
Abstract
Peroxisomes are actively involved in the metabolism of various lipids including fatty acids, ether phospholipids, bile acids as well as the processing of reactive oxygen and nitrogen species. Recent studies show that peroxisomes can regulate cholesterol homeostasis by mediating cholesterol transport from the lysosomes to the endoplasmic reticulum and towards primary cilium as well. Disruptions of peroxisome biogenesis or functions lead to peroxisomal disorders that usually involve neurological deficits. Peroxisomal dysfunction is also linked to several neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease. In many peroxisomal disorders and neurodegenerative diseases, aberrant cholesterol accumulation is frequently encountered yet largely neglected. This review discusses the current understanding of the mechanisms by which peroxisomes facilitate cholesterol trafficking within the cell and the pathological conditions related to impaired cholesterol transport by peroxisomes, with the hope to inspire future development of the treatments for peroxisomal disorders and neurodegenerative diseases.
Keywords
Peroxisome, cholesterol transport, nervous system, peroxisomal disordersIntroduction
Peroxisomes are ubiquitously present in almost all cells in the human body. They can execute a wide array of metabolic functions, including the anabolism of ether phospholipids and bile acids, the catabolism of fatty acids, as well as the dynamic balance of reactive oxygen and nitrogen species. Impairments in the biogenesis and functions of peroxisomes lead to peroxisomal disorders (PDs), which usually manifest as neurological problems in the nervous system and are largely incurable (Figure 1). Recently, there is mounting evidence that peroxisomes can receive cholesterol from the lysosomes and deliver it to the endoplasmic reticulum (ER) or the primary cilium. This role of peroxisome in intracellular cholesterol transport is significant, since cholesterol distribution and levels are aberrant in many cellular and mouse models of PDs. Dyshomeostasis of cholesterol is intimately linked to neurodegenerative diseases as well (Figure 1a). Thus, the purpose of this review is to discuss how peroxisomes facilitate normal cholesterol trafficking and how defects in the process contribute to PDs and neurodegenerative diseases.
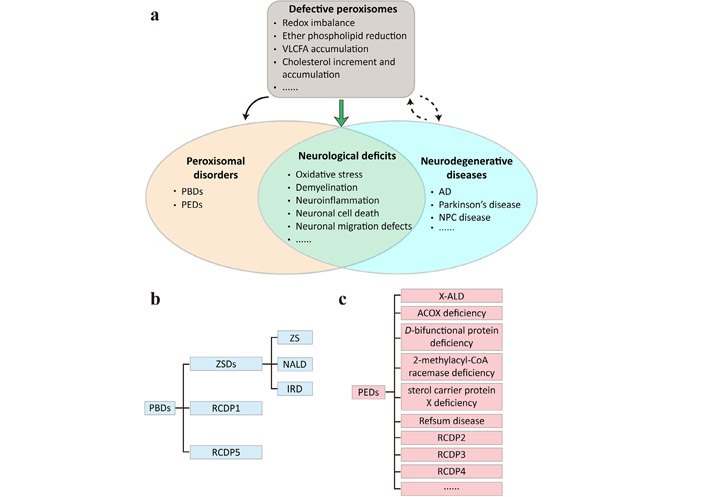
Defective peroxisomes may underlie neurological deficits commonly shared by PDs and neurodegenerative diseases. (a) The peroxisomes play important roles in regulating redox homeostasis and lipid metabolism. Disruptions of peroxisome biogenesis or functions lead to PDs that can be subdivided into (b) peroxisome biogenesis defects (PBDs) and (c) single peroxisomal enzyme deficiencies (PEDs). Peroxisome dysfunction can result, directly or indirectly, from neurodegenerative diseases and may exacerbate disease progression. The commonly shared neurological deficits of PDs and neurodegenerative diseases include, but are not limited to, oxidative stress, demyelination, neuroinflammation, neuronal cell death and neuronal migration defects; (b) PBDs encompass Zellweger spectrum disorders (ZSDs) that can be subdivided into Zellweger syndrome (ZS), neonatal adrenoleukodystrophy (NALD) and infantile Refsum disease (IRD), and rhizomelic chondrodysplasia punctata (RCDP) type 1 (RCDP1) and RCDP5; (c) PEDs are much more heterogeneous and contain X-linked adrenoleukodystrophy (X-ALD), acyl-coenzyme A (CoA) oxidase (ACOX) deficiency, D-bifunctional protein deficiency, 2-methylacyl-CoA racemase deficiency, sterol carrier protein X deficiency, Refsum disease, RCDP2, RCDP3, RCDP4, and more. AD: Alzheimer’s disease; NPC: Niemann-Pick type C; VLCFA: very long-chain fatty acid
In the article, peroxisome functions and cholesterol metabolism in the brain under physiological conditions will be firstly introduced. Then follows how peroxisomes mediate cholesterol transport between the lysosomes and the ER as well as towards the primary cilium. The contributions of peroxisomes and cholesterol metabolism to PDs and neurodegenerative diseases will be discussed at last. These findings on peroxisome-mediated cholesterol transport not only extend our understanding of peroxisomes and intracellular cholesterol trafficking, but also shed light on the potential treatment avenues for PDs and neurodegenerative diseases.
Brief overview of peroxisomes and cholesterol metabolism in the brain
Peroxisomes in the brain
Peroxisomes are single membrane-bound organelles first observed as “microbodies” in mouse kidney cells by Johannes Rhodin with the electron microscope in 1954. They were later isolated from the rat liver and renamed “peroxisomes” by Christian de Duve and co-workers, owing to the inclusion of hydrogen peroxide (H2O2)-producing and -degrading enzymes. It is now known that peroxisomes are ubiquitously present in almost all eukaryotic cells and harbor more than 50 different enzymes catalyzing reactions far beyond H2O2 metabolism. However, the number, size as well as specific functions of peroxisomes vary greatly among different tissues [1]. The brain has less abundant and much smaller peroxisomes than the liver and kidney, with the exact distribution of peroxisomes depending on the detection methods and developmental stages [2]. At the cellular level, peroxisomes are found in both neurons and glial cells including astrocytes, oligodendrocytes, and ependymal cells [3]. In neurons, peroxisomes are largely localized in the somata with a few in the proximal dendrites as well [4].
One of the main functions that brain peroxisomes exert is to initiate ether phospholipid biosynthesis. In this metabolic pathway, peroxisomal dihydroxyacetone phosphate (DHAP) acyltransferase first acylates DHAP, an intermediate of glycolysis, and then alkyl-DHAP synthase exchanges the acyl group for an alkyl group. The resultant alkyl-DHAP is subsequently reduced by acyl/alkyl-DHAP reductase in the peroxisomal and ER membranes, followed by further processing in the ER that eventually leads to the generation of ether phospholipids. Plasmalogens—the most abundant class of ether phospholipids constituting 22% of the total phospholipids in human brains [5]—not only make up the cell membranes and, particularly, the myelin sheaths, but also protect the cells from oxidative stress as well as give rise to secondary messenger molecules such as arachidonic acid or docosahexaenoic acid (DHA) [6]. Defects in the genes encoding DHAP acyltransferase, alkyl-DHAP synthase, and fatty acyl-CoA reductase 1, which provides the acyl/alkyl-DHAP reductase reaction with a fatty alcohol, result in different types of RCDP, a subgroup of PDs that affects multiple organs including the brain, with reduced plasmalogen levels being the main biochemical feature (Figure 1b and c).
Brain peroxisomes are also able to oxidize fatty acids. The substrates for β-oxidation include saturated VLCFAs, polyunsaturated fatty acids, long-chain dicarboxylic acids and many others [7]. The chemical nature of β-oxidation in the peroxisomes is identical to that in the mitochondria with, however, distinct sets of enzymes involved and H2O2 instead of ATP generated. The shortened fatty acids can then be shuttled to mitochondria for additional oxidation and ultimately yield CO2 and H2O. Interestingly, one round of peroxisomal β-oxidation of tetracosahexaenoic acid is in fact the last biosynthetic step of DHA, the major polyunsaturated fatty acid that contributes to the development and normal function of the brain. Peroxisomal β-oxidation also intersects with bile acid biosynthesis primarily in the liver. 3-Methyl-branched chain fatty acids and 2-hydroxylated fatty acids are broken down by the peroxisomes via α-oxidation. Phytanic acid from the human diet first undergoes α-oxidation to produce a methyl group at the carbon 2 position, which then allows β-oxidation to occur. The α-oxidation of even-carbon, 2-hydroxylated fatty acids gives rise to odd-carbon fatty acids in the brain [8]. Impaired peroxisomal β- and α-oxidation cause an accumulation of specific types of fatty acids that can damage the brain. For example, patients with mutations in ACOX1 (encoding the enzyme responsible for the first dehydrogenation step of β-oxidation) or ABCD1 [encoding ATP-binding cassette (ABC) subfamily D member 1, an importer of VLCFAs] and those with mutations in PHYH (encoding phytanoyl-CoA α-hydroxylase, the enzyme involved in α-oxidation) have elevated levels of VLCFAs and branched chain fatty acids, respectively, in the tissues and plasma. Neurological abnormalities are often detected in these patients.
The maintenance of cellular redox state is another important function of peroxisomes. In addition to that from β-oxidation of fatty acids, H2O2 can be generated during D-amino acid degradation by D-amino acid oxidase and D-aspartate oxidase, L-pipecolic acid degradation by pipecolic acid oxidase, as well as by many other peroxisomal oxidases [9]. Peroxisomal superoxide radicals and nitric oxide radicals are produced by xanthine oxidase and nitric oxide synthase, respectively. These reactive oxygen species (ROS) are counterbalanced by several antioxidant enzymes in the peroxisomes. Catalase can degrade H2O2 catalytically by converting it to H2O and O2 as well as peroxidatically by oxidizing a co-substrate such as ethanol, methanol and phenol. Glutathione peroxidase, manganese- or copper/zinc-containing superoxide dismutases are also capable of removing peroxisomal ROS. An imbalance of ROS metabolism in the peroxisomes is closely associated with various neurological diseases and brain aging [10, 11].
The role of peroxisomes in cholesterol biosynthesis has been a matter of controversy for years [12–14]. In fact, convincing evidence supporting a direct involvement of peroxisomes in cholesterol biosynthesis is still lacking, even in a latest study in favor of the viewpoint [15]. It is more likely that peroxisomes contribute to cholesterol homeostasis by transporting the molecule between different organelles and the plasma membrane (PM), which will be discussed in the later sections of this review.
Cholesterol homeostasis in the brain
Cholesterol is the most abundant sterol playing a plethora of functions in mammals. It serves as a basic constituent of cell membranes, a precursor to steroid hormones, bile acids and oxysterols, as well as the lipid moiety of the active Hedgehog and Smoothened proteins. Cholesterol is highly enriched in the brain, representing about 20% of total cholesterol in the body [16]. Brain cholesterol is mainly localized in myelin sheaths as well as on the surfaces of neurons and astrocytes. Synaptic vesicles also contain high levels of cholesterol [17]. The presence of cholesterol imparts specific biophysical properties to myelin and cell membranes. It is also crucial for myelin biogenesis and synaptogenesis [18, 19]. Apart from the nonesterified form, about 1% of the total brain cholesterol is found as cholesteryl esters (CEs) [20].
Cholesterol metabolism in the brain is, like that in the peripheral tissues [21], composed of cholesterol biosynthesis, uptake, export and esterification. However, due to the presence of the blood-brain barrier, neurons and glial cells in the brain are almost completely unable to acquire cholesterol from dietary source and plasma lipoproteins but rely on de novo biosynthesis for cholesterol supply. Overall, brain cholesterol biosynthesis initiates during early embryonic development and continues into the adulthood, reaching a peak at the third postnatal week in mice [22, 23]. In the embryonic brain, neurons actively synthesize cholesterol, using the acetyl-CoA as the substrate, to support new neuron survival and neurite outgrowth [24, 25]. Postnatally, a major fraction of radioactively labeled acetate was found incorporated in lanosterol in neurons whereas in cholesterol in astrocytes [26], suggesting that neurons have a lower capacity to synthesize cholesterol than glial cells. Indeed, it is oligodendrocytes that account for the highest rate of cholesterol biosynthesis during postnatal myelination [18]. At all stages throughout the life and particularly in the adulthood, astrocytes are a major source of cholesterol to neurons so as to support their normal development and functions [27]. By using stable isotope labeling and tracing, cholesterol in adult mouse brain is found to be synthesized primarily via a modified Kandutsch-Russell pathway, in which lanosterol first enters the Bloch pathway and then shifts to the Kandutsch-Russell pathway upon converting to zymosterol, at the absolute rate less than 2% of that of the liver [28]. Defects in cholesterol biosynthesis underlie a number of diseases, with Smith-Lemli-Opitz syndrome caused by mutations in the gene encoding the last enzyme in the Kandutsch-Russell pathway being the most common [29].
Astrocyte-produced cholesterol is taken up by neurons via receptor-mediated endocytosis of apolipoprotein E (ApoE)-containing, high-density lipoprotein-like particles. This process highly resembles low-density lipoprotein (LDL) receptor (LDLR)-dependent uptake of plasma LDL by hepatocytes [30]. In fact, neuronal receptors for ApoE-containing lipoproteins include LDLR, LDLR-related protein, very low-density lipoprotein receptor and ApoE receptor 2, all of which belong to the LDLR family [31]. These receptors bind ApoE via the extracellular ligand binding domains and recruit clathrin and the associated adaptor proteins via the cytoplasmic endocytic motifs, thereby facilitating the internalization of ApoE-containing lipoproteins. In the lysosomes, cholesterol released from ApoE-containing lipoproteins is, through the coordinated actions of NPC1 and NPC2 proteins, inserted into the limiting membranes, from where it is further passed to downstream organelles [32]. Mutations in NPC1 or NPC2 cause NPC disease that features lysosomal cholesterol accumulation in virtually all the tissues and progressive neurodegeneration particularly in the cerebellum [33]. Beyond cholesterol metabolism, LDLR-related protein 1 is also involved in amyloid β clearance and tau uptake in neurons [34, 35], whereas very low-density lipoprotein receptor and ApoE mediate the transduction of the Reelin signaling pathway [36]. Alterations in these ApoE receptor-related pathway contribute to the pathophysiology of AD [37].
Surplus cholesterol in neurons must be properly handled to prevent the potential cytotoxicity associated with cholesterol accumulation. The conversion of cholesterol to 24S-hydroxycholesterol (24S-HC) is a major and unique way for neurons to expel cholesterol [38]. 24S-HC then readily diffuses across the blood-brain barrier into the circulation and is eventually eliminated as the bile. As an agonist of the nuclear transcription factor liver X receptor, 24S-HC can upregulate the expression of genes encoding ABC transporter family members that have been known to mediate cholesterol efflux in the peripheral tissues. Interestingly, multiple ABC transporters, such as ABC subfamily A member 1, ABC subfamily G member 1 and member 4, are present in neurons in the brain [39, 40]. However, how these exporters contribute to cholesterol efflux from neurons still remains unclear, with in vivo results lacking and in vitro results inconsistent [41, 42]. Excess cholesterol can also be esterified by acyl-CoA:cholesterol acyltransferases (ACATs) on the ER and stored in the cell as cytoplasmic lipid droplets. ACAT1 is ubiquitously expressed throughout the body, whereas ACAT2 is primarily restricted to the liver and intestine [43]. In the brains of mice and humans with AD, the levels of CEs are markedly increased [44, 45]. Pharmacological and genetic disruption of ACAT1 effectively reduce amyloid burden and ameliorate cognitive deficits in AD mice through affecting amyloid precursor protein expression, processing, and autophagy-mediated clearance of amyloid β in microglia and tau in neurons [46–52]. Increased 24S-HC and the resultant decreases in amyloid precursor protein levels may also mediate the beneficial effects of ACAT1 ablation on AD [20].
Proper cholesterol transport is crucial for maintaining cellular cholesterol homeostasis as well. In neurons, intracellular cholesterol trafficking involves not only dynamic movements between organelles but also soma-to-axon delivery, since cholesterol synthesis is compartmentalized within the cell body [53, 54]. The mechanisms by which cholesterol is conveyed in neurons are less explored. Nevertheless, in other polarized cells such as hepatocytes and enterocytes, cholesterol moves between membranes by vesicular transport and non-vesicular transport mechanisms [55]. It is possible that neurons may employ similar approaches to mobilize cholesterol. How peroxisomes are engaged in intracellular cholesterol transport in non-neuronal cells is reviewed in the next section.
The role of peroxisomes in intracellular cholesterol transport
Peroxisomes as cholesterol conduits
The readers may notice from the earlier introduction that peroxisomes actively communicate with many other organelles to fulfill their roles in lipid metabolism. Intriguingly, peroxisomes are situated in close proximity (within a distance of 30 nm) to the ER, mitochondria, lysosomes, lipid droplets and more [56]. The contacts between lysosomes and peroxisomes were first identified in an unbiased genome-wide screen that searched for the factors regulating the trafficking of LDL-cholesterol (LDL-C) from lysosomes to the PM [57]. Nine peroxisomal proteins were enriched in the screen and knockdown of each induced cholesterol accumulation in the lysosomes, mimicking the phenotype caused by NPC1 deficiency. By using super resolution structured illumination microscopy and electron microscopy, peroxisomes were found to establish dynamic membrane contacts with lysosomes in an LDL-regulated manner. Such lysosomeperoxisome membrane contacts (LPMCs) could be reconstituted in vitro using lysosomes purified from cells stably expressing NPC1-FLAG-mCherry and peroxisomes from those expressing EGFP-His6-SKL in the presence of cytosol and metabolic energy, and were mediated by lysosomal protein synaptotagmin VII (Syt7) and peroxisomal phospholipid phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] (Figure 2a). Depletion of either Syt7 or peroxisomal PI(4,5)P2 attenuated LPMC formation, impaired in vitro cholesterol transport from lysosomes to peroxisomes and, consequently, increased cholesterol content in lysosomes. Most importantly, robust cholesterol buildup was detected in human patient with various PDs and the mouse model of X-ALD, underscoring a potential contribution of abnormal cholesterol trafficking to PDs.
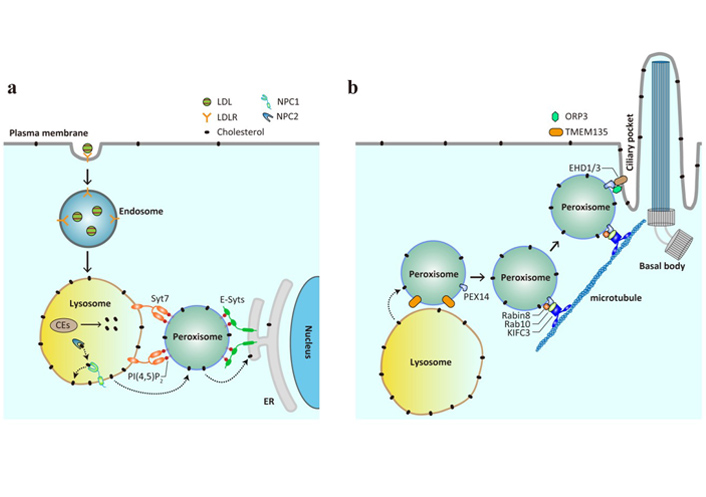
Peroxisomes mediate cholesterol trafficking as conduits and vehicles. (a) LDL is internalized by LDLR on the plasma membrane and delivered from early endosomes to late endosomes and lysosomes, where the carried CEs are hydrolyzed to liberate cholesterol. The lysosomal proteins NPC1 and NPC2 function in concert to insert cholesterol onto the lysosomal membrane, from where it is further transferred to the downstream membranes. Peroxisomes, via PI(4,5)P2, facilitate cholesterol transport from the lysosomes to the ER by interacting with lysosomal protein Syt7 and ER-resident extended synaptotagmins (E-Syts); (b) peroxisomes are able to deliver cholesterol to the primary cilium. The interaction between peroxisomal protein PEX14 and the Rabin8-Rab10-KIFC3 complex allows peroxisomes to move along the microtubules to the primary cilium, and that between PEX14 and the ORP3-EHD1/3 complex mediates the formation of membrane contacts between peroxisomes and ciliary pocket. TMEM135 is likely to be involved in LPMC-mediated cholesterol transport and primary ciliogenesis, since TMEM135 knockdown leads to cholesterol accumulation in lysosomes and defects in ciliogenesis
SKL (also known as peroxisomal targeting sequence 1) is a carboxyl-terminal tripeptide directing protein import into the peroxisomal lumen [58]. One might therefore argue that the nickel beads cannot pull down intact peroxisomes, since the EGFP-His6-SKL recombinant protein should in theory stay inside. While this concern seems legitimate, a later study provided two pieces of evidence showing that the overexpressed SKL-tagged protein is, at least in part, retained on the peroxisomal membrane [59]. First, in the rapamycin-induced protein heterodimerization system, a cytosolic protein could be recruited to the peroxisomal membrane by SKL as effectively as by PEX11a, a known peroxisomal membrane protein. Second, contrasting to the luminal protein catalase that was completely resistant to trypsin digestion till membrane permeabilization, the overexpressed EGFP-His6-SKL protein could be partially digested even in the absence of detergent. These results validated the method of purifying peroxisomes from the EGFP-His6-SKL stable cells.
In agreement with the findings that peroxisomal PI(4,5)P2 is a crucial determinant of LPMC, knockdown of phosphatidylinositol-5-phosphate 4-kinase type-2 α (PIP4K2A), an enzyme catalyzing the synthesis of peroxisomal PI(4,5)P2, reduced LPMC and caused cholesterol accumulation in the lysosomes [60]. Increases in lysosomal cholesterol contents and decreases in ER and PM cholesterol levels were also detected in PIP4K2A-knockout cells. The cholesterol accumulation phenotype, however, was successfully reverted by re-expression of the wild-type form of PIP4K2A in PIP4K2A-knockout cells, but not of the kinase inactive form or the schizophrenia-related form. Mechanistically, reduced peroxisomal PI(4,5)P2 levels account for the effect of PIP4K2A or ABCD1 deficiency on lysosomal cholesterol accumulation [57, 60].
The destiny of LDL-C after reaching the peroxisomes was the next logical question to pursue. Given that peroxisomes and ER are tightly correlated in terms of origin and that LDL-C must be ultimately sensed by the regulatory machineries on the ER membrane [21], whether peroxisomes formed membrane contact sites with the ER for post-lysosomal trafficking of LDL-C was examined using both imaging and biochemical approaches. Indeed, a significant portion of peroxisomes was found juxtaposed to the ER under the confocal and electron microscopes, and such peroxisome-ER membrane contacts (PEMCs) were verified by in vitro reconstitution assay using purified organelles [59]. Interestingly, peroxisomal PI(4,5)P2 that bridged LPMCs also participated in the formation of PEMCs by binding to the ER-resident protein E-Syts. E-Syts are distantly related to Syt7 and encompasses Ca2+-binding C2 domains facing the cytosol, with which peroxisomal PI(4,5)P2 interacts. Ablation of extended synaptotagmins or peroxisomal PI(4,5)P2 significantly decreased PEMCs and caused cholesterol accumulation in the lysosomes instead of peroxisomes. This lysosomal storage of cholesterol is not totally unexpected, since the lysosomes contain multilamellar membranes within the lumen and can therefore accommodate cholesterol that fails to reach the ER due to impaired PEMCs [61]. The direct evidence showing that cholesterol is transported from the peroxisomes to the ER came from the in vitro reconstitution assay, in which peroxisomes from PEX11a-His6-EGFP stable cells pre-exposed to 3H-cholesterol were incubated with the purified ER microsomes and the amounts of 3H-cholesterol in the ER microsomes were measured. Cholesterol transport occurred in the PI(4,5)P2 and E-Syts-dependent manner, suggesting an absolute requirement of PEMCs. More interestingly, peroxisomes could not only form membrane contact sites with both the lysosomes and the ER, but also markedly enhance 3H-cholesterol transfer from the lysosomes to the ER. Altogether, this series of studies convincingly demonstrate that peroxisomes function as an important intermediate organelle during lysosome-to-ER transit of LDL-C.
Peroxisome-mediated cholesterol transport inspires many interesting questions. Since cholesterol can give rise to di- and trihydroxycholestanoic acids that are transported into the peroxisomes for bile acid biosynthesis, LDL-C delivered to the peroxisomes may likely be routed to synthesize bile acids as well. However, this seems not to be the case, since mice lacking Abcd1 or carrying Syt7 point mutations that block Ca2+ binding to the C2 domains displayed similar levels of bile acids to the wild-type mice [59]. It is also worth mentioning that peroxisomes can be tethered to the ER by the peroxisomal membrane protein acyl-CoA binding domain protein 4 or 5 via the interaction with the ER protein vesicle-associated membrane protein-associated proteins [62–64]. This type of PEMCs is irrelevant to LDL-C egress from the lysosomes [59], but regulates the levels of plasmalogen and cholesterol as well as the movements and membrane dynamics of peroxisomes [62, 64]. It is anticipated that more tethers between peroxisomes and the ER will be revealed, with each playing specific roles in lipid metabolism and beyond.
Peroxisomes as cholesterol vehicles
Most peroxisomes undergo random oscillations or exhibit slow motions; however, a small (5–10%) population can travel at fast speed in a saltatory manner over long distances [65, 66]. In mammalian cells, this rapid, directional transport of peroxisomes is microtubule-dependent and requires the coordinated actions of kinesin and dynein motors [67, 68]. Peroxisome movement critically regulates its turnover and functions [69]. In the context of disease pathogenesis, impaired peroxisome trafficking and abnormal peroxisome distribution have been reported in the cells derived from patients with hereditary spastic paraplegia—a devastating neurological disease involving motor neuron degeneration, and with PDs featuring reduced peroxisome abundance, respectively [70, 71].
Two recent studies pinpoint a role of peroxisomes in delivering cholesterol to the primary cilium (Figure 2b) [72, 73]. The primary cilium is a specialized organelle that protrudes from the surface of almost all mammalian cells, including neurons and astrocytes, for sensing and transducing external physicochemical stimuli, among which include the Hedgehog signaling that is essential for development and maturation of the brain and other tissues [74]. The work from Maharjan and colleagues [72] was stemmed from our previous findings that loss of peroxisomal protein TMEM135 reduced LPMCs and induced cholesterol accumulation in the lysosomes [57]. The authors detected that TMEM135 deficiency substantially reduced the ciliation of serum-starved human retinal pigment epithelial cells, which are widely used to study ciliogenesis. These observations are in accordance with the long-held view that the ciliary membrane is highly enriched in cholesterol [75], and also with the results showing shortened primary cilium in other models of altered cholesterol metabolism [76–78]. Examination of each of the early steps of ciliogenesis revealed that TMEM135 ablation specifically impaired the trafficking of the small GTPase Rab8 to the centrioles, where it is activated by the guanine nucleotide exchange factor Rabin8. As a result, the recruitment of intraflagellar transport protein 20 to the centrioles and subsequent ciliary membrane elongation and ciliogenesis were suppressed. Notably, all these deficits in TMEM135-depleted cells were effectively rescued by the addition of cholesterol in complex with methyl-β-cyclodextrin, suggesting a requirement of cholesterol for primary cilium formation. Since the inactive form of Rab8 is preferentially localized to peroxisomes [79], which fail to receive cholesterol upon TMEM135 deficiency [72], it is reasonable to speculate that peroxisomes provide cholesterol for Rab8-harboring vesicles for their transport to the centrosome. The carried cholesterol may, together with Rab8 vesicles, be ultimately assembled into the ciliary membrane [80].
The study by Miyamoto and colleagues [73] provides direct evidence showing that peroxisomes can function as a mobile carrier delivering cholesterol to the primary cilium. These authors started with the observations that both ciliary cholesterol and Hedgehog signal transduction were reduced in primary fibroblasts from patients with ZS—a severe form of PDs caused by PEX mutations (Figure 1b)—as well as in cultured epithelial cells lacking PEX1 or PEX4. They attributed these ciliary phenotypes to reduced cholesterol supplied by peroxisomes for two reasons. First, the majority of peroxisomes were lost upon PEX deficiency; second and more importantly, LDL whose carried cholesterol must pass through several organelles, including peroxisomes, prior to reaching the ciliary membrane was less potent to rescue the Hedgehog signaling compared with direct cholesterol supplementation via the cholesterol/methyl-β-cyclodextrin complex. Under different imaging microscopes, peroxisomes were visualized to form dynamic contacts with the ciliary pockets through the interaction between PEX14 and EHD1 and 3. PEX14 is a peroxisomal membrane protein with multiple roles, one of which is to mediate the long-range movement of peroxisomes by binding tubulin [66]. EHD 1 and 3 have been shown to localize to the ciliary pocket membrane and function upstream of Rab8 in promoting ciliary vesicle formation [81]. Consistent with these previous results, microtubules were found to be essential for both the peroxisome-primary cilium contacts and ciliary cholesterol supply. These peroxisome-primary cilium contact sites were potentiated by oxysterol binding homology protein-related protein 3, a sterol transfer protein co-residing with PEX14 and EHD1 at the ciliary pocket.
With respect to the molecular mechanisms of peroxisome movement, the authors found that Rabin8 and the downstream effector Rab10, via interaction with PEX14, participated in peroxisome transport to primary cilium (Figure 2b). Ablation of Rabin8 or Rab10 reduced peroxisome dynamics and the contacts between peroxisomes and primary cilia, whereas forced targeting of peroxisomes to the ciliary pockets and over-expression of constitutively active Rab10 restored ciliary cholesterol levels in Rabin8-depleted cells. The constitutively active form of Rab8 was ineffective because Rabin deficiency prevented GTP loading on Rab8, as shown in the other study [72]. They further demonstrated that KIFC3—a C-terminal kinesin of the kinesin-14 family involved in peroxisome positioning [82]—could simultaneously interact with PEX14 and Rab10, thereby anchoring the peroxisomes on the microtubule networks, with the C-terminal motor domain of KIFC3 directing their transport towards to the ciliary pockets. Based on all these results, the authors proposed that the peroxisomal supply of cholesterol for primary cilium was a three-step process, in which peroxisomes with cholesterol were first attached to microtubules via the PEX14-Rab10-KIFC3 complex, followed by long-range movement directed and powered by the Rabin8-Rab10-KIFC3 complex, and finally the peroxisome-primary cilium contacts formed between PEX14 and EHD1/3 and secured by ORP3.
The discovery that peroxisomes can receive and mobilize cholesterol is worthy of discussion. Among all possible post-lysosomal routes that LDL-C may choose [55], the lysosome-peroxisome-ER membrane contacts undoubtedly provide a more direct route allowing ER-resident regulatory machineries to sense exogenous cholesterol rapidly, with peroxisomes adding a layer of regulation while serving as a safeguard to buffer the steep cholesterol gradient between lysosomes and the ER. Apart from forming contacts with the ER, peroxisomes can carry cholesterol over long distances to primary cilium [73]. The hitchhiking of highly motile, pre-existing organelles for cholesterol transport is novel in terms of the canonical picture of intracellular cholesterol transport involving vesicular and non-vesicular mechanisms, and also of the hitchhiking motion of many other cargos, such as RNA granules and messenger ribonucleoprotein complexes [83, 84], where cargos and carriers are separate entities. Peroxisomes are good cholesterol carriers since most are aligned with microtubules with motor proteins pre-quipped [85]. Moreover, peroxisomes may have already received cholesterol from lysosomes via LPMCs. The transport of peroxisomes along microtubule does not necessarily contradict with their contacts with lysosomes and ER because different organelles can attach to each other for co-migration [86]. Lysosomes may also be a perfect candidate due to their mobility and capacity for cholesterol. In fact, lysosome-derived vesicles have been shown to mobilize LDL-derived cholesterol in a Rab8a-Myosin5b-actin dependent process to the PM [87]. Such organelle/vesicle-mediated cholesterol movement can be particularly important in neurons with complex morphology, and may contribute to PDs and neurodegenerative diseases when disrupted.
Peroxisome deficits and impaired cholesterol metabolism in brain pathology
Peroxisome deficits in PDs and other neurodegenerative diseases
PDs are a group of inherited diseases in which peroxisome biogenesis or functions are impaired (Figure 1). At least 14 different PEX genes that encode peroxins for peroxisome assembly, proliferation and fission, and 11 genes that encode enzymes or transporters of peroxisomal metabolic pathways have been identified, based on which PDs are further divided into PBDs (Figure 1b) and PEDs (Figure 1c) [88]. ZSDs are a prototype of PBDs and comprise ZS, NALD and IRD—3 forms that share overlapping symptoms presented by several organs of the body but differ in disease severity and age of onset [89]. RCDP1 and RCDP5 also belong to PBDs and are caused by mutations in PEX7 and PEX5 that specifically encodes the long isoform, respectively [90]. PEDs are more heterogeneous in terms of genetic causes and clinical presentations. X-ALD is a prevalent PED caused by mutations in the ABCD1 gene that encodes a transporter mediating the import of VLCFAs into the peroxisomes. It has three major forms including cerebral adrenoleukodystrophy that may appear in childhood, adolescence or adulthood, adrenomyeloneuropathy, and adrenal insufficiency. Males are more susceptible to X-ALD and can present one or more of the forms beginning early in life, whereas most females only develop adrenomyeloneuropathy in the 4th decade and later [91]. Other PEDs associated with impaired fatty acid β-oxidation include ACOX deficiency, D-bifunctional protein deficiency, 2-methylacyl-CoA racemase deficiency and sterol carrier protein X deficiency. Refsum disease is the result of defects in α-oxidation of phytanic acid, whereas the remaining three RCDP types (RCDP2, RCDP3, RCDP4) are caused by mutations in the genes encoding enzymes involved in plasmalogen biosynthesis. This topic has been reviewed in detail elsewhere [92].
Despite genetic and phenotypic complexity, neurological deficits are widely presented in most PDs (Figure 1a). With respect to brain pathologies, patients with ZS and the less severe NALD exhibit cerebral malformations and heterotopic positioning of cerebellar Purkinje cells as a result of defective neuronal migration [93]. Delayed myelination has also been observed in some cases of ZSDs using cerebral magnetic resonance imaging [94, 95]. Childhood cerebral adrenoleukodystrophy, as the most severe form of X-ALD, features degenerative myelin loss in the cerebral white matter that can become inflammatory over time and leads to severe cognitive deterioration and even premature death [96]. Moreover, post-developmental neuronal degeneration, such as the loss of Purkinje cells and granular neurons in the cerebellum of some RCDP patients, also represents a class of brain pathology in PDs [97]. For more aberrant phenotypes of PD brains, the readers are referred to previous reviews [2, 3]. The consequences of such brain abnormalities include hypotonia, seizures, developmental delays, and hearing and vision loss, which all serve as the clinical manifestations of PDs [98].
Beyond PDs, peroxisomal dysfunction has been linked to some neurodegenerative diseases, such as AD, Parkinson’s disease, amyotrophic lateral sclerosis, and multiple sclerosis [3, 99, 100]. An intimate interplay between these two is not surprising, given that the brain is highly enriched (50–60% of dry weight) in lipids and that the peroxisomes actively metabolize the substances of pivotal structural and functional roles to the brain cells. Alteration of these peroxisome-related lipids may contribute to the development, if not the onset, of aforementioned neurodegenerative diseases. Indeed, reduced levels of DHA and DHA-containing ethanolamine plasmalogens have been detected in the brain of AD and Parkinson’s disease patients [101, 102]. Conversely, the levels of VLCFAs are elevated in the cortical areas of human AD brains and multiple sclerosis grey matter [103, 104]. Moreover, peroxisomal dysfunction can also cause oxidative stress that explains for neurodegeneration seen in AD and Parkinson’s disease.
Impaired cholesterol metabolism in PDs and other neurodegenerative diseases
The involvement of cholesterol metabolism in PDs has been examined using fibroblasts derived from PD patients, immortalized cells lacking PEX genes, and mouse models mimicking PDs. A series of studies in the 1990s first evaluated cholesterol biosynthesis by examining the incorporation of radioactive precursors into cholesterol in cultured PD fibroblasts and peroxisome-deficient Chinese hamster ovary cells [105–109]. However, no consensus was reached on whether cholesterol biosynthesis was affected in the absence of functional peroxisomes. Cholesterol uptake and esterification were similarly investigated by exposing cultured cells to LDL [106, 110]. All these early results should be interpreted with caution not only because of artificial in vitro culture conditions but also because they may merely be the indirect consequences of peroxisomal dysfunction, which is now known can induce ER stress and impair mitochondrial redox balance [111, 112]. Later, two mouse models for human ZS—Pex2 and Pex5 knockouts that survive for limited, yet different, periods after birth [113, 114]—and one for X-ALD—Abcd1 knockouts [115]—were developed and the effects of peroxisomal deficiency on cholesterol homeostasis were assessed in vivo. Unfortunately, inconsistent cholesterol phenotypes were observed in these different models. Whereas early postnatal Pex2-deficient mice showed reduced cholesterol levels in the plasma and reduced cholesterol content but increased expression activities and expression of cholesterolgenic enzymes in the liver [116], no significant changes were detected in Pex5-deficient newborns [117, 118]. Also, while Abcd1-deficient mice displayed reduced expression of 3-hydroxy-3-methylglutaryl-CoA reductase, the rate-limiting enzyme of the cholesterol biosynthetic pathway, in the liver with the elevated levels of plasma cholesterol [119], those lacking the gene encoding L-bifunctional protein, which catalyzes the second and third step of fatty acid β-oxidation in the peroxisomes, exhibited increased levels of 3-hydroxy-3-methylglutaryl-CoA reductase proteins but similar or decreased cholesterol biosynthesis in the liver [120]. Even with the availability of additional mouse models resembling both PBDs and PEDs [98, 121], there has been only evidence showing the correlation, but not causal relationship, between disturbed cholesterol biosynthesis and PDs.
The contribution of intracellular cholesterol transport to PDs was first brought to attention by the previously mentioned paper on LPMCs [57]. ABCD1 deficiency reduces peroxisomal PI(4,5)P2 levels and therefore impairs LPMC formation. In Abcd1-knockout mice, robust cholesterol accumulation was observed in the cerebellum and adrenal gland at 7 months of age, well in advance to the locomotion deficits seen at 20 months and later. These results suggest that defective cholesterol transport may be a cause rather than consequence of X-ALD. In a broader context, the accumulation of cholesterol has been detected in the fibroblasts from patients with X-ALD, ZS, IRD, RCDP2 and RCDP3 [57, 73, 122], as well as in the cultured cells deficient in several other genes involved in PBDs and PEDs (Xiao et al., unpublished results). Based on these findings, it is reasonable to speculate that defective cholesterol trafficking is a common feature of PDs. As a result of cholesterol sequestration within the lysosomes, peroxisomes are short of cholesterol, which may exacerbate peroxisomal dysfunction. Reduced amounts of cholesterol arriving at the ER also result in activation of the sterol regulatory element-binding protein pathway as reported in Pex2−/− mice [116]. Moreover, lysosomal cholesterol accumulation causes detrimental effects on the cells through STING activation and other mechanisms [123, 124]. Impaired cholesterol transport to primary cilium leads to ciliopathies in PDs as discussed earlier [72, 73]. Hence, abnormal intracellular cholesterol transport may accelerate PD progression and manipulating cholesterol movement can be a new way to treat PDs.
Cholesterol imbalance is frequently implicated in many neurodegenerative diseases, as discussed in detail by several excellent previous reviews [47, 125]. However, there have been controversies whether increased or decreased levels of blood cholesterol are associated with neurodegeneration. For example, higher, similar, or lower plasma cholesterol have all been reported in the AD and Parkinson’s disease cohorts relative to their corresponding controls [126]. These seemingly contradictory results are partly attributed to the differences in disease progression. It is also of note that the alteration of cholesterol levels in circulation is independent of that in the brain and may therefore not a good indicator of brain abnormalities in AD and Parkinson’s disease. Direct measurements of cholesterol levels in the cerebrospinal fluid may be more reliable in predicting neurodegeneration in the brain. NPC disease is a neurovisceral disorder with highly variable neurological and systemic symptoms. Cholesterol derivatives including cholestane-3,5,6-triol and 7-ketocholesterol and those generated from cholestane-3,5,6-triol, such as 3β,5α,6β-trihydroxycholestan-(25R)26-oic acid, 3β,7β-dihydroxycholest-5-en-(25R)26-oic acid and 3,5,6-trihydroxycholanic acid, are elevated in the NPC patients [127–130]. Interestingly, the C27 metabolites are drastically increased in some PDs as well [131]. These results can be explained by the facts that the conversion of C27-bile acids to C24-bile acids occurs in peroxisomes, and that the activities of peroxisomal enzymes are already compromised prior to the onset of symptoms in Npc1 mutant mice [132]. They also highlight a close interplay between peroxisomal dysfunction and neurodegeneration. Although it is unclear whether LPMC-mediated cholesterol transport to peroxisomes is defective in other neurodegenerative diseases, this route undoubtedly holds great promise as a novel strategy for the treatment of PDs and NPC disease.
Conclusions
Starting from an organelle simply involved in H2O2 metabolism to the one actively regulating lipid metabolism and inflammation, our understanding of the peroxisomes has kept evolving thanks to the decades of active investigation. However, what we know about the peroxisomes still lags far behind other subcellular organelles such as the lysosomes and mitochondria. With respect to the role in cholesterol metabolism, it is now clear that peroxisomes can transport cholesterol from the lysosomes to the ER as well as carry cholesterol all the way to primary cilium. Failure to do so leads to cholesterol accumulation in the lysosomes, hyperactivation of the sterol regulatory element-binding protein pathway, ciliopathies and, a step further, drive the development of PDs. Strikingly, lysosomal cholesterol accumulation is also a prominent feature of the NPC disease. The presence of aberrant cholesterol distribution in these widely different diseases raises several questions. How do peroxisomes manage to handle multiple lipid-metabolizing pathways, and how does disruption of peroxisomal functions affect cholesterol egress from the lysosomes? How might peroxisome-mediated cholesterol transport facilitate repair of neurons and neural circuits in pathological settings? How is cholesterol dynamically transported among different organelles within the cell? How are cholesterol levels in local membrane precisely controlled? These questions need particularly to be assessed in neurons, given the neurological phenotypes commonly observed in PDs and NPC disease. Furthermore, future studies are necessary to determine whether promoting cholesterol transport is a universal strategy to relieve symptoms of PDs, NPC disease, and other neurodegenerative diseases.
Abbreviations
| ABC: | ATP-binding cassette |
| ABCD1: | ATP-binding cassette subfamily D member 1 |
| ACATs: | acyl-coenzyme A:cholesterol acyltransferases |
| ACOX: | acyl-coenzyme A oxidase |
| AD: | Alzheimer’s disease |
| ApoE: | apolipoprotein E |
| CEs: | cholesteryl esters |
| CoA: | coenzyme A |
| DHA: | docosahexaenoic acid |
| DHAP: | dihydroxyacetone phosphate |
| ER: | endoplasmic reticulum |
| E-Syts: | extended synaptotagmins |
| H2O2: | hydrogen peroxide |
| IRD: | infantile Refsum disease |
| LDL: | low-density lipoprotein |
| LDL-C: | low-density lipoprotein-cholesterol |
| LDLR: | low-density lipoprotein receptor |
| LPMCs: | lysosome-peroxisome membrane contacts |
| NALD: | neonatal adrenoleukodystrophy |
| NPC: | Niemann-Pick type C |
| PBDs: | peroxisome biogenesis defects |
| PDs: | peroxisomal disorders |
| PEDs: | single peroxisomal enzyme deficiencies |
| PEMCs: | peroxisome-endoplasmic reticulum membrane contacts |
| PIP4K2A: | phosphatidylinositol-5-phosphate 4-kinase type-2 α |
| PI(4,5)P2: | phospholipid phosphatidylinositol 4,5-bisphosphate |
| PM: | plasma membrane |
| RCDP: | rhizomelic chondrodysplasia punctata |
| RCDP1: | rhizomelic chondrodysplasia punctata type 1 |
| ROS: | reactive oxygen species |
| Syt7: | synaptotagmin VII |
| X-ALD: | X-linked adrenoleukodystrophy |
| VLCFAs: | very long-chain fatty acids |
| ZS: | Zellweger syndrome |
| ZSDs: | Zellweger spectrum disorders |
| 24S-HC: | 24S-hydroxycholesterol |
Declarations
Author contributions
BLS and JL conceived the manuscript. JX and JL wrote the manuscript. BLS edited the manuscript. All authors contributed to the writing and revision of the manuscript and approved the final version.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by grants from the National Natural Science Foundation of China (31771568 and 31690102) and China Postdoctoral Science Foundation Grant (2021M6924782). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2021.