the overproduction and underexcretion episodic effects of OSA on hyperuricemia cease so that a blood test taken during waking hours could miss the SUA peaks. And if MSU has precipitated recently, which removes much SUA to form the MSU crystals, the measurement of SUA would be greatly undervalued [42].
All the diseases recognized as comorbidities of hyperuricemia and gout [8] are consequences of OSA, plus many more. All the consequences of OSA that this author has found to be presented in medical journal literature have been updated in Table 2. They are listed in three categories: those for which there is evidence of their reversibility just by resolving the OSA; those which are irreversible even after OSA has been resolved; and those whose reversibility has not yet been determined. For those in the reversible column, where there is evidence of how much time was needed after OSA resolution for reversibility to be evident, that time period is included in Table 2.
OSA consequences and their reversibility just by resolving OSA
| Usually reversible (at least partially) | Reversibility undetermined | Usually irreversible |
|---|---|---|
| Gout flares—immediately [43] | Coronary [75]/peripheral [76, 77] artery disease | Residual MSU with hyperuricemia [91, 92] |
| Atrial fibrillation—6 months [44] | Dyslipidemia/atherosclerosis [78] | Myocardial infarction [93] |
| Hypercoagulability—6 months [45, 46] | Congestive heart failure [79] | Ischemic stroke [94, 95] |
| Chronic kidney disease—3 months [47–49] | Autoimmune diseases [80, 81] | Type 2 diabetes [96, 97] |
| Ventricular mechanical dysfunction—3 months [50] | Chronic migraine [82] | Cancer [98–103] |
| Hypertension—6 months [51–53] | Hypothyroidism [83] | Mitral valve disease [104] |
| Nonalcoholic fatty liver disease [54, 55] | Systemic inflammation [84] | Aortic valve stenosis [105] |
| Excessive daytime sleepiness [56] | Erythrocytosis/polycythemia [85] | Interstitial lung disease [106, 107] |
| Nocturia [57] | Insomnia [86] | - |
| Cognitive impairment—1 year [58–60] | Keratoconus [87] | - |
| Depression—4 months [61, 62] | Female infertility [88] | - |
| Erectile dysfunction [63–65] | Glaucoma [89] | - |
| Endothelial dysfunction—3 months [66] | Inflammatory bowel disease [90] | - |
| Gastroesophageal reflux [67–71] | - | - |
| Rate of Alzheimer’s disease progress [72] | - | - |
| Telomere shortening—6 months [73, 74] | - | - |
-: blank cell
Readers who previously were not fully aware of the perils of OSA, hopefully are now convinced of the perilousness of allowing the continuation of unresolved OSA just by the number and variety of entries in Table 2, and that many of them are life-threatening.
The beginning of hyperuricemia is often diagnosed by a blood test to detect an elevated concentration of SUA. That may be an indicator that SUA is elevated, but it is not a reliable indicator. SUA is too variable as a result of a recent meal, alcohol imbibement, diuretic medication, or the OSA effects on SUA mentioned at the end of the section Pathophysiology of hyperuricemia and gout from OSA. A more reliable indicator of excessively high SUA episodes would be the formation of significant concentrations of crystals of MSU, the precipitate from OSA-induced jumps in SUA supersaturation, which remain for a very long time once they have been formed. It should be noted that Figure 1 shows that nascent MSU is the same marker used by the immune system as a danger signal to activate an innate antipathogen immune response, reacting to the likelihood that too many cells are dying too quickly [108].
As mentioned by Abrams [109], one method which has been demonstrated as effective for detecting MSU crystals is dual energy computed tomography (DECT). DECT has a very fine resolution so that a clear image of each MSU crystal may be formed. A major drawback of DECT is the exposure of the patient’s body to a high level of X-rays, a drawback which would be exacerbated if DECT were to be used repeatedly on the same patient, as would be the case should such inspection become standard with every periodic physical exam before OSA has been diagnosed. Furthermore, clear imaging of the crystals is more than required. All that is required is reliable detection, which can be achieved much more safely, but without such fine resolution, by ultrasonic inspection of the extremities [109], looking for the classic double contour displayed from the ultrasonic reflection of MSU crystals.
Another benefit of ultrasonic inspection is that it can be employed by the examining physician at the point-of-care [110], without the need to involve a radiology specialist. One can foresee that many new physicians will have training in the use of ultrasound, and be able to use it effectively in their physical exams of patients.
Gout has been reported to have so many of the same life-threatening metabolic syndrome comorbidities known to be consequences of long-term untreated OSA (e.g., cardiovascular diseases, kidney disease, hypertension, type 2 diabetes [111, 112]), some of which, including gout, may be reversible just by resolving the OSA [113] (See Table 2). Gout may first manifest long before these other life-threatening consequences of OSA. Because OSA is so largely underdiagnosed [15], gout may be the first indicator, and an early warning, of its presence. Life-threatening diseases have similarly been reported for asymptomatic hyperuricemia [8, 19–23].
The benefit of ULT is controversial for preventing these comorbidities and their mortality. At least one paper has demonstrated that claim [114] to some extent for gout, but it requires ULT dosage to target SUA concentration < 0.36 mmo/L (equivalent to 6.0 mg/dL). Without that target SUA concentration, other papers refute ULT’s mortality benefit or present ULT risks: increasing the dosage of allopurinol was found to have no benefit for reducing the risk of major cardiovascular events or all-cause mortality in gout patients [115, 116]; a meta-analysis found no difference in all-cause mortality between gout patients using allopurinol vs. using no ULT [117]; examining the multi-ethnic study of atherosclerosis (MESA) database found cardiovascular disease to be more prevalent in those gout patients using ULT than not [118]; ULT did not prevent the development of cardiovascular disease in Japanese patients with asymptomatic hyperuricemia [119]; older adults using allopurinol are subject to persistent physical disability [120]; and those who are hypersensitive to allopurinol often have serious reactions with high mortality [121]. Febuxostat was found to further increase the risk of major cardiac events and mortality [122, 123]. The controversies of ULT benefit for chronic kidney disease are reviewed by Goldberg et al. [124] and Kohagura et al. [125].
A trial, reported to be mostly successful in using allopurinol dose escalation to achieve a target SUA concentration of 0.36 mmo/L, was conducted in people with gout over an average period of 6.5 years per completing participant, reporting that 60 of the 183 original participants (33%) had died during the study [126]. This report demonstrates that gout risks premature death, even with targeted ULT treatment. Unresolved OSA also risks premature death [127, 128], and is likely the underlying major reason for the risk of premature death with gout and hyperuricemia. Dr. Nathaniel Marshall, the lead author of the second SA mortality study [128], commented that his study results showed that SA has about the same effect on mortality as getting 18 years older [129].
Alternatively, resolving OSA early enough reduces the risk of developing most of these life-threatening diseases. Although unresolved OSA has been shown to elevate the risk for cardiovascular disease [79], the rate of cardiovascular events has been shown to be the same in those with OSA resolved by continuous positive airway pressure (CPAP) vs. individuals with no OSA [130]. CPAP also has been shown to reduce the recurrence of atrial fibrillation [44], to restore cardiac mechanical function [50], and to reverse resistant hypertension [51–53]. CPAP also has been shown to ameliorate the progression of chronic kidney disease, and to reverse it in some cases [47–49]. Unfortunately, CPAP used to treat OSA usually has not improved type 2 diabetes [96, 97], although it may improve insulin sensitivity. Insulin insufficiency from pancreatic beta cell dysfunction and mass reduction remains to keep hemoglobin A1c elevated and diabetes still a threat. Perhaps the earlier resolution of OSA would have resulted in better improvement of type 2 diabetes.
Following effective OSA treatment, the OSA-induced flares of hyperuricemia and gout would cease because OSA’s overproduction and underexcretion episodic effects on hyperuricemia no longer occur, and the reduced GFR often reverses within three months of effective treatment for OSA [47, 49]. After OSA has been resolved so its elevation of SUA is discontinued, the residual MSU stores throughout the body from previous OSA hypoxia would dissolve very slowly, replenishing elevated SUA’s depletion until they do [131, 132]. Their dissolution may be accelerated by ULT.
Up until now, modern medicine has equivocated about the need to treat asymptomatic hyperuricemia. Of course, the treatment of hyperuricemia has tacitly envisioned urate lowering drugs as the treatment to be used. This author propounds diagnostic testing for OSA, followed by its resolution as warranted, as a necessary inclusion in the treatment for hyperuricemia. That should provide the benefit of significant risk reduction for the development, and even reversal, of the diseases listed in Table 2. Maintaining OSA resolution also should prevent the return of hyperuricemia, even after urate lowering drugs have been discontinued.
The information in this paper indicates that neither gout nor hyperuricemia should be considered to be a systemic disease when resulting from OSA. They each should be considered to be a symptom of the systemic disease of OSA.
This paper has: (1) demonstrated the reasons that most hyperuricemia, whether asymptomatic or gouty, would be caused by OSA’s hypoxic episodes; (2) presented the perils of leaving OSA unresolved; and (3) propounded the use of periodic ultrasonic examination to screen for the onset of OSA by detecting significant agglomerations of MSU crystals, leading to diagnostic testing and effective treatment for OSA. Further study is needed to determine the sensitivity and specificity of ultrasonic detection of MSU crystals for OSA screening, as described more fully in [109].
Further studies also are needed to determine: (1) the prevalence of OSA in a cohort of randomly selected gout patients; (2) a comparison of OSA resolution vs. ULT for prevention of future gout flares; and (3) a comparison of OSA resolution vs. ULT for prevention of gout’s premature mortality. But neither people with gout nor people with hyperuricemia should be left at risk of premature death until the studies are completed.
In the meantime, the onset of gout or diagnosis of hyperuricemia is a warning that should lead to OSA’s prompt diagnosis, and treatment when indicated, thereby preventing continued development of OSA with later life-threatening and often irreversible consequences [133] (see Table 2), leading to a shortened lifespan. Even though only one clinical study of this approach has been performed, a few medical practitioners have followed this approach, finding that most of their tested gout patients were diagnosed with OSA. Resolving OSA not only can prevent further flares of gout and hyperuricemia, but if accomplished early enough, also can extend the length and quality of life.
APO: apolipoprotein
CPAP: continuous positive airway pressure
DECT: dual energy computed tomography
GFR: glomerular filtration rate
HIF‑1α: hypoxia‑inducible factor‑1α
IL-1β: interleukin-1β
MSU: monosodium urate
OSA: obstructive sleep apnea
SA: sleep apnea
SUA: serum uric acid
UA: uric acid
ULT: urate lowering therapy
The supplementary material for this article is available at: https://www.explorationpub.com/uploads/Article/file/100715_sup_1.pdf.
The author gratefully acknowledges the information gleaned from interchanges with Dr. Richard Johnson, nephrologist; Dr. Peter Delannoy, biochemist; and Dr. Steven Weintraub, pulmonologist.
BA: Conceptualization, Investigation, Writing—original draft, Writing—review & editing.
The author declares that there is no conflict of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.