
Abstract
Neuroinflammation plays a key role in the pathogenesis of post-cardiac arrest (CA) brain injury. Innate immune cells sense a variety of danger signals through pattern-recognition receptors and evoke rapidly after ischemic challenge, triggering inflammatory responses and amplifying brain damage. A programmed cell death (PCD) pathway is activated after ischemic and/or inflammatory stimuli, leading to the elimination of the damaged cells. However, PCD also regulates inflammatory responses flexibly. The present review aimed to summarize the mechanisms of inflammatory responses, including the biology of immune cells, the innate immune recognition that initiates the inflammation, and the immunomodulatory effects of PCD following CA. Promising therapeutic approaches of targeting inflammatory responses to alleviate brain injury and improve neurological outcomes after CA are also reviewed.
Keywords
Cardiac arrest, neuroinflammation, innate immune recognition, programmed cell death, microgliaIntroduction
Cardiac arrest (CA) represents one of the most devastating medical emergencies and is a leading cause of mortality and morbidity in China and worldwide [1]. Based on the 2020 CA Registry to Enhance Survival (CARES) registry, the incidence of emergency medical services (EMS)-treated out-of-hospital CA (OHCA) in people of any age is 88.8 individuals per 100,000 population [2]. Despite advancements in the “chain of survival” by health staff and EMS, as well as increased awareness of cardiopulmonary resuscitation (CPR), just 24.0% of patients who have an OHCA survive until hospital admission, and 9.0% survive to hospital discharge [2]. Even if the return of spontaneous circulation (ROSC) is achieved, the post-CA syndrome is associated with a high mortality rate after hospitalization [3, 4]. Post-CA syndrome is characterized by hypoxic-ischemic brain injury, systemic ischemia-reperfusion injury, myocardial dysfunction, and persistent precipitating pathology [4]. Hypoxic-ischemic brain injury after resuscitation is the principal cause of death and poor neurological outcomes in the OHCA population [5, 6], taking on the character of delayed neuronal death. Previous studies have shown that most neurons lose function over time [4], but the mechanisms are not fully understood. The “two-hit” model was proposed to describe the pathophysiology of post-CA brain injury, which is defined by primary (ischemic) injury caused by the immediate cessation of cerebral blood flow during CA and secondary (reperfusion) injury caused after resuscitation [7]. Secondary injury, in which neuroinflammation has been widely recognized as playing an essential role, further amplifies the primary brain injury, causing delayed neuronal death and poor neurological outcomes [6].
Neuroinflammation is characterized by multiple pathological processes. Glial cells are activated during neuroinflammation, accompanied by the infiltration of peripheral immune cells as well as the release of pro-inflammatory mediators, including cytokines and adhesion molecules [8]. Specifically, the activation of the innate immune system has been shown to play a critical role in all phases of post-CA brain injury. Innate immune cells, such as microglia/macrophages, elicit and release cytotoxic substances [9, 10], which initiate inflammatory responses after ischemia [8]. Active innate immune responses and the subsequent release of pro-inflammatory mediators activate programmed cell death (PCD). PCD not only is the outcome of the cells suffering the inflammatory injury but also fuels the inflammatory responses via pro-inflammatory intracellular contents released in lytic types of PCD such as pyroptosis [11] and necroptosis [12], inducing a vicious cycle leading to excessive inflammation. Here, existing evidence on the immune and inflammatory mechanisms underlying post-CA brain injury are described and emerging pharmacological interventions focused on anti-inflammation will also be introduced.
Pathophysiology of post-CA brain injury
Post-CA brain injury occurs in stages. Primary injury is rapidly triggered by cessation of cerebral blood flow within minutes to hours after CA, leading to oxygen, glucose, and ATP depletion, causing the dysfunction of Na+/K+ adenosine triphosphatase (ATPase), resulting in a massive influx of sodium and water and intracellular cytotoxic edema. Loss of plasma membrane potential due to the dysfunction of Na+/K+ ATPase triggers the opening of voltage-gated ion channels, causing intracellular calcium overload [6, 7]. During resuscitation, the restoration of oxygen oxidative phosphorylation limits ongoing hypoxic damage, but also promotes the generation of reactive oxygen species (ROS), which damages DNA, proteins, and lipids, further aggravating intracellular damage [6, 13]. Secondary injury after the restoration of cerebral blood flow generally occurs over hours to days after resuscitation, microglia/macrophages are activated and circulating leukocytes migrate into the central nervous system (CNS), amplifying brain damage through the production of ROS, matrix metalloproteinases (MMPs), and cytokines/chemokines, further leading to blood-brain barrier (BBB) breakdown [14], brain edema, and neuronal death. Consistent with the course of secondary injury, clinical data also showed that brain injury following CA is often devastating beyond 72 h post-arrest, indicating the essential role of secondary injury in the pathogenesis of post-CA brain injury [15].
Synopsis of the biology of immune cells after CA
Following the restoration of blood flow, innate and/or adaptive immunity components are activated, and neuroinflammatory responses are induced. Microglia, the resident macrophages in the CNS, play an initiative role in these inflammatory cascades. Activated microglia further recruit peripheral immune cells into the brain, including peripheral myeloid cells and lymphocytes.
Microglia
Microglia, the resident innate immune cells in the brain, are activated rapidly after CA. Activated microglia polarize between a classically activated (M1, pro-inflammatory) phenotype or an alternatively activated (M2, anti-inflammatory) phenotype [16–18]. Microglia change dynamically and temporally into different phenotypes in different phases of injury, contributing to either tissue damage or repair in various brain diseases [19–21]. Pro-inflammatory cytokines and components, including interleukin-1β (IL-1β), IL-6, IL-12, MMP-9, and tumor necrosis factor-α (TNF-α), are secreted by activated M1 microglia [22]. M1 microglia also release chemokines like C-C motif chemokine ligand 5 (CCL5) [23] and C-X-C motif chemokine ligand 10 (CXCL10) [24] to recruit peripheral immune cells [25]. Hence, they are deeply involved in cytotoxicity, acute inflammatory responses, and BBB permeabilization. M2 microglia were considered to play a beneficial role in neuroinflammation, expressing IL-4, IL-10, IL-13, brain-derived neurotrophic factor (BDNF), and transforming growth factor-β (TGF-β) and participating in wound healing, anti-inflammatory responses, phagocytosis of debris, and extracellular matrix (ECM) protection [25, 26]. In a murine model of CA and CPR, the M1 microglia marker, CD16/32, and inducible nitric oxide synthase (iNOS) dramatically elevated after CA/CPR in brains [27]. The expression of M2 microglia markers TGF-β, arginase1 (Arg1), and IL-10 also increased in the acute phase after CA/CPR [27]. Previous studies demonstrated that activated microglia with an M2 phenotype improve neuronal survival and outcomes in a model of CA/CPR [28, 29].
However, emerging evidence has shown that microglia/macrophages act in multivariate responses which let us hard to define them as simply “M1” and “M2” [30]. These two rigid activation states are only observed in vitro [17]. Recent single-cell transcriptomics data showed that microglia often co-express M1 and M2 markers [31]. Thus, M1/M2 classification might be an outdated concept that fails to fully address microglia’s function [31–33]. Microglia express a highly plastic phenotype and function, displaying multivariate states in the CNS [30]. Integrative analyses of single-cell transcriptome and multi-omics data have identified different microglial states [34], including disease-associated microglia (DAM) [35], microglial neurodegenerative phenotype (MGnD) [36], and proliferative-region-associated microglia (PAM) [37]. In future studies, using as many layers of complexity as possible to describe different states of microglia in post-CA brain injury should be considered.
Peripheral myeloid cells
Individuals suffering from a systemic inflammatory response after OHCA represent a significant increase of pro-inflammatory cytokines in blood [38]. Peripheral myeloid cells are activated by danger/damage-associated molecular patterns (DAMPs) and pro-inflammatory cytokines from systemic circulation. Several studies have shown that peripheral myeloid cells invade the CNS parenchyma after CA in murine models [39, 40]. Chemokines suchCCL2 [41] or CCL5 [23] released by microglia, BBB leakage [39], and DAMPs released from injured brain regions may all contribute to the infiltration of peripheral myeloid cells in the brain. However, the roles played by peripheral myeloid cells in post-CA brain injury are still indeterminate. Recent studies have reported that the crosstalk between peripheral inflammation and neuroinflammation in ischemic stroke via triggering receptor expressed on myeloid cells 1 (TREM1), an inflammatory type I membrane receptor expressed on myeloid lineage cells, leads to detrimental brain infiltration [42, 43]. In experimental models of various diseases, TREM1 magnifies the pro-inflammatory responses by synergizing with classical pattern-recognition receptors (PRRs) [44–46]. Clinical data also show that plasma levels of TREM1 increase after CA [47], indicating that the peripheral TREM1 expressed by peripheral myeloid cells might involve in the pathogenesis of post-CA neuroinflammation, although the underlying mechanisms are incompletely understood.
Lymphocytes
Following ROSC, CD4+ and CD8+ T lymphocytes accumulate in the brain [39, 48–50]. In a mouse model of CA/CPR, lymphocytes infiltrated the CNS within 3 h after resuscitation and maintained for at least 72 h [48]. Most infiltrating lymphocytes are pro-inflammatory CD4+ T cells, producing pro-inflammatory cytokines, including TNF-α and interferon-γ (IFN-γ) [48]. Functional T cell deletion via T cell receptor α (TCRα) knockout significantly attenuated brain injury following CA/CPR [48]. Lymphocyte biology in post-CA neuroinflammation is worthy of further investigation.
Innate immune recognition ignites neuroinflammation after CA
As the resident innate immune cells in the brain, microglia use multiple PRRs to sense a variety of host-derived DAMPs and pathogen-associated molecular patterns (PAMPs) (Figure 1) [51–53]. There is growing evidence to indicate that innate immune recognition results in developing or accelerating inflammation in the CNS after CA.
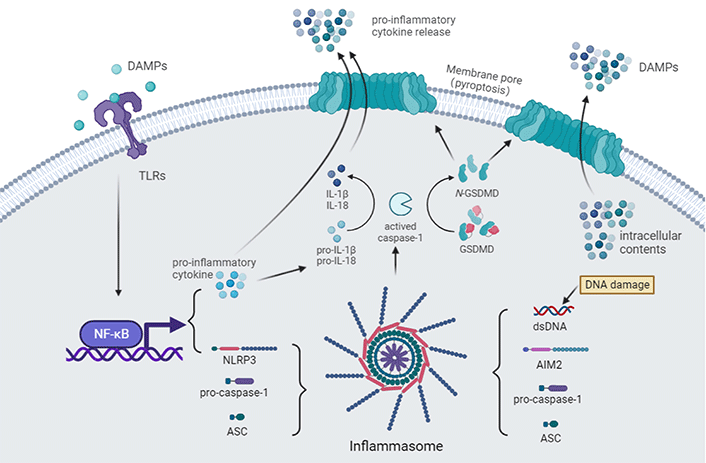
Innate immune recognition after CA. The toll-like receptor (TLR) pathway is activated after binding with DAMPs [e.g., high mobility group box-1 (HMGB1)], leading to the expression of pro-inflammatory cytokines and nucleotide-binding and oligomerization domain (NOD)-like receptor (NLR) family pyrin domain (PYD) containing 3 (NLRP3) via activating nuclear factor-κB (NF-κB) signaling [54]. Accompanied by cellular perturbations (e.g., icon flux, mitochondrial damage, lysosomal damage, and metabolism), NLRP3, apoptosis-associated speck-like protein (ASC), and pro-caspase-1 assemble to form the NLRP3 inflammasome. The generation of double-stranded DNA (dsDNA) by DNA damage after an ischemic insult can also activate the absent in melanoma 2 (AIM2) inflammasome [55, 56]. Pro-caspase-1 undergoes self-cleavage and becomes its active form. Active caspase-1 further activates pro-inflammatory cytokines [57–59]. Active caspase-1 also cleaves gasdermin-D (GSDMD), unmasking the N-terminal domain, which forms pores in the plasma membrane, leading to cell swelling and pyroptosis, enabling the release of pro-inflammatory cytokines and other intracellular contents that act as DAMPs in the extracellular environment [60]
TLRs signaling
TLRs are members of the toll/IL-1 receptor (TIR) family. To date, 10 human functional TLRs and 13 murine TLRs have been identified [55, 61]. Microglia express a wide range of TLRs that respond to various CNS-damaging insults via PRR-mediated signaling [62, 63]. TLR4 is the first TLR discovered in humans and the most studied member of the TLR family [64]. TLR4 activation leads to the expression of cytokines such as IL-1β, TNF-α, cyclooxygenase 2 (COX-2), and iNOS via activating NF-κB signaling [65, 66]. A significant increase level of HMGB1, a key ignition molecule for TLR4, had been reported in both resuscitated patients and animals [54, 67]. HMGB1 is a non-histone DNA-binding protein that localizes at the nucleus [68]. In extreme pathogenic, immunoinflammatory, and environmental danger conditions, HMGB1 is released to the extracellular milieu and triggers inflammatory responses through binding to TLR4 [69]. Previous studies showed that the inhibition of the TLR4 signal restored BBB integrity, downregulated the expression of inflammatory cytokines, and protected the brain in models of spontaneously hypertensive [70], intracerebral hemorrhage [71], and ischemic stroke [72]. In a CA/CPR model, blocking the HMGB1/TLR4/NF-κB pathway with an HMGB1 blocking peptide significantly attenuated dendritic damage and neuronal death in the hippocampal CA1 while reducing neuroinflammation [54]. At present, the inhibition of TLR4 can be a promising therapeutic strategy for post-CA brain injury.
NLR signaling
NLRs, a subgroup of PRRs located in the cytosolic compartment of cells [73], are classified into four subfamilies according to their characteristic N-terminal domains, among them the NLRP family has been identified to play a critical role in innate immune and inflammatory responses and [74], to cope with the presence of noxious stimuli in the cytosol, NLRs and other proteins assembled a high-molecular-weight multiprotein complex [75, 76]. The most well-characterized inflammasome is the NLRP3 inflammasome. NLPR3 is expressed and induced after cerebral ischemia, mainly in microglia [77]. Upon activation, NLRP3 generally complexes with ASC, the adaptor protein, via PYD-PYD interaction. The caspase recruitment domain (CARD) at the C-terminal of ASC further recruited pro-caspase-1, resulting in self-cleavage and activation of caspase-1. Consequently, active caspase-1 activates pro-inflammatory cytokines, including IL-1β and IL-18 [57–59]. Diverse cellular perturbations trigger NLRP3 activation, including icon flux, mitochondrial damage, lysosomal damage, and metabolism [78].
NLRP3 inflammasome activated in the brain after CA [40]. Depending on different causes of CA, NLRP3 was induced at different time courses after resuscitation. In the asphyxia-induced and electric-induced CA model, the protein level of NLRP3 was significantly up-regulated within 6 h after resuscitation in microglia [40, 79], while 72 h in potassium-induced CA [80]. It has been suggested that high extracellular K+ can block the activation of NLRP3 [81]. Different extracellular K+ levels may be conducive to explaining the diversity of NLRP3 biology in different models. Co-immunoprecipitation assay has revealed the interactions among NLRP3, ASC, and caspase-1 at 12 h after asphyxia-induced CA, which implied that CA activates the assembly of NLRP3 inflammasome [40].
MCC950, a potent and specific NLRP3 inflammasome inhibitor [82], has shown its efficacy in the CA murine model. MCC950 treatment attenuated inflammatory responses, suppressing the pro-inflammatory cytokine release [40, 80]. The improvement of neurologic function and survival rate was observed after MCC950 treatment in both the asphyxia-induced and the potassium-induced models [40, 80]. Interestingly, the neuroprotective effect of some neuroprotective therapies is dependent on the inhibition of NLRP3 inflammasome. Glibenclamide, a sulfonylurea that was generally considered to improve neurological outcomes via reducing brain edema in animal and preclinical studies [41], has been recognized that its neuroprotective effect is independent on preventing brain edema, while through the inhibition of K+ efflux, which activated NLRP3 inflammasome [83]. It emphasizes that exploring the underlying mechanism of NLRP3 inflammasome activation may advance our identification of putative targets for intervention after CA.
AIM2-like receptor signaling
AIM2 is a member of the augmenter of liver regeneration (ALR) protein family, characterized by an N-terminal PYD [pyrin and hematopoietic expression, IFN-inducible nature, and nuclear localization (HIN) domain (PYHIN)] domain and a C-terminal HIN domain [84]. Generally, AIM2 is activated by cytoplasmic dsDNA to defend against the infection of DNA viruses and intracellular bacterial pathogens [85], while it can also sense self-DNA released from dying cells after CNS injury [55, 56]. Once dsDNA binds to the HIN domain of AIM2, the PYD domain can recruit the adapter protein ASC during inflammasome assembly [86]. Like the assembly of NLRP3 inflammasome complexes, the AIM2-ASC complex recruits the effector protein, caspase-1, resulting in self-cleavage and activating pro-inflammatory cytokines [86].
The messenger RNA (mRNA) and protein level of AIM2 is elevated and synchronizes with the upregulation of inflammatory factors in the cortex after CA in a rat model [87]. It indicates that AIM2 inflammasome may be involved in the pathogenesis of post-CA brain injury, although there is no direct evidence about AIM2 inflammasome assembly. Interestingly, compared with NLRP3, AIM2 is mainly expressed in neurons rather than microglia [87]. Although the differential expression of AIM2 is associated with sterile inflammatory responses in the brain, the roles of AIM2 in post-CA neuroinflammation remain largely unknown.
PCD module inflammatory responses
PCD was generally considered as the end point event of CNS injury. However, PCD also module inflammatory responses via various signal pathways. In response to pathogenic, immunoinflammatory, and environmental danger, PCD pathways are activated to eliminate the damaged cells, including neurons, which are traditionally considered unrenewable. Based on the integrity of the plasma membrane, PCD pathways can be divided into lytic or non-lytic cell death [88]. Lytic types of PCD ultimately result in the rupture of the cell. The typical forms of lytic PCD are pyroptosis and necroptosis. Intracellular content released from lytic cells acts as DAMPs in the extracellular space. DAMPs trigger inflammatory responses via stimulating PRRs, while non-lytic forms of PCD, such as apoptosis, represent the coordinated disintegration of dying cells and are generally considered immunologically silent [88]. The complicated interactions between inflammatory responses and PCD make investigating the mechanism underlying neuroinflammation more challenging.
Apoptosis
Apoptosis is considered to be a typical, caspase-dependent PCD. Apoptosis can be triggered by extrinsic pathways, such as the activation of the death receptor, or intrinsic pathways, such as the B cell lymphoma-2 (BCL-2)-regulated apoptotic pathway [89]. Consequently, the initiator caspase-8 and 9 were proteolytically activated. Then activated caspase-8 and 9 cleave the effector caspase-3 and 7. The effector caspases cleave hundreds of cellular proteins, including polyadenosine-diphosphate-ribose polymerase (PARP), leading to the fragmentation of DNA and the exposure of phosphatidylserine (PtdSer) on the outer layer of the cell membrane of a dying cell [88].
After an ischemia insult induced by CA, apoptotic neuronal death occurs within 24 h [41, 90]. However, the pharmacological intervention focused on hindering the apoptosis pathway has failed to improve neurological outcomes and neuronal cell death after CA [91].
In the past few years, the clearance of apoptotic cells, also known as efferocytosis, was recognized to play an important role in maintaining routine tissue homeostasis [92]. Surface-exposed PtdSer on apoptotic cells is recognized by either direct PtdSer receptors, which bind PtdSer directly, or indirect PtdSer receptors that cannot engage PtdSer directly but use bridging molecules [for example, milk fat globule epidermal growth factor 8 (MFG-E8)] that bind PtdSer (Figure 2) [92]. During acute tissue injury, apoptotic cells accumulate beyond the normal rate, leading to the insufficiency of apoptotic cell clearance. Apoptotic cells undergo secondary necrosis while failing to be clean at the early stages of death, fueling inflammation by releasing intracellular contents [88, 92].
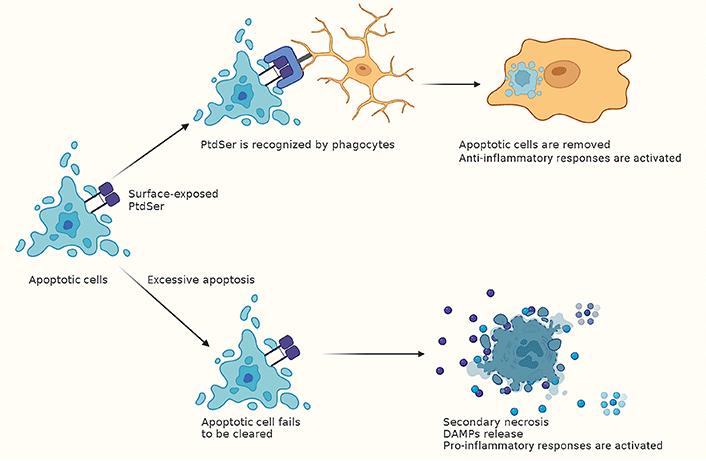
The clearance of apoptotic cells module inflammatory responses after CA. Phagocytes recognize surface-exposed PtdSer on apoptotic cells [92]. Then the apoptotic cells are removed, and anti-inflammatory responses are activated. While after CA, the number of apoptotic cells increases beyond the normal rate, leading to the insufficiency of apoptotic cell clearance. This can lead to secondary necrosis, which results in the release of DAMPs and activating pro-inflammatory responses [88, 92]
After CA, the accumulation of extensive cellular debris and cell corpses conducing to overwhelming neuroinflammation and the enlargement of secondary injury. Promoting clearance function toward damaged cells and cellular debris may be indispensable for timely eliminating the source of inflammation. In an animal study of CA/CPR, flufenamic acid treatment significantly optimizes the phagocytic capacity of microglia/macrophages, accompanied by the reduced accumulation of damaged cells in the brain and better outcomes [27]. This suggests that the clearance of apoptotic cells is likely critical in designing apoptotic cell-based therapies for post-CA brain injury.
Pyroptosis
Pyroptosis is a lytic form of cell death induced by the activation of inflammasome [40, 93]. Pyroptosis is inherently inflammatory due to the release of pro-inflammatory intracellular contents such as IL-1β and IL-18 [11]. As previously mentioned, inflammasome assembling results in pro-caspase-1 self-cleavage and activation [73, 74]. Activated caspase-1 cleaves GSDMD, the terminal executioner of pyroptosis, into an N-terminal and a C-terminal fragment. The N-terminal GSDMD fragments then oligomerize in the plasma membrane, leading to rapid plasma membrane rupture. Caspase-11 in murine and caspase-4 and 5 in humans can also cleave GSDMD and activate pyroptosis [87].
The increased protein levels of cleaved caspase-1 and cleaved GSDMD were reported in the brain after resuscitation, while no significant change in the protein levels of cleaved caspase-11 [40]. The assembly of NLRP3 inflammasome was observed at 12 h after CA through co-immunoprecipitation. NLRP3 and caspase-1 were mainly co-localized in microglia instead of other types of cells after CA [40]. Inhibiting activated caspase-1 by acetyl-tyrosyl-valyl- alanyl-aspartyl-chloromethylketone (Ac-YVAD-cmk) or NLRP3 by MCC950 prevents microglial pyroptosis and neuroinflammation and ameliorates neurological injury after CA [40]. These findings suggest that microglia pyroptosis aggravates sterile inflammation after CA in the brain.
Necroptosis
Necroptosis is a lytic, inherently inflammatory, caspase-independent PCD. Necroptosis can be induced by multiple innate immune signaling pathways, including death receptors, Z-DNA-binding protein 1 (ZBP1), and TLRs [94]. TNF receptor activation is the most well-recognized type of programmed necrosis [94]. Once the necroptosis pathway is activated, the receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-containing proteins, including RIP kinase 1 (RIPK1), TIR-domain-containing adapter-inducing IFN-β (TRIF), and ZBP1, form functional amyloid with RIPK3, leading to the oligomerization, autophosphorylation, and activation of RIPK3. Consequently, phosphor-RIPK3 phosphorylates the pseudokinase domain of mixed lineage kinase domain-like protein (MLKL). Phosphor-MLKL oligomerize and is trafficked to the plasma membrane, resulting in loss of membrane integrity [94, 95], and releasing DAMPs into the cellular surroundings.
Studies have reported necroptosis occurs in the brain after CA [96–98]. In a rat model, the expressions of RIPK1, RIPK3, and phosphor-MLKL proteins were up-regulated after 24 h of resuscitation [96]. In the swine model of CA, the phosphorylated MLKL expression level in brain tissues increases significantly [97]. Interestingly, targeted temperature management (TTM), the only therapy that has shown a profit in clinical trials [98], inhibits phosphorylated MLKL expression [97], indicating its multiple targets protectivity.
Ferroptosis
Some other types of PCD pathways also involve in neuroinflammation after CA. Ferroptosis, an iron-dependent cell death characterized by accumulation of free iron, lipid peroxidation, and plasma membrane damage [99], occurs in the brain after CA. Ferroptosis-related proteins [e.g., glutathione peroxidase 4 (GPX4) and acyl-coenzyme A synthetase long-chain family member 4 (ACSL4)] increased after resuscitation [100], accompanied by elevated ROS production [101]. The inhibition of ferroptosis via baicalein treatment relieved brain injury after ROSC [101]. Ferroptosis causes the release of DAMPs, which alert immune cells [99]. As the research on the involvement of ferroptosis in the inflammatory response after resuscitation is still insufficient, more evidence is needed to prove the link between ferroptosis and post-CA neuroinflammation.
Anti-inflammatory strategies for post-CA brain injury: preclinical studies and clinical translation
At present times, some developing therapies that prevent excessive inflammatory responses after CA emerged, including glibenclamide, minocycline, MCC950, and propofol.
Glibenclamide
Glibenclamide treatment is a potential pharmacological therapy that successfully achieved improved functional outcomes after CA in preclinical studies [102–104]. Glibenclamide regulates microglia activation and the neuroinflammatory response in the brain after CA through TLR4/NLRP3 signaling [102], improving early electrophysiologic recovery, coma recovery, arousal, and brainstem function after CA with decreased number of ischemic neurons [103]. Emerging evidence showed that glibenclamide directly prevents neuroinflammation by inhibition of K+ efflux which activates the NLRP3 inflammasome [81]. The comparable efficacy between glibenclamide treatment and TTM alone has been reported, suggesting that glibenclamide treatment is an alternative approach for post-CA brain injury [41, 104]. When given along with TTM, glibenclamide treatment substantially attenuates histological injury [41]. These preclinical results suggest that glibenclamide treatment can be a well-promising pharmacological therapy, and further exploration is needed.
Minocycline
Minocycline, a broad-spectrum tetracycline family antibiotic, is highly lipophilic with excellent penetration into cerebral tissues [105, 106]. It selectively inhibits activated microglia polarization to a pro-inflammatory (M1) state [107] and exerts anti-inflammatory functions. The preclinical studies show that even low doses of minocycline have significant anti-inflammatory effects [108], but the treatment time is important because of the nature of delayed secondary neurodegeneration and neuroinflammation nature. Only early minocycline administration shows efficacy in stroke models [107–109]. The efficacy of minocycline in post-CA brain injury remains controversial. In rat models of CA/CPR, minocycline fails to improve neurological and histological outcomes [110, 111]. However, minocycline attenuates microglial response in a mouse model and reduces neuronal death after CA [112]. Further experiments are required to clarify the safety and efficacy of minocycline treatment after CA.
MCC950
MCC950, a potent and selective NLRP3 inflammasome inhibitor targeting NLRP3 [80], significantly prevents microglial apoptosis, alleviating brain damage after CA [40]. In mouse models of CA/CPR, MCC950 treatment improves neuro-functional recovery (or neurological deficits score) and the survival rate [78, 113]. It also decreases the plasma concentrations of IL-1β (by suppressing IL-1β mRNA levels) [113] and neuron-specific enolase [113].
However, a phase II clinical trial for rheumatoid arthritis shows a concerning metabolism and toxicity of MCC950, which increases serum liver enzyme levels [114], while no evidence was found in vitro. MCC950 has more therapeutic results in animal models of autoimmune diseases, cardiovascular diseases, metabolic diseases, and other diseases [114], but its functions in brain injury after CA still have potential. Inhibition of NLRP3 inflammasome with MCC950 could be a promising therapy to improve outcomes after CA/CPR.
Propofol
Evidence from in vitro [115] and in vivo [116] studies indicate that propofol can modulate microglial activation and has potential anti-inflammatory functions. Propofol induces the anti-inflammatory treatment by suppressing microglial activation after CA and TNF-α and IL-1β release, likely via the purinergic ligand-gated ion channel 7 receptor (P2X7R)/phosphor-p38 (p-p38) pathway [117]. Another study shows propofol might reduce microglia activation and neurotoxicity by inhibiting extracellular vesicle release [118]. These shreds of evidence suggest that propofol may be a new treatment for neuroinflammation after CA.
Conclusions
In summary, significant progress has been made in understanding the inflammatory responses in the CNS after CA. Recent work [19, 38–41] has begun to clarify the complex roles of different immune cells in neuroinflammation, while the crosstalk between immune cells and nonimmune cells, neuroinflammation, and peripheral inflammation remains to be determined. Regarding the immunomodulatory effects of different PCD pathways, there are still several unresolved questions, such as how innate immune recognition interacts with PCD pathways, how PCD pathways affect the prognosis of post-CA brain injury, and how different immune cell subpopulations sense PCD and then amplify or attenuate the subsequent inflammatory responses. The investigation of associations among immune cells, innate immune recognition, and PCD in neuroinflammation after CA will be helpful in applying the theoretical framework to the diagnosis and therapy.
Abbreviations
| AIM2: |
absent in melanoma 2 |
| ASC: |
apoptosis-associated speck-like protein |
| BBB: |
blood-brain barrier |
| CA: |
cardiac arrest |
| CCL5: |
C-C motif chemokine ligand 5 |
| CNS: |
central nervous system |
| CPR: |
cardiopulmonary resuscitation |
| DAMPs: |
danger/damage-associated molecular patterns |
| dsDNA: |
double-stranded DNA |
| GSDMD: |
gasdermin-D |
| HIN: |
hematopoietic expression, interferon-inducible nature, and nuclear localization |
| HMGB1: |
high mobility group box-1 |
| IFN-γ: |
interferon-γ |
| IL-1β: |
interleukin-1β |
| MLKL: |
mixed lineage kinase domain-like protein |
| NF-κB: |
nuclear factor-κB |
| NLR: |
nucleotide-binding and oligomerization domain-like receptor |
| NLRP3: |
nucleotide-binding and oligomerization domain-like receptor family pyrin domain containing 3 |
| OHCA: |
out-of-hospital cardiac arrest |
| PCD: |
programmed cell death |
| PRRs: |
pattern-recognition receptors |
| PtdSer: |
phosphatidylserine |
| PYD: |
pyrin domain |
| RIPK1: |
receptor-interacting protein kinase 1 |
| ROS: |
reactive oxygen species |
| ROSC: |
return of spontaneous circulation |
| TLR: |
toll-like receptor |
| TNF-α: |
tumor necrosis factor-α |
| TREM1: |
triggering receptor expressed in myeloid cells 1 |
| TTM: |
targeted temperature management |
Declarations
Author contributions
YZ and ZL equally contributed to: Conceptualization, Writing—original draft, Writing—review & editing. SP and KH: Conceptualization, Supervision, Funding acquisition. KZ, YC, JC, and MAN: Writing—review & editing. All authors read and approved the submitted version.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China [No. 81871030, 82072133, 82171345]; the Guangzhou Science and Technology Plan Project [202206010032]; and the Guangdong Basic and Applied Basic Research Foundation [2021A1515010922, 2021A1515011017]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2023.