 Review
Review

Affiliation:
1Biophysics and Structural Genomics Division, Saha Institute of Nuclear Physics, A CI of Homi Bhabha National Institute, Kolkata 700064, India
†These authors contributed equally to this work.
ORCID: https://orcid.org/0009-0003-9661-6992
Affiliation:
1Biophysics and Structural Genomics Division, Saha Institute of Nuclear Physics, A CI of Homi Bhabha National Institute, Kolkata 700064, India
†These authors contributed equally to this work.
ORCID: https://orcid.org/0009-0002-8212-253X
Affiliation:
2Chemistry Department, Indian Institute of Technology Bombay, Maharashtra 400076, India
Email: piyalimajumder0407@gmail.com
ORCID: https://orcid.org/0000-0002-6538-9315
Affiliation:
1Biophysics and Structural Genomics Division, Saha Institute of Nuclear Physics, A CI of Homi Bhabha National Institute, Kolkata 700064, India
Email: debashis.mukhopadhyay@saha.ac.in
ORCID: https://orcid.org/0000-0002-4516-2260
Explor Neurosci. 2024;3:1–26 DOI: https://doi.org/10.37349/en.2024.00033
Received: May 25, 2023 Accepted: November 30, 2023 Published: February 20, 2024
Academic Editor: Ryszard Pluta, Medical University of Lublin, Poland
The article belongs to the special issue Alzheimer’s Disease
Receptor tyrosine kinases (RTKs) are known to perform versatile roles in disease landscapes, which determine the fate of the cell. Although much has been discussed from the perspective of proliferation, this review focuses on the impact of RTK-mediated signaling and its role in cytoskeletal degradation, the penultimate stage of cellular degeneration. In the case of degenerative diseases such as Alzheimer’s disease (AD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD), age-related macular degeneration (AMD), and type 2 diabetes mellitus (T2DM), RTK signaling has been reported to be perturbed in several studies. The implications of downstream signaling via these receptors through canonical and noncanonical pathways alter the status of actin filaments that provide structural integrity to cells. Degenerative signaling leads to the altered status of rat sarcoma (Ras), Ras homologous (Rho), Ras-related C3 botulinum toxin substrate (Rac), and cell division control protein 42 (Cdc42), the best-characterized components of the cytoskeleton remodeling machinery. RTKs, along with their diverse adaptor partners and other membrane receptors, affect the functionality of Rho family guanosine triphosphate hydrolases (GTPases), which are discussed in this review. To conclude, this review focuses on therapeutic strategies targeting RTKs and Rho GTPase-mediated pathways that can be more effective due to their combined multifactorial impact on neurodegenerative cascades.
Cell surface receptor proteins such as receptor tyrosine kinases (RTKs) act as the gateway of signal transduction into the cell [1]. Extracellular ligands bind to the respective receptors and mediate transphosphorylation to initiate the downstream signaling pathways. This across-the-membrane signaling is indispensable for the normal functioning of a cell, such as maintaining cell growth, cell-to-cell communications, and development [2]. Additionally, this RTK-mediated signaling, along with several other growth factors (GFs) and downstream molecules, plays a pivotal role in neuronal function and development. For example, neurotrophins and other GFs that are expressed in very limited amounts are extremely crucial in regulating neuronal development, plasticity, and survival [3]. By binding to their respective receptors, such as the tropomyosin receptor kinase (TRK) family RTK and p75 neurotrophin receptor (p75NTR), the neurotrophins activate several downstream signaling pathways, including mitogen-activated protein kinase (MAPK), phosphoinositide 3 kinase (PI3K), and members of the cell division control protein 42 (Cdc42)/rat sarcoma (Ras) homologous (Rho)/Ras family [4]. Mice lacking TRKA receptors suffer severe sensory neuropathies and death [3]. TRKA also regulates the expression and activity of several survival proteins, such as B-cell lymphoma-extra large (Bcl-XL), caspase-9, and Bcl-2-associated X protein (Bax). Fibroblast GF receptor (FGFR) and epidermal GF receptor (EGFR) are crucial for neuronal development and survival. EGFR-deficient mice showed degeneration in different parts of the brain, neuronal apoptosis, and delayed glial fibrillary acidic protein (GFAP) expression [5]. All these studies imply that RTK signaling is important in maintaining the level of survival proteins and axon growth by regulating gene transcription, synthesis, and degradation.
Most of these signaling pathways are tightly regulated by different post-translational modifications (PTMs), such as phosphorylation, acetylation, SUMOylation, ubiquitination, and glycosylation [6], and their dysregulation results in neuronal defects [7]. PTMs are also responsible for the buildup of misfolded proteins in neurodegenerative disorders, which leads to endoplasmic reticulum (ER) stress, unfolded protein responses, and deregulation of downstream signaling pathways [6]. Studies have shown that decreased H3 and H4 acetylation results in decreased neurotrophic factor expression and increased deposition of amyloid β (Aβ) plaques, neurofibrillary tangles, and Lewy bodies in Alzheimer’s disease (AD) and Parkinson’s disease (PD) [6]. Overload of Aβ in AD causes disruption in alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid subtype glutamate receptor (AMPAR) trafficking and subsequent deficits in long-term potentiation (LTP) [8]. AMPARs belong to a class of glutamate receptors that are widely present in neurons and glial cells and are important in the regulation of synaptic plasticity [9]. There are multiple reports regarding PTMs of the C-terminal tail of the AMPAR subunit, and increased acetylation of AMPAR results in a significant deficit in synaptic plasticity [10]. Again, tubulins which are one of the main components in neuronal cytoskeleton are also regulated by several PTMs [11]. For example, phosphorylation of serine 172 in neuronal β-tubulin regulates microtubule dynamics and neuronal function whereas, mutation of this residue results in migration defects, and brain dysgenesis. Apart from that, other modifications of tubulin like acetylation, polyglutamylation, and tyrosination are also associated with microtubule dynamics [11].
Perturbation of the RTK-mediated signaling, therefore, results in several diseases, including proliferative processes like cancer as well as many degenerative diseases. Degeneration results from the progressive loss of structure and function of the affected cells owing to genetic, hereditary, or other factors. In the context of degenerative disorders, RTK-mediated signaling leading to cytoskeleton disintegration is important, as the regulation of RTKs remains the determining factor behind the fate of the cell in many cases. Recent studies have revealed that cytoskeleton rearrangements and membrane lipid characteristics are essential to determine receptor association in the absence of ligands [1]. Simultaneously, numerous extracellular ligand-mediated RTK signaling pathways result in cytoskeletal loss. For example, the PI3K-protein kinase B (AKT) and Rho-Rho-associated coiled-coil kinase (ROCK) pathways are major pathways for controlling cytoskeleton dynamics, cell adhesion, and cell motility [12]. Cytoskeleton disassembly has been reported in many degenerative diseases, ultimately leading to cell death. Rho family proteins serve as key regulators for this modulation of actin cytoskeleton dynamics [13]. Several RTKs, such as mesenchymal-epithelial transcription factor (c-MET), platelet-derived GF receptor (PDGFR), anaplastic lymphoma kinase (ALK), and receptor related to tyrosine kinase (RYK), are involved in cytoskeletal remodeling through different signaling pathways and downstream effector proteins, the dysregulation of which leads to degenerative disease scenarios. Previous studies from [14] showed that the downregulation of RTKs ALK and RYK through respective microRNAs (miRNAs) results in upregulation of the transcription factor paired box 4 (PAX4) and consequent upregulation of GF receptor-bound protein-2 (Grb2) and NADPH oxidase 4 (NOX4), leading to loss of cytoskeletal proteins such as tubulin, vimentin, α-smooth muscle actin, and stathmin1 via the Grb2/PI3K/AKT/RhoA/Rac1/Cdc42/cofilin pathway. However, the reversal of cytoskeletal degradation occurs upon Grb2 overexpression, which is a common adapter for both RTKs. A microarray-based study identified PAX4 as a top candidate that is involved in neurodegeneration in PD, Huntington’s disease (HD), and AD [15]. Impaired RTK signaling has also been reported in PD. α-Synuclein aggregation, which is a pathological hallmark of PD, specifically inhibits brain-derived neurotrophic factor (BDNF)/TRKB signaling by binding to the kinase domain of the TRKB receptor, suppressing the lipid raft distribution of the BDNF/TRKB complex, and trafficking the receptor, leading to dopaminergic neuronal death [16]. The RhoA/ROCK axis is also implicated in dopaminergic neuron degeneration in PD, and there is a reduced messenger RNA (mRNA) level of the Cdc42 protein in PD patients, which is involved in cytoskeletal reorganization [17]. Apart from these, other common neurodegenerative disorders include amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), which are not only characterized by progressive neuronal loss and protein aggregation in specific parts of the brain but also show chronic inflammation mediated by activated microglia and astrocytes [18]. Dysregulated erythropoietin-producing hepatoma (Eph)/ephrin signaling has been reported in these diseases [18]. Eph/ephrin bidirectional signaling not only helps in neuronal communication but also mediates neuron-glia crosstalk [19]. In ALS, higher levels of ephrin type-A receptor 4 (EPHA4) are associated with motor neuron degeneration, and knockdown of EPHA4 reverses the condition and rescues axonopathy [20]. Dysregulation of cytoskeletal proteins is also associated with ALS, but the underlying mechanism linking RTK dysregulation with cytoskeletal degeneration is not well studied thus far. It is assumed that this cytoskeletal dysregulation in these neurodegenerative disorders is a consequence of impaired RTK signaling, which acts as a cue for the normal functioning of the cell. This review therefore primarily focused on the RTK-mediated regulation of cytoskeletal organization under different degenerative scenarios.
RTKs represent a major class of cell surface receptors, and thus, signaling through RTKs has been extensively studied in cancers. Many such RTKs are also involved in the pathophysiology of several degenerative diseases. All RTKs share a similar structure comprising of an extracellular domain, which is the ligand-binding site, a single hydrophobic transmembrane α-helix, and an intracellular region consisting of the juxtamembrane (JM) domain and cytosolic domain with tyrosine kinase activity [21]. Based on the structure of the extracellular domain and the ability to bind different ligands, the RTK family has been divided into 20 classes (see Figure 1) [22]. Among the 90 tyrosine kinase genes that have been reported in humans, 58 encode RTK proteins [23]. The remaining 32 are categorized as soluble nonreceptor tyrosine kinases, which are further divided into nine subfamilies. They are intracellular cytoplasmic proteins and play significant roles in cell signaling. Examples include Janus kinase (JAK), FES, Abelson murine leukemia viral oncogene homolog 1 (Abl), activated Cdc42 kinase 1 (ACK), spleen tyrosine kinase (SYK), Tec protein tyrosine kinase (TEC), focal adhesion kinase (FAK), Src, and C-terminal Src kinase (Csk) [24].
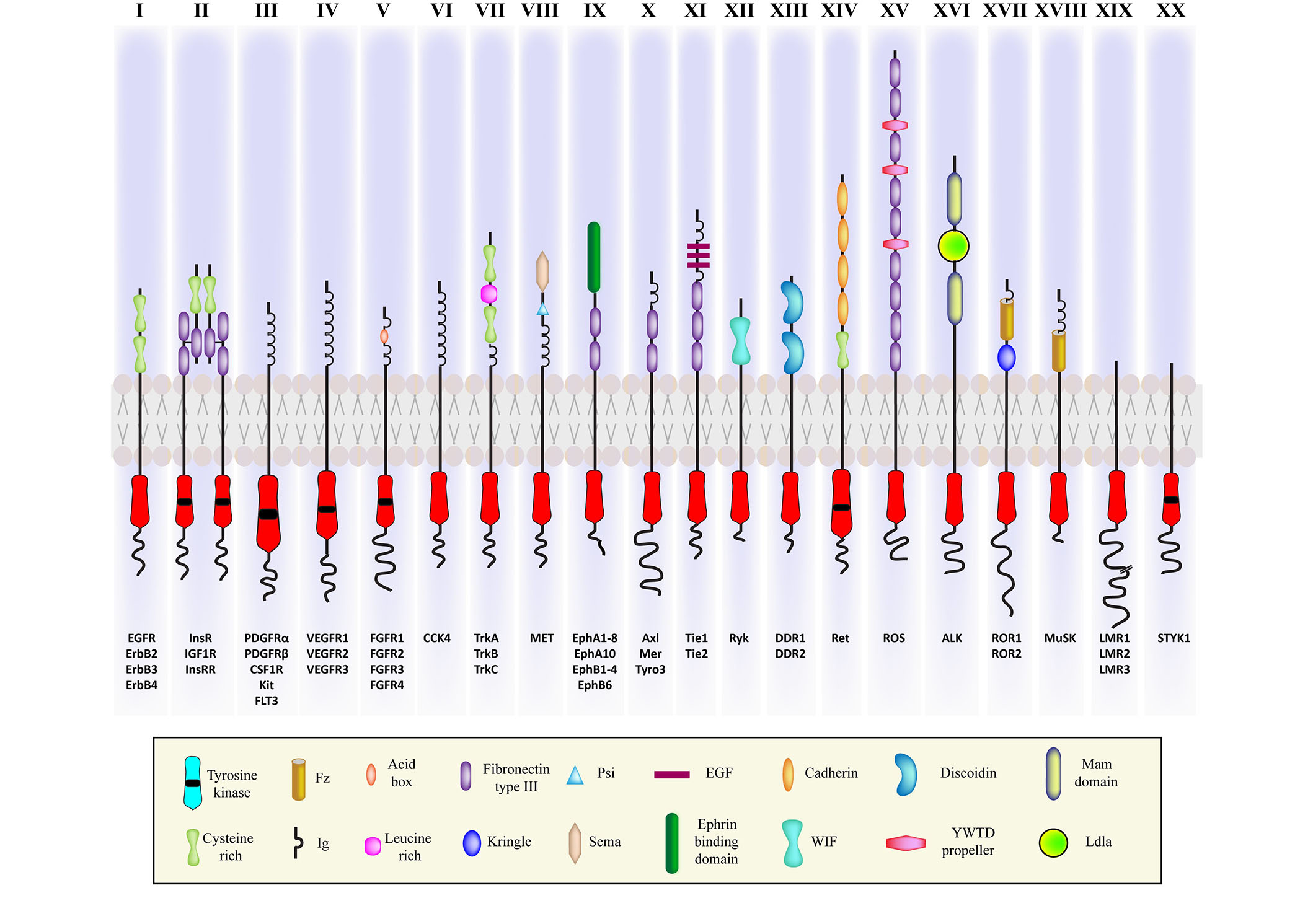
Illustration of the representatives of twenty classes of RTKs with variable cytosolic and ligand binding extracellular domains. ErbB: erythroblastic leukemia viral oncogene homolog; InsR: insulin receptor; IGF1R: insulin (INS)-like GF 1 receptor; InsRR: InsR-related receptor; CSF1R: colony stimulating factor 1 receptor; Kit: Kit proto-oncogene, receptor tyrosine kinase; FLT3: fibromyalgia syndrome-like tyrosine kinase 3; VEGFR: vascular EGFR; CCK4: colon carcinoma kinase 4; MET: mesenchymal-epithelial transition; Mer: proto-oncogene tyrosine kinase; Tyro3: tyrosine protein kinase receptor 3; Tie 1: tyrosine kinase with immunoglobulin-like and epidermal GF (EGF) like domain 1; DDR: discoidin domain receptor; Ret: rearranged during transfection protein; ROS: ROS proto-oncogene; ROR: receptor tyrosine kinase-like orphan receptor; MUSK: muscle specific kinase; LMR: lemur tyrosine kinase; STYK: serine/threonine/tyrosine kinase; Fz: Frizzled; Ig: immunoglobulin; PSI: plexin-semaphorin-integrin; WIF: Wnt inhibitory factor; YWTD: Tyr-Trp-Thr-Asp domain; Ldla: low-density lipoprotein receptor class A
Note. Adapted with permission from “Cell signaling by receptor tyrosine kinases,” by Lemmon MA, Schlessinger J. Cell. 2020;141:1117–34 (https://doi.org/10.1016/j.cell.2010.06.011). c2010 Elsevier Inc.
Structural studies of RTKs have provided a clear picture of their activation via ligand binding [22]. However, there is a difference in the mode of activation among the different classes of RTKs. Most RTKs are monomeric [25], and ligand binding induces receptor dimerization [21], except INS and IGF1R receptors, which exist as dimeric complexes even before ligand binding [26]. Four modes of RTK dimerization have been reported thus far [2]. For TRKA [nerve GF (NGF) receptor (NGFR)], activation is completely dependent on the ligand without any direct contact between its extracellular regions. For some RTKs, dimerization is completely receptor-mediated without any physical contact between the two activating ligands (e.g., the ErbB family). In Kit [stem cell factor (SCF) receptor], activation involves the binding of ligand homodimers to two receptor molecules that interact among themselves across a dimer interface. The other mode involves accessory molecules in addition to ligand binding and direct receptor-receptor contact, as in the FGFR family [2]. Without ligand stimulation, RTKs remain in an inactive state by autoinhibitory mechanisms [27].
Upon ligand binding, RTK signaling leads to the activation of its tyrosine kinase activity and autophosphorylation of the tyrosine residues in its cytosolic domain and often amplifies signals [28]. Some of the proteins with which the activated receptor may interact are guanosine triphosphate hydrolases (GTPases) activating protein (GAP), phospholipase C-γ, and Src-like nonreceptor tyrosine kinases, among others. These proteins may directly bind to phosphotyrosine residues or indirectly bind through adaptor proteins such as InsR substrate 1 (IRS1), factor receptor substrate 2 (FRS2), and Grb2-associated binding protein 1 (Gab1), among others [22]. Adaptor proteins, although lacking in enzymatic activities, act as important scaffolds that drive key signaling events and often help in signal amplification [29].
The adaptors have been categorized based on their functional and structural aspects. One class comprises docking proteins having multiple tyrosine phosphorylation sites to bind downstream effector proteins such as fibroblast growth FRS2 and Gab, whereas the other class has only Src homology 3 (SH3) and/or SH2 domains that bind to signaling proteins without phosphorylation sites [30]. For example, the effector proteins neural Wiskott-Aldrich syndrome protein (NWASP) and p21 activated kinase (PAK), which are involved in the organization of the cytoskeleton, physically associate with non-catalytic region of tyrosine kinase adaptor protein 1 (Nck) through the SH domain [31]. The interaction between the signaling proteins with phosphotyrosine binding motifs results in the activation of the canonical signaling pathways, most importantly those including MAPK, PI3K, JAK-signal transducer of activators of transcription (STAT), and protein kinase C (PKC), which are implicated in cell cycle control, metabolism, differentiation, and proliferation.
Another class of proteins is the Rho GTPases, which are molecular switches that regulate diverse cellular signaling pathways. Among many receptors, G-protein coupled receptors (GPCRs) are known to activate G-proteins directly. Although most G-protein-mediated signaling pathways are triggered by GPCRs through activation of the family of heterotrimeric G-proteins, another superfamily of G-proteins (Ras) is also involved, which includes the Ras, Ras-associated binding protein (Rab), adenosine diphosphate (ADP) ribosylation factor (Arf), Ras-like nuclear protein (Ran), and Rho families [32]. Rho family proteins have been studied thus far due to their involvement in regulating actin cytoskeleton reorganization and exhibiting interactions with a vast number of effector proteins, such as serine/threonine kinases, lipid kinases, and adaptor proteins, that modulate several cell processes. Reportedly out of 58 RTK genes encoded by the human genome, almost every RTK is known to activate at least one of the Rho family members [32]. In some cases, the activators of Rho GTPases, Rho guanine exchange factors (GEFs), also act as connectors between RTKs and the Rho family of proteins.
Reportedly, many RTKs undergo nuclear translocation by various mechanisms. Recently, fragmentation of the intermediate filament lamin was shown to be a potential contributing factor to such translocation events in the abundance of Aβ42, as observed by Islam et al. [33]. Cathepsin L functions as a protease to damage the nuclear boundary by fragmenting lamin B1. The study validates the results in transgenic mice and primary neurons, which makes it imperative to understand the nuclear translocation alteration in degenerative laminopathies. Upon receptor activation and ligand binding, several RTKs undergo proteolytic cleavage to shed their extracellular domain followed by subsequent processing of the transmembrane domain by the γ-secretase complex, releasing the intracellular domain from the cell membrane [34], resembling the amyloidogenic fate of amyloid precursor protein (APP). Intracellular RTK fragments can also be generated by other mechanisms, such as caspase-dependent cleavage and alternative mRNA splicing. Aside from the active RTK fragment, certain RTKs can also translocate into the nucleus as holoreceptors in response to various stimuli [34], each having a known transport mechanism, as listed in Table 1. Understandably nuclear localization of the RTKs should have a significant role in determining the cell fate, besides their signaling modalities. A comprehensive view of the RTKs and their by-far-known translocation mechanisms have been portrayed in Table 1.
A list of 20 subfamilies of human RTKs along with their family members and respective extracellular and intracellular domains
| Family name | Members | Ligands | Extracellular domain | Intracellular domain | γ-secretase cleavage | Nuclear localization status | Mechanism of nuclear localization |
|---|---|---|---|---|---|---|---|
| EGFR | ErbB1, ErbB2, ErbB3, ErbB4* | EGF, transforming GF A (TGFA), heparin-binding EGF-like GF (HBEGF), betacellulin (BTC), Amphiregulin (AREG), epiregulin (EREG), epigen (EPGN) | 2-Cysteine rich domains | JM domain, tyrosine kinase domain, a C-terminal regulatory region | Yes | Yes | Clathrin mediated endocytosis [35], retro-translocation by ER-associated trafficking machinery [36], nuclear localization signals (NLS) sequence [34] |
| InsR | InsR*, IGF1R*, IRR | INS-like GF-1 (IGF-1), IGF-2, INS | 2-Chains α and β, 2-leucine rich domain separated by a cysteine rich domain, 3-fibronectin type III (FNIII) domains | Tyrosine kinase domain | Yes | Yes | The possible mechanism includes SUMOylation/via microtubules with the help of some proteins [p150Glued, AREG, importin-β, ran-binding protein 2 (RanBP2)] or clathrin mediated endocytosis [37], NLS |
| PDGFR | PDGFRα, PDGFRβ, Kit, fibromyalgia syndrome-like tyrosine kinase 3 ligand (FLT3L), colony stimulating factor 1 (CSF1)* | Platelet-derived GF (PDGF), PDGF-A, PDGF-AA, PDGF-AB, PDGF-AB/BB, PDGF-B, PDGF-BB, PDGF-C, PDGF-CC, PDGF-D, PDGF-DD | 5-Ig-like domains | JM domain, tyrosine kinase containing a long kinase insert domain region, a C-terminal extension | Yes | Yes | Mediated by clathrin coated pits, β-importin, via TATA element modulatory factor 1 (TMF1) positive Golgi vesicles [38] |
| VEGFR | VEGFR1*, VEGFR2*, VEGFR3* | Vascular endothelial GF (VEGF)-A, VEGF-B, neuropilin-1, placental GF (PlGF), VEGF-C, VEGF-D | 7-Ig-like domains | JM domain, tyrosine kinase domain | Yes | Yes | Mediated by VEGF stimulation and complex formation with tissue transglutaminase II for endothelial cells [39], endocytosis [40] |
| FGFR | FGFR1, FGFR2, FGFR3*, FGFR4* | Fibroblast GF (FGF) 1–22 | 3-Ig-like domains | JM region, tyrosine kinase domain | Yes | Yes | Importin-β-mediated interferons (INFS) mechanism [34] clathrin mediated endocytosis [41] |
| CCK | CCK4/PTK7* | Coreceptor for Wnt signaling | 7-Ig domains | Catalytic domain lacking tyrosine kinase activity | Yes | Yes | - |
| NGFR | TRKA*, TRKB*, TRKC | NGF, BDNF, Neurotrophin 3 (NT3), NT4 | 3-Leucine repeats flanked by cysteine clusters and 2-Ig-like domains | Tyrosine kinase domain | Yes | Yes | Mediated by Carrier vesicles [42], NLS, and phosphorylation dependent process [43], mediated by importins [44] |
| Hepatocyte GF receptor (HGFR) | MET*, Recepteur d’ origine nantais (RON) | Hepatocyte GF (HGF) | Sema domain, a PSI domain, and 4-Ig-like plexins transcription factors (IPT) domains | JM domain, tyrosine kinase domain, carboxy-terminal tail region | Yes | Yes | Mediated by Gab1 and importin-β, NLS [43], integral trafficking from ER to the nuclear envelop transport (INTERNET) [34] |
| Erythropoietin-producing human hepatocellular receptor (EPHR) | EPHA1, EPHA2*, EPHA3, EPHA4*, EPHA5*, EPHA6, EPHA7*, EPHA8, EPHA10, EPHB1, EPHB2*, EPHB3*, EPHB4*, EPHB6* | Ephrin A (1–5), ephrin B (1–3) | Cysteine-rich domain and 2 FNIII repeats | Tyrosine kinase domain | Yes | Yes | pH-Dependent nuclear localization signal [34] |
| Axl | Axl*, Mer*, TYRO3* | Growth arrest specific gene 6 (Gas 6) | 2-Ig-like domains and 2-FNIII domains | Tyrosine kinase domain | Yes | Yes | NLS [45] |
| Tie | Tie*, TEK | Angiopoietin (ANG) GFs (ANG1, ANG2, ANG4) | 2-Ig-like, 1-EGF, 3-FNIII domains | Tyrosine kinase domain | Yes | Yes | Caveolin mediated nuclear translocation [46] |
| RYK | RYK* | Wnt1, Wnt3a | Leucine-rich domain with a WIF-type Wnt binding region | Tyrosine kinase domain lacking kinase activity | Yes | Yes | Chaperone mediated, suppressor of Mek null 1/2 (smek1/2) functions as a chaperone and regulates nuclear translocation [47] |
| DDR | DDR1, DDR2 | Collagen (I, IV, V, VI, VIII), collagen (I, III, X) | Discoidin domain, discoidin-like domain, extracellular juxtamembrane region | JM domain, tyrosine kinase domain | - | Yes | Stimulation with collagen leads to interaction with Sec61 translocon subunit beta (SEC61B) and results in nuclear translocation [48] |
| ROS | ROS | - | 6-FNIII like domains | Tyrosine kinase domain | - | - | - |
| Leukocyte tyrosine kinase (LTK) | LTK, ALK | ALK and LTK ligand 1 (ALKAL1), ALKAL2 | 2-Meprin A-5 protein and receptor protein phosphatase mu (2-MAM) domains 1-Ldla motif, cysteine rich and glycine rich domains | JM domain, tyrosine kinase domain | - | - | - |
| ROR | ROR1, ROR2 | Wnt5a | Ig-domain, cysteine-rich domains, and kringle-like domains | Tyrosine kinase domain, proline-rich, serine-threonine rich domains | - | Yes | Cleavage releasing mechanism in addition to Ran-GTPase pathway [45] |
| MUSK | MUSK* | Agrin | 3-Ig-like and 1-cysteine rich frizzled-like domain | A JM domain, a kinase domain, and a short cytoplasmic tail | Yes | - | - |
| LMR | Apoptosis associated tyrosine kinase (AATYK), AATYK, AATYK3 | A short extracellular domain containing leucine residues | Tyrosine kinase domain | - | - | - | |
| Ret | Ret | Glial cell line-derived neurotrophic factor (GDNF), neuturin, artemin, persephin, growth differentiation factor-15 (GDF-15) | 4-Cadherin like domain, a cysteine rich domain | JM domain, kinase domain, a C-terminal tail | - | Yes | - |
| STYK1 | STYK1 | - | It has a truncated extracellular domain, a single transmembrane domain | A relatively short intracellular domain possessing tyrosine kinase activity | - | - | - |
RTKs lie at the core of complex intertwined signaling networks, tightly regulating a plethora of intracellular signaling pathways. Some of the major players are well-studied, especially in proliferative processes such as cancer. Knowledge about their involvement in neurodegeneration is rather recent, and interestingly, the mechanism is noncanonical in many cases.
The MAPK signaling pathway is well studied and serves as a model for the RTK signaling paradigm [51]. MAPK cascades are evolutionarily conserved and ubiquitously expressed in eukaryotes. The cascade consists of three core kinases [mitogen-activated protein 3 kinase (MAP3K), MAPK kinase (MAPKK), and MAPK] with other upstream and downstream components and plays a significant role in cell growth, proliferation, and differentiation. Extracellular signal-regulated kinase 1/2 (ERK1/2), c-Jun N-terminal kinase (JNK), p38, and ERK5 are the four different MAPK cascades that have been identified [52]. When RTKs are stimulated by external factors, they activate MAPKs in a multi-step process. For example, after EGFR activation by its respective ligand, the adaptor protein Grb2, along with son of sevenless (Sos), binds to its cytoplasmic receptor tail. Translocation of Sos to the plasma membrane allows its access to Ras, which in turn is activated, and its subsequent interaction with serine-threonine kinases ultimately leads to MAPK activation [53]. Studies have shown that aberrant MAPK signaling contributes to the pathophysiology of several degenerative disorders, such as AD, PD, and ALS [54]. Several kinases, including p38, JNK, and ERK, are responsible for hyperphosphorylation of the tau protein, which leads to neurofibrillary tangle formation in the AD brain. Additionally, Aβ aggregates, another hallmark of the AD brain, activate microglial macrophages, resulting in ROS production and proinflammatory cytokines, which trigger the JNK and p38 signaling cascades, ultimately resulting in neuronal death [54].
The PI3K/AKT pathway is a highly conserved, tightly regulated multistep process [55] and is crucial for various aspects of cell growth and survival in both physiological and pathological conditions, such as cancer [56]. GPCRs and RTKs, when activated, recruit class I PI3Ks that enable the phosphorylation of phosphatidylinositol-(4,5)-bisphosphate (PIP2) to phosphatidylinositol-(3,4,5)-trisphosphate (PIP3) [57]. The accumulation of PIP3 permits AKT to localize to the plasma membrane, and its successive activation is followed by phosphorylation on serine 473 and threonine 308 by mammalian target of rapamycin complex 2 (mTORC2) and 3-phosphoinositide-dependent kinase 1 (PDK1). AKT serves as an important downstream mediator of PI3K signaling, which regulates several putative intracellular pathways, including mTORC1-mediated cellular proliferation, apoptosis, and metabolic networks. PI3K/AKT signaling is further implicated in the regulation of ROS production [57]. Disruption of PI3K/AKT signaling largely contributes to the pathogenesis of several degenerative diseases, such as AD, PD, and HD. Glycogen synthase kinase-3beta (GSK-3β) is an important downstream effector of AKT and can phosphorylate many endogenous substrates, such as transcription factors and proteins involved in cellular metabolism. It has been reported that dysregulated PI3K/AKT/mTOR signaling can enhance GSK-3β activity, leading to hyperphosphorylation of the tau protein and neurofibrillary tangle (NFT) formation in AD patients [58].
In the inositol (1,4,5) triphosphate (IP3)/diacylglycerol (DAG) pathway, calcium ions (Ca2+) perform a very diverse function inside the cell, and calcium signaling is known to regulate a plethora of intracellular processes, including metabolism, gene regulation, and autophagy [59]. Ligand (such as acetylcholine, thyroid-stimulating hormone) binding and subsequent activation of either GPCRs or RTKs leads to phospholipase C (PLC) activation [60]. The SH2 domain of the activated phospholipase binds to specific phosphotyrosines of the activated receptors, positioning the enzyme close to the substrate PIP2, where it is cleaved by PLC-β, generating two secondary messengers, IP3 and DAG [61]. IP3 binds and opens ligand-gated calcium channels on the membrane of the ER and stimulates the release of Ca2+. Ca2+ controls various downstream targets, including PKC [60]. It facilitates PKC binding and activation by DAG. Various known PKC isoforms act on different functional proteins, such as cytoskeleton proteins, transcription factors, and enzymes. PKC activation phosphorylates several downstream substrates, such as glycogen synthase and the calmodulin-binding protein neurogranin [60]. Ca2+ plays an essential role in neuronal cells in maintaining synaptic plasticity. Evidence suggests that dysregulated Ca2+ signaling is the driving force behind neurodegeneration in AD, PD, and HD [62]. Alterations in ER-Ca2+ signaling interfere with the unfolded protein response, which in turn results in increased protein load and stress in the ER, as reported in the case of AD. It also hinders the normal proteolytic cleavage of APP, resulting in increased Aβ42 fragments and hyperphosphorylated tau, the pathological hallmarks of the AD brain [63].
Apart from these major signaling pathways, several other pathways are also activated by RTKs. The JAK-STAT pathway is important for a large group of cytokines and GFs to regulate critical cellular events, such as immune system development, mammary gland formation, and stem cell maintenance. One of the vital roles of the JAK-STAT pathway is transmitting signals from the cell membrane receptor to the nucleus [64]. Various proteins, including suppressors of cytokine signaling (SOCS), protein inhibitors of activated STATs (PIAS), and protein tyrosine phosphatases (PTPs), modulate this pathway by determining the duration of the signaling cascade [64]. Aberrant JAK-STAT signaling reportedly results in autoimmune disorders, cancer, and other neuroinflammatory diseases, such as AD and PD.
Transforming GF beta (TGFβ) signaling occurs through the canonical small mothers against decapentaplegic (SMAD) pathway that regulates the expression of several hundred genes, implicating many pathways, such as ERK/MAPK and PI3K/AKT, which are involved in cell polarity and cytoskeleton dynamics [65, 66]. The TGFβ pathway has several implications in the pathophysiology of AD. Studies have shown that TGFβ1 levels increase in cerebrospinal fluid and within Aβ plaques in AD patients. Impaired SMAD3 signaling has also been observed to be associated with Aβ accumulation and neurofibrillary tangle formation. However, in contrast to its neurotoxic and deleterious role, the neuroprotective role of TGFβ has also been studied in AD [67].
RhoA/ROCK signaling is crucial in regulating actin cytoskeleton dynamics, cell shape, adhesion, and migration [68]. Rho GTPases, also known as “molecular switches”, are involved in the regulation of several cellular signaling pathways. Rho GTPases fall under the Ras superfamily of small GTPases and consist of three well-characterized members, Cdc42, Rac, and Rho. Among several effectors of Rho, ROCK mediates a large proportion of signals from Rho and modulates actin cytoskeleton dynamics [69]. Aberrant Rho/ROCK signaling has been studied in a proliferative scenario for a very long time, but recent evidence has shown its involvement in several neurodegenerative diseases as well. For example, ROCK activation is reported to increase β-secretase activity and promote Aβ production [70]. At the same time, ROCK is also responsible for the formation of NFTs, and hyperphosphorylated tau is an essential component of NFTs [70]. Furthermore, RhoA can regulate ROS [71]. A major imbalance in ROS production versus antioxidative defense has been observed in cases of AD, diabetes, age-related macular degeneration (AMD), PD, and HD. Type 2 diabetes mellitus (T2DM), also known as type III AD, is characterized by INS resistance and loss of pancreatic β cells, and Rho/ROCK signaling plays an important role. Decreased Rac1 signaling in skeletal muscles has been associated with INS resistance in INS-resistant obese and T2DM human subjects [72].
Emerging evidence shows that the plasma membrane is a compartmentalized entity with boundaries [12]. The role of its detailed structure in regulating the cytoskeleton, and vice versa, largely depends on constant crosstalk that includes Rho GTPases and creates the plausible link between external cues mediated by RTKs and intracellular cytoskeletal remodeling and degradation, as observed in the case of degenerative disorders (see Figure 2). It is conventionally assumed that receptors dimerize or oligomerize in response to ligand stimulation; however, studies show that in the absence of ligands, they can form active homodimers or heterodimers provided that the cytoskeleton provides the scaffold [1]. This ligand-independent association is affected by cytoskeletal rearrangements and lipid composition and, in some cases, by noncanonical ligands, such as in the case of Aβ activating InsR [73]. The diffusion metrics of the receptors can functionally change oligomerization, ligand-independent phosphorylation, and cytoskeletal organization probabilities. The extracellular ligands that activate respective RTKs trigger endocytosis of receptors that involve actin cytoskeleton components that mobilize the process by trapping or anchoring. Receptor-ligand interactions imperatively trigger downstream signaling pathways, and depending on differences in signal activation duration, magnitude, and associated ligands, the effectiveness of signal transduction varies [74–76]. The intricate signaling networks thus formed by several RTKs and supporting cytoskeletal elements along with downstream Rho GTPases are crucial to be considered together in neurodegeneration.
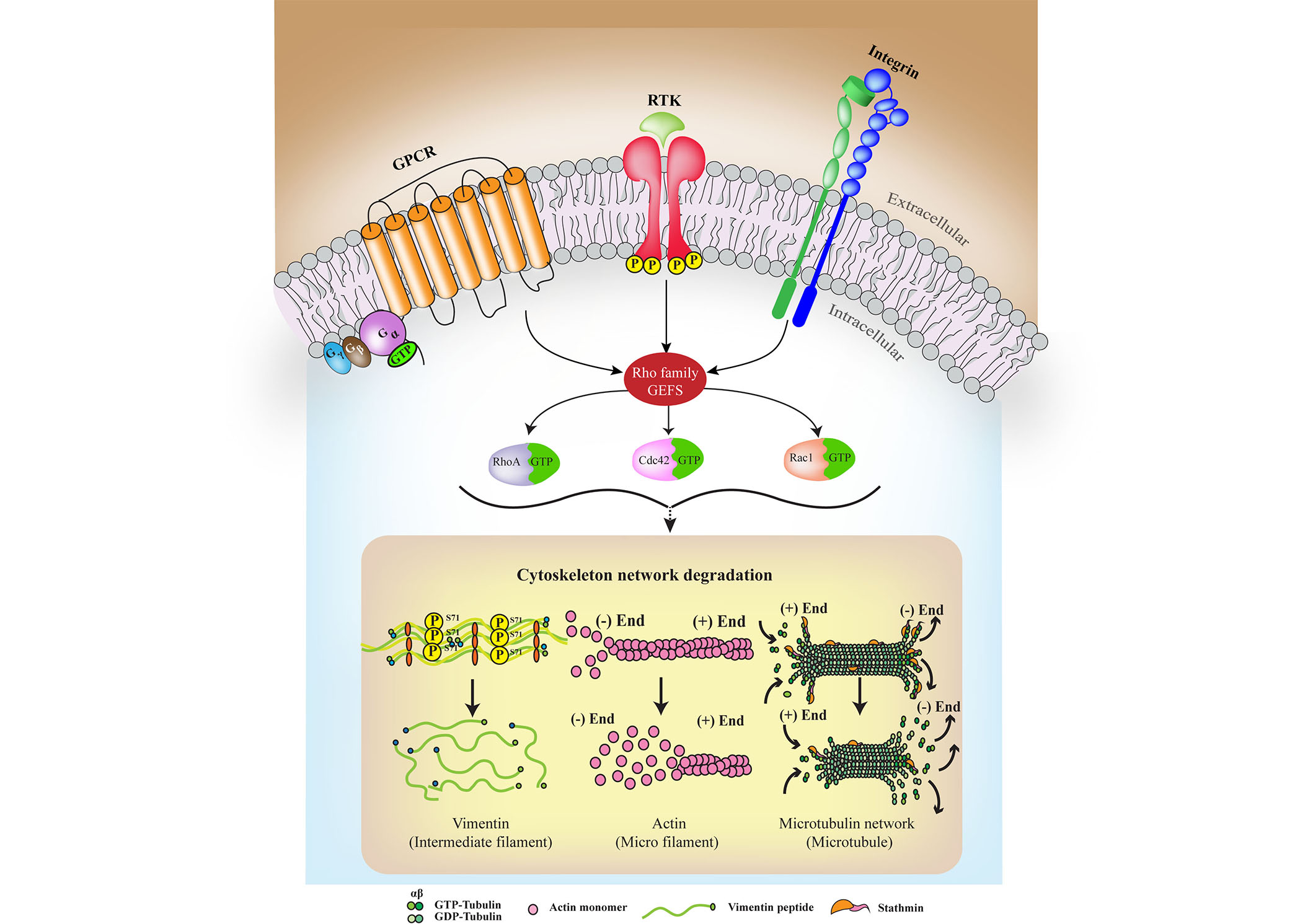
A characteristic simplified model is used to represent the crosstalk among RTKs, GPCRs, and integrins (ITGs) that converge at Rho GEFs to act upon the cytoskeleton. The mechanism of interaction varies among RTKs and their respective GPCR pathway members, which participate in the crosstalk process; however, their roles converge to lead to the ultimate fate of degeneration. Primarily, the Gα displaces from the Gβ-γ complex upon GPCR activation due to ligand binding and a guanosine triphosphate (GTP) is hydrolyzed to guanosine diphosphate (GDP). It is then that different signaling adaptors bridge the GPCRs with RTKs at the membrane or through overlapping signaling partners. Sustained signaling waves can then leads to cytoskeletal degradation. P: phosphorylation
For example, ligand-activated EGFRs might escape lysosomal degradation and transmit signals through AKT, mTORC, and ERK1/2. They, in turn, can translocate into the nucleus to perform cytoskeleton remodeling, migration, or cross-talk with other pathways, depending on the influencing factors. The spatiotemporal localization of the receptors and effectors thus influences the cytoskeleton. The EGFR signaling pathway also interfaces with several other signaling pathways, including TGFβ1 and FAK signaling, which are implicated in cancer and other disease pathogenesis. The combination of EGF and TGFβ1 signaling is reportedly important for infection [77]. EGFR is activated through ligands, and the process can be disrupted by cluster of differentiation 99 (CD99) activation [78]. Activation of CD99 can inhibit EGF-mediated activation of EGFR by impairing PTPN12-dependent c-Src/FAK activation. The role of EGFR in cytoskeletal modification is further illustrated through direct interaction with cytoskeleton-associated post-synaptic density-95, disks-large and zonula occludens-1 (PDZ) and Isl1 and Mec-3 (LIM) domain protein 1 (PDLIM1) and is verified through in vitro experiments [79]. Cell model-based data from the group, generated through coimmunoprecipitation (co-IP) and in situ proximity ligation assays (PLA), shed light on the possibility of integrating RTK signaling with cytoskeletal elements.
ITGs induce ligand-independent phosphorylation of RTKs through crosstalk mechanisms and are responsible for coupling the extracellular matrix (ECM) to the actin cytoskeleton through their association with paxillin (PXN) and vinculin (VCL) to form focal adhesions [80]. ITGs are a major connecting point for the RTK signaling cascade and other surface elements with the cytoskeleton system. ITGs can facilitate neuron-astrocyte interactions through the Thy-1/cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB)-binding protein (CBP)/Csk/Src complex, which causes neurite retraction through the Rho/ROCK pathway [81]. Perturbations to the system can result in several diseases, including neurodegeneration. The crosstalk between ITGs and different GF receptors, all of which are RTKs, such as IGF1R, PDGFR, c-MET, and EGFR, is evident [82]. ITGs determine cell adherence and localize focal contacts; they colocalize with cytoskeletal molecules and talin along with signaling molecules such as Src and FAK. They can function as downstream or upstream mediators of the EGFR pathways [83, 84]. Interestingly, cells have been observed to sense the environment through ECM and ITG-associated complexes. This allows them to prepare for S-phase or mitosis depending on external cues [85], thus connecting the cytoskeleton to ITGs and cell cycle re-entry (CCR) observed in various neurodegenerative scenarios [86].
The RTK c-MET is of interest in terms of cytoskeletal remodeling since it can control the actin cytoskeleton through the key migratory signal output Rac1. c-MET traffics to a perinuclear endosome (PNEs) to facilitate sustained Rac1 signaling. The study focuses on cancer and primary cells to elucidate a broader general mechanism. In PNEs, c-MET requires PI3K and the Rac-GEF Vav2 to elicit its function [87]. Fascinatingly, MET relies on CD44v6 for signaling and internalization followed by stimulation. MET-induced signaling bridges the cascade involving CD44v6 and cytoskeletal remodeling at once through ezrin, radixin, and moesin (ERM) proteins, which can then bind to actin and activate Ras [88]. Furthermore, Abl kinases and MET share a unique activation mechanism, and Abl kinases are important for the functionality of MET as an independent receptor in fibroblasts [89].
Cytoskeletal remodeling is also crucial for the formation of polarized actin microdomains known as dorsal ruffles, and PDGFR is involved in this process. The WASP-interacting protein (WIP)-Nck signaling complex interacts with RTKs to promote actin remodeling in fibroblasts and shows that WIP involvement might be important for migration [90]. Actin remodeling is crucial for degenerative processes, and thus, dorsal ruffles are important to mention here. Although their mechanisms are not completely experimentally understood, the involvement of circular dorsal ruffles in the bistability mechanism of actin polymerization is known [91]. Furthermore, PDGFR stimulation can induce Rac1, RhoA, and Cdc42 activation. PDGF induces the activation of Sos1 to activate Rac1. Rho GEFs, Vav2, and alpha-PAK-interactive exchange factor (αPix) are associated with this RTK, and thus, their roles need to be studied in detail in neurodegenerative scenarios.
Eph receptors, along with their membrane-tethered ephrin ligands, regulate diverse cell-cell interactions and bidirectional signaling. These pathways regulate cytoskeletal dynamics. Mainly EphrinBs are modified to allow higher-order clustering as well as bidirectional signaling. EphB1 receptor stimulation activates Nck/NF-κB-inducing kinase (NIK) pathways through an interaction mechanism that upregulates JNK and mediates attachment and cell migration. EphB1 also upregulates FAK, which plays an important role in ITG signaling, thus correlating cytoskeletal remodeling with RTK signaling [92]. Furthermore, EphrinB activation leads to phosphorylation of Disabled 1 (Dab1), a regulator of Reelin (RELN) signaling through the Src family of kinases, which leads to functional loss of EphrinBs and induction of aberrant neuronal migration in neurons. RELN binds to the EphB extracellular domain, which is associated with apolipoprotein E receptor 2 (ApoER2) in neurons. From the developmental perspective, the RELN pathway is essential for neuronal migration, and several defects arise in EphB knockouts, which elucidates the importance of this RTK in the formation and maintenance of laminated structures in the brain [93]. Chanda et al. [94] illustrates that EPHA2 is deregulated by Aβ through direct and/or indirect mechanisms along with its downstream effectors, including CREB and p38. The study focuses on a chain of events that arise due to cytosolic and nuclear Aβ, which interact with mature miRNAs and noncoding RNAs, respectively, to control the EPHA2 signaling cascade [94]. Importantly, this study focuses on the ability of EPHA2 to alleviate Aβ-induced cytotoxicity and shows a novel mechanism that is disrupted in AD.
Furthermore, recent studies have shown substantial overlaps in signaling cascades as well as transactivation in RTKs and GPCRs, such as angiotensin receptor 2 and TRKB in the medial prefrontal cortex, 5-hydroxytryptamine receptor 1A (5-HT1A) and TRKB in the cerebral cortex, muscarinic acetylcholine receptor (mAChR) and FGFR1 in hippocampal neurons, and EGFR and adenosine A1 receptor in cortical neurons [95]. Antidepressants often target one of these receptors and affect another based on this crosstalk in the brain, further affecting signaling cascades. In addition, FGFR1-5-HT1A hybrids can also form in astrocytes, showing a wide range of crosstalk possibilities that affect neuronal plasticity, cognition, and cytoskeletal degeneration [96]. Transactivation by GPCRs that can interact with amyloid produced in neurodegenerative scenarios further causes concurrent waves of Ca2+ signaling, which hyperphosphorylates ERK, MAPK, and p38MAPK and activates PKA and PKC in disease scenarios. In the case of astrocytes, these events lead to the hyperphosphorylation of GFAP and vimentin, which together with RhoA-activated stress fiber formation leads to cytoskeletal degradation [97]. FGFR on astrocytes can control membrane growth and dynamically sculpt interactions among neurons and glia [98], thus facilitating the hyperphosphorylation of neurofilament subunits [neurofilament light (NFL), neurofilament medium (NFM), neurofilament heavy (NFH)] in neurons and astrocytes as a result of crosstalk among receptors, and downstream signaling can thus influence cytoskeletal fate in neurodegenerative scenarios [99].
Interestingly, ALK and RYK, involved in the Wnt/β-catenin pathway, have also been reported to impact the cytoskeleton in AD as well as in T2DM. Downregulation of ALK/RYK was correlated with Grb2. α-Tubulin expression was checked and found to be downregulated, which reflects poor cellular structural integrity [100]. The double knockdown of these RTKs leads to a further reduction in α-tubulin levels, and overexpression of Grb2 is reported to significantly increase the expression of vimentin, α-tubulin, α-stathmin, and α-smooth muscle actin (α-SMA), which eventually assists in recuperating the cytoskeletal structure of SHSY5Y and HepG2 cells [101].
ROR1 levels in AD have been reported to be downregulated by miR-146a and miR-34a, which correlates positively with the cytoskeleton disruption observed in the disease [102]. Additionally, ROR1 overexpression leads to aberrant neurite outgrowth in SHSY5Y cells. Even in the presence of Aβ, ROR1 could elongate the neurites and successfully enhance the f-acting-actin ratio.
While discussing crosstalk possibilities, it is pertinent to mention membrane lipid rafts (MLR). They aid signal transduction through the formation of scaffolds for receptors such as RTKs, GPCRs, and downstream enzymes, including adenylyl cyclases, endothelial NO synthase (eNOS), nitric oxide synthase-3 enzyme (NOS3), ion channels, and other important molecules, thus creating provisions for crosstalk [103]. Such structures are important in the sense that they not only control endocytic processes, receptor recycling, and functional phosphorylation but also interact with several JM regions of RTKs to facilitate clustering. MLR and caveolae further assist in signaling and can affect cellular structure indirectly [104] or directly by bringing these molecules spatially closer. Caveolin-1 (Cav1) and flotillin-2 (Flot2), both associated with caveolae, have also been associated with RTKs, eliciting the possibility of crosstalk and collaborative control of intracellular processes from the membrane [105, 106].
In summary, the crosstalk possibilities with membrane proteins for RTKs, downstream mediators, and regulation by noncoding RNAs are a few of the known ways through which these receptors influence cytoskeletal remodeling and ultimately induce apoptosis. Apoptosis is typically represented by cell contraction, plasma membrane blebbing, chromatin condensation, and DNA fragmentation [107], and such changes in cell morphology can only be achieved by cytoskeleton reorganization and caspase-mediated digestion of cytoskeleton components [108] such as actin filaments, microtubules, and intermediate filaments. Apoptotic signaling often activates Rho/ROCK pathways. The Rho pathways downstream allow for actin reorganization, which is essential for degradation and proliferation.
The Rho family of GTPases is composed of 20 members; however, three members have mostly been studied concerning RTKs, Cdc42, Rac1, and RhoA. Three general classes of proteins further regulate Rho family signaling: GEFs, GAPs, and GDP dissociation inhibitors (GDIs) [89]. Rho GTPases are directly and indirectly regulated by ROS/reactive nitrogen species (RNS), which are generated in degenerative scenarios. Broadly, two mechanisms are involved in the RTK-mediated activation of Rho GTPases that lead to degeneration. In the first type of mechanism, Rho GTPases are indirectly activated in the downstream signaling of RTKs. For example, the autophosphorylation of PDGFRB and EPHB2 induces the docking of the SH2 domain of the P85 subunit of PI3K. This event signals PI3K to recruit α-Pix, which mediates the activation of Cdc42 and Rac1 [109]. The tethering of the Nck/PAK complex with RTK may also be involved in the recruitment of α-Pix by a PAK-α-Pix interaction [110]. In the second mechanism, Rho GTPases are directly activated by RTKs through Rho GEFs when RTKs and Rho GTPases reside in proximity to the plasma membrane. For example, the PDZ domain of leukemia-associated RhoGEF (LARG; Rho GEF) interacts with the C-terminal domain of IGF1R and mediates INS-like GF (IGF) signaling by activating RhoA [111]. Rho/ROCK signaling opposes the Rac pathway and acts as a pro-apoptotic pathway. The levels of both are upregulated before neuronal death, and ROCK inhibitors have been shown to reduce cell loss in the rat retina. However, constitutively active Rac1 mutants also led to neuronal death [112]. These studies attempted to elucidate the connection between RTKs and Rho family GTPases in the context of neurodegenerative signaling. Additionally, we performed bioinformatics analysis using the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways tool to visualize (see Figure 3) the convergence of the milieu of RTK signaling molecules on Rho GTPases concerning cytoskeletal remodeling cascades [113]. KEGG is a conglomerate of 16 databases and thus provides systems information about pathways through the integration of the BRITE hierarchy. A human-specific pathway map with the organism prefix “hsa” for the actin cytoskeleton with “RTK” as the search term and colored reported Rho GTPases involved in the mechanism. The KEGG othologs (KO) identifiers are converted to gene identifiers in the reference pathway. In the context of neurodegeneration, several illustrated connections are yet to be understood, and a particular RTK axis, i.e., individual differential involvement of RTK along with downstream mediators, is yet to be established. However, it is important to appreciate the complexity of the pathways to holistically assess the importance of studying these pathways in detail in neurodegenerative cascades.
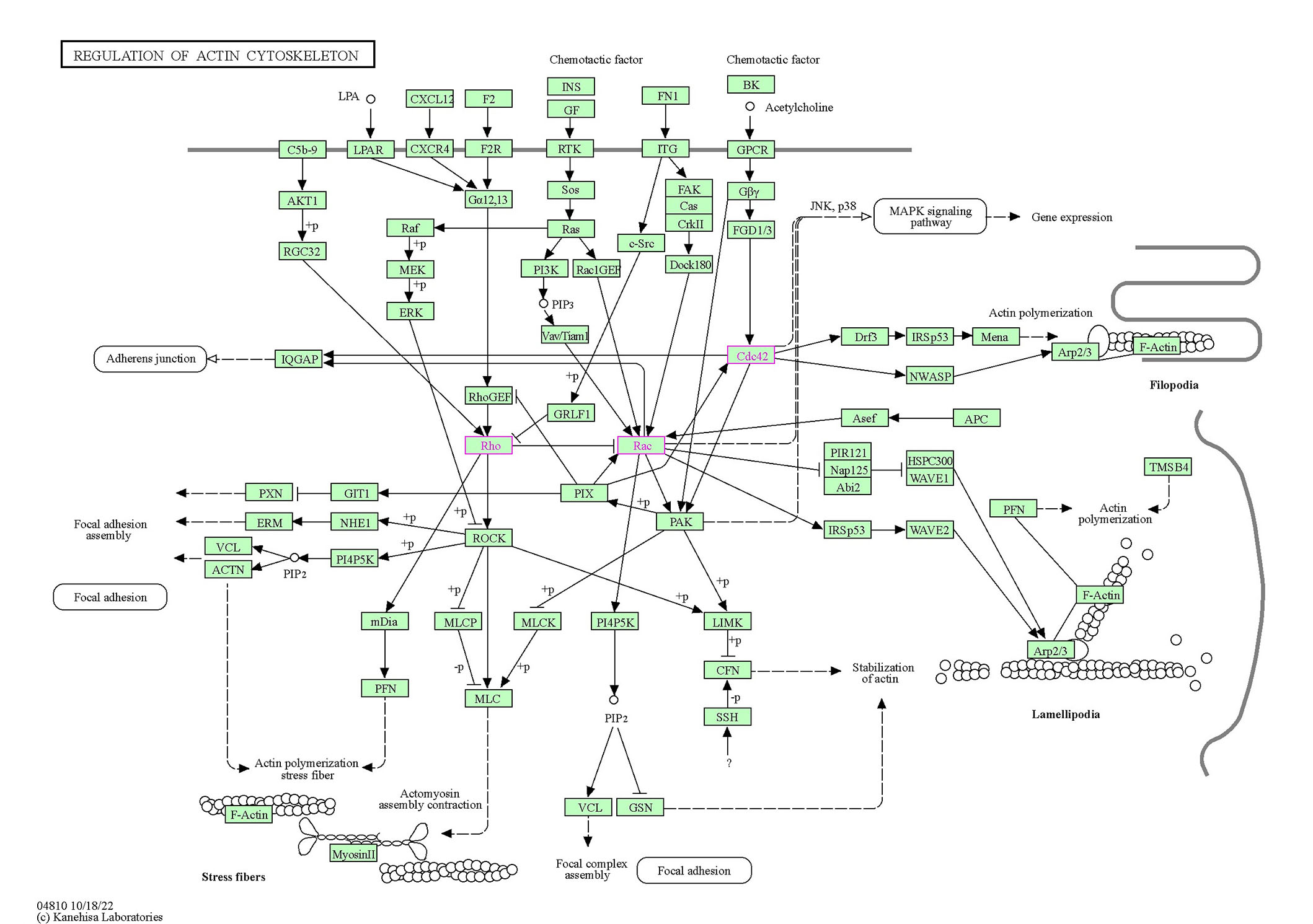
The signaling cascades that originate at the cell surface, involving RTKs and GPCRs directly and/or indirectly, lead to Rho, Rac, and Cdc42 activation. These Rho GTPases, through their respective downstream effectors, influence cytoskeleton remodeling and degeneration. The Rho GTPases are colored pink and in green rectangular boxes. All other interacting partners are in rectangular boxes with important functions of the cytoskeleton in transparent rounded rectangles. The membrane is indicated by a bold line on which the RTKs and other receptors coexist. Dotted arrows indicate the indirect link of the molecules to functions or partially explored mechanisms of action. Solid arrows indicate a molecular interaction leading to activation, and +p on the arrows indicates a phosphorylation event due to the interaction, -p on the arrows indicates a dephosphorylation event. Arrows with flat heads indicate inhibition. LPA: lysophosphatidic acid; CXCL12: C-X-C chemokine ligand 12; F2: coagulation factor II; FN1: fibronectin 1; BK: bradykinin; C5b-9: complement C5a,b, C6, C7, C8, and C9; LPAR: Lysophosphatidic acid receptor; CXCR4: C-X-C chemokine receptor type 4; F2R: coagulation factor II receptor; RGC32: response gene to complement 32; FGD1/3: faciogenital dysplasia 1/3; IQGAP: IQ motif-containing GTPase activating protein 1; GRLF1: glucocorticoid receptor DNA-binding factor 1; GIT1: G protein-coupled receptor kinase interacting protein-1; NHE1: Na(+)/H(+) exchanger isoform-1; ACTN: actinin; PI4P5K: phosphatidylinositol-4-phosphate 5-kinase; mDia: mammalian Diaphanous; MLCP: myosin light chain phosphatase; PFN: profilin; MLC: myosin light chain; GSN: gelsolin; MLCK: myosin light chain kinase; LIMK: LIM domain kinase; CFN: cellular fibronectin; SSH: Shingshot homolog; Drf3: Diaphanous-related formin-3; Mena: mammalian-enabled protein; Asef: average saturation efficiency filter; APC: adenomatous polyposis coli; PIR121: p53-inducible mRNA 121; HSPC300: haematopoietic stem progenitor cell 300; Nap125: nucleoid-associated proteins; WAVE1: WASP-family verprolin homologous protein 1; Abi2: Abl interactor 2; Arp2/3: actin-related protein 2/3; TMSB4: thymosin beta4
Note. Adapted with permission from the Kanehisa laboratories [Internet]. Kanehisa Laboratories; c1996–2023 [cited 2023 Jun 7]. Available from: http://www.genome.jp/kegg/
The pathways depicted above are holistic possibilities for interactions in terms of signaling, which can potentially lead to neurodegeneration. However, neurodegenerative scenarios are often associated with aging and genetic predispositions [114], where there is a notable reduction in ligands, GFs, and hormones that can influence the activity of the RTK or indirectly regulate through transactivation via ITGs or GPCRs. In such scenarios, the pathological hallmarks of the disease often determine the cytoskeletal fate. The toxic Aβ that aggregates in AD often blocks cellular communication and hyperphosphorylates signaling molecules through toxic sustained signaling, eventually causing the degradation of actin [115]. The three main downstream effectors of the cytoskeleton highlighted in pink (Figure 3) are altered in AD. Rac1 has been shown to be directly associated with memory loss in animal and human studies, with contrasting results indicating complex interactions that are yet to be understood in their entirety [116]. However, the relationship has been shown to be bidirectional and impacts astrocytes in the process. Rac1 regulates Aβ production through the upregulation of γ-secretase, whereas Aβ induces the colocalization of Rac1 with actin filaments, leading to degradation [116]. Rac1 is also an important mediator of the ROS in astrocytes observed in AD [117]. In autism spectrum disorder (ASD), however, alterations in Rac1 activity were due to several autism-risk gene mutations, including fragile X mental retardation 1 (Fmr1), neurexin 1 (Nrx1), neuroligin 4 (Nlg4), and tuberous sclerosis 1 (Tsc1) [118]. In contrast, Cdc42 is abnormally upregulated in neurons but downregulated in other cells in AD patients [119]. In microglia, Cdc42 and Rac1 activation ameliorated migration triggered by Aβ [120]. Activation of FAK/Rac1/Cdc42-GTPase signaling ameliorates impaired microglial migration response to Aβ42 in triggering receptor expressed on myeloid cells 2 loss-of-function murine models. In microglia, Cdc42 and Rac1 activation ameliorated migration triggered by Aβ [121]. In ASD, thousand-and-one kinase 2 (TAOK2) deficiency causes dosage-dependent impairments in cognitive processes, structure, and function and further results in RhoA downregulation [122].
RTKs play major roles in signaling and degenerative pathways, which could be regulated through their interactions with other downstream effectors and crosstalk with membrane proteins. Furthermore, there are specific RTKs capable of binding to Aβ directly, such as EPHA4, EPHB2, and InsR among the known proteins [123]. Several small molecule drugs targeting RTKs have been developed to combat cancer, and thus, repurposing them for a degenerative scenario could be a viable option. Alectinib, brigatinib, ceritinib, crizotinib, entrectinib, and lorlatinib target several RTKs, including ALK, TRKA/B/C, ROS1, HGFR, IGF1R, Ret, and InsR [124]. The application of such targeted therapies toward downstream signaling pathways of RTKs, such as MEK, where binimetinib, colimetinib, and trametinib have been approved for cancer treatment [125], could be another viable option to target early events in dementia, including CCR, among others [126–128].
RTK immunotherapy alone, however, might not be the way to develop potent therapeutics, and instead, a combined therapy might be more effective due to the multiple layers of RTK interactions [129]. For instance, in choroidal neovascularization (CNV), AMD, a pathological output of neovascular AMD (nAMD), VEGFR2, and FGFR1 are dysregulated. Brivanib (a dual RTK inhibitor of the aforementioned inhibitors) has yielded positive results in cell and animal models. In mouse CNV, brivanib injection blocked the phosphorylation of FGFR1 and VEGFR2. It reduced CNV leakage without any notable toxicity [130]. In vitro experiments confirmed that the drug inhibits proliferation, formation of tubes in microvascular endothelial cells, and migration. Another group reported similar importance of RTKs in AMD in terms of retinal pigment epithelium (RPE) degeneration. RTK ligands, including BDNF, HGF, GDNF, milk fat globulin protein E8 (MFG-E8), and thrombospondin type 1/2 (TSP1/2), were identified as key molecules that could rescue phagocytic dysfunction in AMD [131].
In the case of degenerative diabetic retinopathy, PDGFRβ, VEGFR1, and IGFR have been implicated, and targeting them in a novel retinopathy mouse model has clarified the contribution of the dysregulation toward the vascular phenotype observed in the disease. Thus, targeting RTK signaling involved in disease progression might be a key step toward amelioration [132].
Gefitinib is a selective EGFR inhibitor that has been explored in epithelial tumors. In vivo studies have shown that gefitinib extensively inhibits the growth of human tumor cell-derived cell lines in nude mice. It has been shown in clinical trials that it can improve cancer prognosis. The prospects of such EGFR inhibitors in the brain and the use of the blood-brain barrier (BBB) to understand this has been explored by Tavassoly et al. [133]. However, the need for combinatorial inhibitors emerges here as well, since amplification of MET has been shown to lead to gefitinib resistance, which indicates crosstalk possibilities [134].
Alternatively, targeting Rho GTPases could be a viable option, and it has been explored to a limited extent. Rac1 inhibitors [NSC23766, 6-mercaptopurine (6-MP), EHT1864] have been tested on hippocampal neurons, primary neurons, and SHSY5Y [135]. EHT1864 disrupts Rac1-mediated downstream signaling and γ-secretase cleavage upstream, thus impairing Aβ signal transduction entirely. However, in guinea pigs, the inhibitor only led to a 30% reduction in Aβ production [136]. NCS23766 and 6-MP are promising inhibitors of Rac1 that act on the PI3K/PDK1/novel PKCs (nPKCs)/Rac1 axis and have been tested on primary neurons as well as organotypic cultures of the hippocampus and entorhinal cortex. They markedly reduce apoptosis. However, the entire pathway through which death signaling is triggered by Aβ remains unexplored, and thus, their effectiveness as drugs cannot be ascertained entirely [137]. RhoA/Rac inhibitors have been tested (Y27632, SR3677, HA-1077) and reported to be effective in decreasing Aβ production in different AD models, including SHSY5Y cell lines, primary cortical neurons from embryos, and mouse models [135]. The ROCK inhibitors (Y27632, HA-1077, and H-1152P) were tested by Leuchtenberger et al. [138], and their results concluded that the inhibitors do not directly lower Aβ1-42 production, but they work on photorefractive keratectomy (PRK) to exert their positive effects. Herskowitz et al. [139] showed that fasudil (HA-1077), the only commercially available ROCK inhibitor, is nonselective. Since ROCK1 and ROCK2 function differently, SR3677 could be better at targeting APP phosphorylation at the T654 position in vivo by ROCK2 [139]. It is important to mention non-steroidal anti-inflammatory drugs (NSAIDs) here since they have been studied now for a while and are known to reduce ROCK activity that allows for preferentially lowering the amyloidogenic pathway of APP production [140] in mouse models of AD. However, further research to characterize the mechanism of action and underlying nonspecific impacts is needed to underscore the applicability of Rho GTPase inhibitors for therapeutic purposes.
The PI3K/AKT pathway is an important downstream signaling cascade for several RTKs. Hence, controlling it is projected to be a therapeutic strategy for the degeneration observed in intervertebral disc degeneration (IDD) and ocular neovascularization [141, 142]. Several disorders stem from RTK upregulation, and thus, a direct mechanism of targeting pathways such as PI3K or, for that matter, the receptors might be more successful. Sprouty proteins could be an answer to this conundrum since they act as inhibitors of GF-dependent neuronal and glial pathways [143]. Downregulating Sprouty 2 could be of therapeutic value through small interfering RNA (siRNA)-mediated therapy or gene therapy, as it is upregulated in neurological disorders. Its downregulation has also improved nerve regeneration possibilities in the peripheral nervous system [144]. However, there is enough evidence indicating the importance of cytoskeletal stability for neurodegenerative disorders [145], and research on RTK involvement in generating signaling cascades that affect cytoskeletal fate is still lacking for the development of effective therapies. Further studies focusing on this avenue of research can potentially lead to the development of effective therapeutic strategies.
Holistically, the diverse and widespread RTK signaling cascade allows for varied roles to be combined under the same umbrella, which leads to varied degenerative signaling with the penultimate fate of cytoskeletal disruption and death. External cues assimilated by the surface receptors and their consequential downstream players, such as those of the following cascades, IP3-DAG, PI3K, MAPK, and JAK-STAT, allow for degenerative disorders to progress with time. RTKs might interact with a multitude of other receptors, such as ITGs and GPCRs, on the membrane front, and the possibility of ligand-dependent or ligand-independent signaling has gradually become clear. RTKs have even been shown to perform nuclear shuttling in parts or as holoreceptors, as in the case of IGF1R. Furthermore, this review focused on the convergence of these promiscuous mechanisms leading to Rho GTPases, which, along with Rho-mediated activation of Rac, form the final point where all divergences dissipate and their unified roles in affecting cytoskeletal fate emerge. Cytoskeletal remodeling has been previously implicated and linked to several RTKs, and thus this review intends to unify and develop a coherent concept that allowed us to focus on therapeutic possibilities. Some therapies encompassing RTKs have been discussed here; however, understanding the convergence better would allow us to develop combinatorial regimes that would perhaps increase the chances of success manifold. Several Food and Drug Administration (FDA)-approved GPCR targets exist, and thus, understanding the crosstalk could pave the prospect of developing more effective therapies for degenerative diseases in the future.
AATYK: apoptosis associated tyrosine kinase
Abl: Abelson murine leukemia viral oncogene homolog 1
AD: Alzheimer’s disease
AKT: protein kinase B
ALK: anaplastic lymphoma kinase
ALS: amyotrophic lateral sclerosis
AMD: age-related macular degeneration
AMPAR: alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid subtype glutamate receptor
ANG: angiopoietin
APP: amyloid precursor protein
Aβ: amyloid β
BDNF: brain-derived neurotrophic factor
Cdc42: cell division control protein 42
CNV: choroidal neovascularization
Csk: C-terminal Src kinase
DAG: diacylglycerol
DDR: discoidin domain receptor
EGF: epidermal growth factor
EGFR: epidermal growth factor receptor
Eph: erythropoietin-producing hepatoma
EPHA4: ephrin-type A receptor 4
ER: endoplasmic reticulum
ErbB: erythroblastic leukemia viral oncogene homolog
ERK1/2: extracellular signal-regulated kinase 1/2
FAK: focal adhesion kinase
FGF: fibroblast growth factor
FGFR: fibroblast growth factor receptor
Gab1: growth factor receptor-bound protein-2-associated binding protein 1
GEFs: guanine exchange factors
GF: growth factor
GPCRs: G-protein coupled receptors
Grb2: growth factor receptor-bound protein-2
HD: Huntington’s disease
Ig: immunoglobulin
IGF1R: insulin-like growth factor 1 receptor
InsR: insulin receptor
IP3: inositol (1,4,5) triphosphate
ITGs: integrins
JAK: Janus kinase
JNK: c-Jun N-terminal kinase
LTK: leukocyte tyrosine kinase
MAPK: mitogen-activated protein kinase
MET: mesenchymal epithelial transition
mTORC2: mammalian target of rapamycin complex 2
MUSK: muscle specific kinase
Nck: non-catalytic region of tyrosine kinase adaptor protein 1
NFT: neurofibrillary tangles
NLS: nuclear localization signals
PAK: p21 activated kinase
PD: Parkinson’s disease
PDGF: platelet-derived growth factor
PDGFR: platelet-derived growth factor receptor
PDK1: 3-phosphoinositide-dependent kinase 1
TGFβ: transforming growth factor beta
PI3K: phosphoinositide 3 kinase
PKC: protein kinase C
PTMs: post-translational modifications
Ras: rat sarcoma
RET: rearranged during transfection
RELN: Reelin
Rho: rat sarcoma homologous
ROCK: rat sarcoma homologous-associated coiled-coil kinase
ROR: receptor tyrosine kinase-like orphan receptor
RTKs: receptor tyrosine kinases
RYK: receptor related to tyrosine kinase
SH3: Src homology 3
Sos: son of sevenless
STAT: signal transducer of activators of transcription
STYK: serine/threonine/tyrosine kinase
T2DM: type 2 diabetes mellitus
TGFA: transforming growth factor A
TRK: tropomyosin receptor kinase
VEGF: vascular endothelial growth factor
VEGFR: vascular endothelial growth factor receptor
PS: Conceptualization, Writing—original draft, Writing—review & editing. RD: Conceptualization, Writing—original draft, Writing—review & editing. PM: Conceptualization, Writing—original draft. DM: Conceptualization.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
The research is funded by the Department of Atomic Energy, Government of India [RSI4002]. The funders did not have any role in study design, collection of data and analysis, preparation of the manuscript or the decision to publish.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Janusz Wiesław Błaszczyk
Tatsushi Yuri ... Hisashi Nojima
Danqing Xiao, Chen Zhang
Carlos Gutierrez-Merino
Julius Mulumba ... Yong Yang
Felipe P. Perez ... Maher Rizkalla
Ezra C. Holston
Jorge Medeiros
Ryszard Pluta
