
Abstract
Neuropathic pain (NP), which is difficult to treat, remains a heavy burden for both individuals and society. The efficacy of current treatments is insufficient. The pathophysiology of NP is still not fully elucidated, and there is a need to explore new therapeutic targets to develop more effective treatment strategies. Recent studies showed that thrombospondin 4 (TSP4) protein expression is increased in the spinal cord following nervous system injury and that blocking or inhibiting this increase improves NP. In this review, it has been aimed to present the evidence for the role of TSP4 in the mechanisms of NP development and to evaluate the therapeutic potential of TSP4 blockade in the treatment of NP.
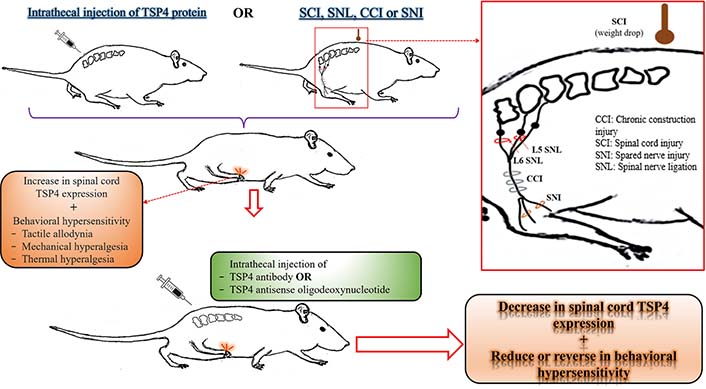
The relationship between TSP4 protein level in the spinal cord and behavioral hypersensitivity in rodents. This illustration has been created to collectively reflect the results of experimental animal studies investigating the relationship between the TSP4 levels in the spinal cord and NP. For more detailed information on related studies, please see the “
Keywords
Astrocyte, dorsal root ganglion, neuropathic pain, nerve injury, rodent, spinal cord, thrombospondin 4Introduction
Neuropathic pain (NP) is a type of pain that can occur due to any injury to the nervous system and has a high rate of chronicity. It is a critical health problem. The quality of life of patients with NP decreases considerably because various comorbidities such as anxiety, depression, and sleep disorders often accompany pain [1–3]. In addition, it also constitutes a significant economic burden at the social level as it increases the use of health services and general health costs [4].
The etiology of NP has a broad spectrum including diabetes mellitus, surgery, trauma, herniated discs, nerve compression syndromes, multiple sclerosis, stroke, human immunodeficiency virus infection, herpes zoster virus infection, various cancers, antineoplastic drugs, etc. [1, 4–6]. It is estimated that the prevalence of NP in the general population ranges from 6.9% to 10% [7]. It is also predicted that the probability of encountering NP in the clinic will increase due to the prolongation of human lifespan and the resulting increase in the incidence of chronic diseases [8, 9]. In addition, evidence has been found that the severe acute respiratory syndrome coronavirus 2 virus, which causes coronavirus disease of 2019 (COVID-19) infection, can cause neurological involvement [10–13]. Therefore, it is thought that some of the patients infected with COVID-19 may develop NP within weeks or months or exacerbation in patients with NP [14].
The treatment of NP is different from the treatment of other chronic pain, such as nociceptive or nociplastic pain, where there is no nervous system damage [15, 16]. For example, non-steroidal anti-inflammatory drugs, which produce significant analgesic efficacy in inflammatory pain, are often useless in patients with NP [15]. Pharmacological agents currently used as first-line drugs in treating NP are gabapentinoids containing gabapentin and pregabalin, tricyclic antidepressants such as amitriptyline, imipramine, nortriptyline, desipramine and clomipramine, and duloxetine, a serotonin-noradrenaline reuptake inhibitor [17–21]. Drugs recommended for second or third-line treatment of NP or indicated in special conditions are opioids (morphine, oxycodone, fentanyl, and tramadol), venlafaxine (a serotonin-norepinephrine reuptake inhibitor), some anticonvulsants (carbamazepine and lamotrigine), topical agents (lidocaine and capsaicin) and, botulinum toxin A. However, they have insufficient efficacy and are not specific for NP [22, 23], leading to the inability of most patients with NP to be treated appropriately and adequately [24, 25]. A recent observational study reported that the effects of several internationally recommended drugs for treating NP varied from 30% to 50% in “ideal” subjects selected from positive clinical trials. Therefore, this rate is likely much lower considering the real-life conditions and the entire patient population [26]. In addition, many of these drugs have many undesired effects. Especially those administered systemically may cause symptoms related to the central nervous system (CNS), such as dizziness, drowsiness, insomnia, impaired attention/concentration/thinking, and/or peripheral adverse effects such as nausea, vomiting, constipation, and dry mouth. The inability of patients to tolerate some of these adverse effects complicates compliance with treatment. In order to avoid these side effects, prescribing drugs in suboptimal doses [27] or not increasing the dose [22] may also cause inadequate treatment. The potential for addiction and the development of tolerance to the effects of opioids significantly limit their use [28].
For all these reasons, developing more effective strategies for treating NP is needed. Although knowledge about the mechanisms underlying the pathophysiology of pain is increasing, the complex network of mechanisms and interactions that occur in the peripheral nervous system (PNS) and CNS complicates the precise determination of the pathophysiology of NP. Thrombospondin 4 (TSP4) may be one of the fundamental mechanisms in NP initiation and/or maintenance. A better understanding of the role of TSP4 in the pathophysiology of NP may help develop new strategies for treating NP. Therefore, we reviewed the role of TSP4 in the pathophysiology of NP, which is caused by peripheral nerve injury (PNI), and the therapeutic potential of TSP4 blockage treating NP.
Method
We searched in the “PubMed” database by using the keywords “thrombospondin 4” and “neuropathic pain”. In our search in July 2022, we found 13 publications. From these studies, we shared the findings of 10 original studies investigating whether there is an association between intrathecal administration of TSP4 or spinal TSP4 expression and the development of behavioral hypersensitivity following an NP model in animals. All of these studies are listed in Table 1. We also extensively searched for in vitro studies and other literature data that could contribute to this research, by searching the “ScienceDirect” and “Web of Science” databases without any exclusion criteria.
General characteristics and structures of TSPs
TSPs are extracellular, oligomeric, multidomain and calcium (Ca2+)-binding glycoproteins [29]. Endothelial cells, platelets, fibroblasts, muscle cells, astrocytes, neurons, and many other cells may express one or more isoforms of TSP [30–36]. Their expressions are prominent, especially during growth and development. In adulthood, although they are not normally abundant except in rapidly cycling tissues such as bone [37], their expressions often increase considerably in response to various pathologies [38]. TSPs mediate cell-cell interactions and cell signaling at cell surfaces and in the extracellular matrix (ECM), and they mediate a variety of processes such as inflammation, immune response, wound-healing, osteogenesis, angiogenesis, vessel wall biology, connective tissue organization, synaptogenesis, and tumor growth [29, 31, 32, 39].
Firstly, TSPs were discovered by Baenziger et al. [40] in 1971. Later, it was found to have more than one species. Today, it is known that the TSP superfamily consists of five members with trimeric (group A, Figure 1A) or pentameric (group B, Figure 1B) structures [34, 41, 42].
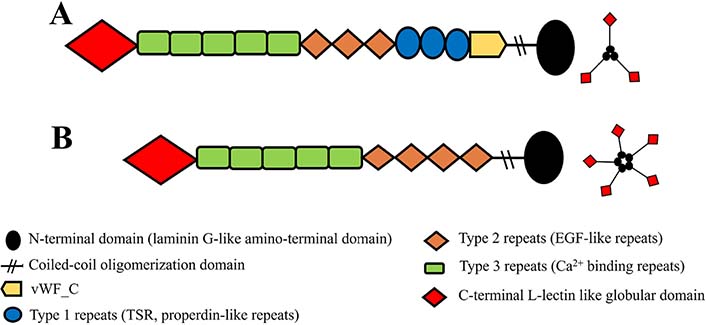
Simple representation of domain architectures of TSP glycoproteins. A. Domain architectures of group A (TSP1 and TSP2) and its trimeric structure; B. domain architectures of group B (TSP3, TSP4, TSP5), and its pentameric structure. EGF: epidermal growth factor; vWF_C: von Willebrand type C domain
Note. Adapted from “Thrombospondins and synaptogenesis,” by Wang B, Guo W, Huang Y. Neural Regen Res. 2012;7:1737–43 (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4302456/#ref1). CC BY-NC.
The sequences of all members of the TSP family have the highest similarity in the carboxyl regions, including type 2 repeats, type 3 repeats, and the globular carboxy-terminal domain (53–82%), while the lowest in the amino-terminal domain (approximately 25%) [39]. The common functions attributed to all TSPs at the cellular level are primarily based on their interactions with ECM components and glycosaminoglycans, and their promoting Ca2+-dependent cell attachment [29]. These similar effects of TSPs are thought to be mediated by carboxyl regions with high sequence homology. Unlike group A, group B lacks the vWF_C and type 1 repeats (also known as properdin-like repeats). Another major structural difference is the number of type 2 repeats known as EGF-like repeats. Group A has three and group B has four EGF-like repeats [39, 43]. In addition, other minor structural variations are thought to give them special functions. Their distributions differ considerably in each structure and system and, each has different expression patterns in developing tissues and throughout life [34].
Relationships of TSPs with the nervous system
Astrocytes, one of the glial cells, modulate synapse formation and synaptic transmission in the CNS [44–46]. TSPs are a component of the intercellular signaling network mediated by astrocytes in the CNS [47, 48], and astrocyte-derived TSPs are considered an essential synaptogenic signal in synaptogenesis [31, 48–50]. They also support the neurite outgrowth in PNS and CNS [36, 42, 47, 48, 50, 51].
The contribution of TSPs to synaptogenesis is associated with the interaction of their type 2 repeats (EGF-like) domain with the vWF_A of the α2δ1 subunits of the voltage-gated Ca2+ channel receptor (Cavα2δ1) [49, 52]. Cavα2δ1 has also been defined as the major neuronal receptor of TSPs in the CNS [49]. Although EGF-like domains differ in number between groups A and B [39, 43], all TSPs secreted by astrocytes have the potential to play a role in synaptogenesis [48, 49], particularly in excitatory glutamatergic synaptogenesis [29, 53], but not inhibitory gamma-aminobutyric acid (GABA)ergic [46].
It has been determined that at least four TSP isoforms (TSP1–4) are secreted by astrocytes [54]. Among them, TSP1, 2, and 4 are three TSP isoforms that mainly contribute to the process of synaptogenesis. TSP3 and TSP5, predominantly produced in muscle and bone tissues, contribute very little to synaptogenesis [42]. Under physiological conditions, TSP1 and TSP2 are secreted by immature astrocytes mainly during the growth and development period and support synaptogenesis [48, 55, 56], while TSP4 is expressed from mature astrocytes and plays a role in the synaptic plasticity and, supports the maintenance and stability of synapse formation in the adult brain [36, 42, 57]. Therefore, TSP4 is thought to represent the adult isoform of TSP in the CNS [58].
TSP4, firstly identified by Lawler and colleagues [59], is expressed in motor and sensory neurons, neuromuscular junctions, and synapse-rich structures such as the cerebellum and retina [36]. In addition to the nervous system, it is also expressed in the vascular endothelium, skeletal and smooth muscles, and heart [33, 36, 59, 60]. However, it is thought to be a major neural TSP because of its expression and distribution in the nervous system, its relationship with synapse-rich layers, and its support for neurite outgrowth [36]. Under physiological conditions, TSP4 derived from mature astrocytes supports the protection of synaptic structures in the adult CNS [36, 42, 57]. However, it is also intensely secreted by immature astrocytes in the CNS as a response to nerve injury, and it has a critical role in injury-induced abnormal synaptogenesis leading to spinal sensitization and NP [41, 61]. TSP4 may also potentially participate in synaptic formation in the PNS because of its presence at neuromuscular junctions [31].
TSP4 and NP
The majority of peripheral nerves are composed of sensory (afferent), motor, and autonomic (efferent) neurons [62]. Therefore, neurodegenerative changes resulting from PNI may lead to long-term undesirable consequences such as sensorimotor dysfunction and NP [63].
NP is a clinical reflection of changes in the nervous system that result in sensitization of the PNS and CNS [2], and these changes differentiate NP from other types of chronic pain [6]. TSP4 may be a potential one of the fundamental mechanisms in NP initiation and/or maintenance. Therefore, a better understanding of the role of TSP4 in the pathophysiology of NP may help develop new strategies for treating NP. Therefore, we reviewed the role of TSP4 in the pathophysiology of NP caused by PNI. Peripheral and central changes contributing to NP have been visualized in Figure 2.
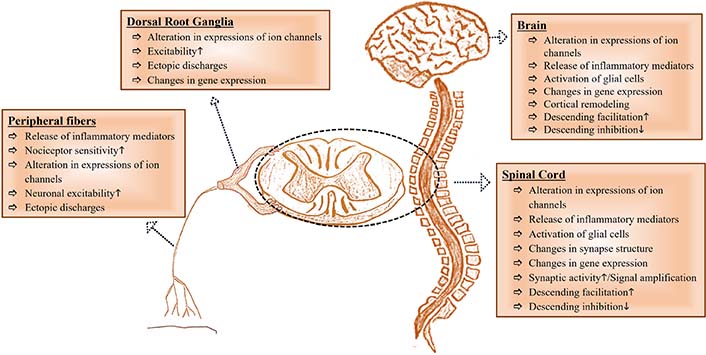
A glance at peripheral and central changes contributing to NP. It has been inspired by [23]. The upward arrow (↑) represents “increase” and, the downward arrow (↓) represents “decrease”
TSP4 and mechanisms contributing to peripheral sensitization
Peripheral sensitization can be briefly summarized as “the increased response of peripheral nociceptors due to the effect of inflammatory mediators released around by injured cells”. However, it is not that simple. Under physiological conditions, it is rare for healthy primary afferent neurons to reach the stimulus threshold without a stimulus input [64]. However, changes that occur as a result of any nerve injury may cause to increase in the nociceptor sensitivity, a decrease in the pain thresholds, an increase in membrane excitability (neuronal hyperexcitability), and changes in synapse formation. Therefore, activation of unmyelinated C fibers and thin myelinated A-δ fibers, which are nociceptive afferent fibers, by mechanical, thermal, and chemical stimuli that normally require high nociceptor thresholds indicates a potential tissue injury [6].
Following a PNI, neurotrophic factors, inflammatory mediators, leukotrienes, and cytokines are released at the injury site [65, 66]. This condition, which is responsible for the induction of peripheral sensitization [67], increases Ca2+ influx to nociceptive terminals, causing activation of secondary messenger systems, conformational changes in various proteins, and increased expression of the ion channels. Sodium (Na+) channels are important for neuronal and other excitable membranes [64]. PNI induces the expression of Na+ channels which have a lower stimulation threshold, in the injured neuron areas. Thus, the stimulation thresholds decrease, the responses to particular stimuli increase, and repetitive stimulations, also known as ectopic discharges, cause spontaneous depolarization [1, 2, 23, 64, 66, 67]. The expressions of other ion channels, such as Ca2+-activated potassium channels that affect neuronal excitability [68], may also change.
In many neurons, Ca2+ influx triggers low-threshold spikes that trigger an action potential burst mediated by Na+ channels via low-voltage-activated (LVA) channels [69], while it induces the release of neurotransmitters from synapses via high-voltage-activated (HVA) channels [70]. Both PNI [in mice with L4/5 spinal nerve ligation (SNL)] and in vitro TSP4 incubation (in naive mice) reduced HVA Ca2+ current (ICa) and increased LVA ICa in dorsal root ganglion (DRG) neurons. However, neither HVA nor LVA ICa changed in DRG neurons of TSP4 gene knockout mice with SNL [71]. Additionally, sympathetic neurons in the DRG, located in the proximal nerve trunk, sprout, especially following complete nerve transection, and form a structure called “neuroma” containing both afferent C fibers and efferent postganglionic sympathetic C fibers that secrete noradrenaline and adrenaline [66]. Sensitivity of sensory neurons and nociceptors, peripheral ends of primary sensory afferent fibers to catecholamines and other proinflammatory mediators, increases. Interestingly, healthy neighboring nociceptors also may contribute to peripheral sensitization by producing ectopic discharges [2, 66, 72].
TSP4 may contribute to peripheral sensitization in the NP process because of its presence at neuromuscular junctions [31]. Indeed, detecting the gene encoding the TSP4 protein (THBS4) activation in astroglia/satellite cells, Schwann cells, and primary sensory neurons of transgenic animals with L5 SNL supports this view [73]. It has also been suggested that TSP4, which is synthesized in neurons and glial cells following PNI, can be secreted into an extracellular space on DRG neurons in an autocrine manner, and the TSP4 level rises in DRG [73].
Injury or inflammation of the peripheral nerve causes upregulation of Cavα2δ1 in both DRG [49, 74–77] and the spinal cord [78, 79]. Both gabapentin and pregabalin are Cavα2δ ligands [78, 80, 81], and Cavα2δ1 is also the main receptor that mediates many effects of TSPs in nerve systems [49]. It has been shown that gabapentin blocked the effects of TSP4 on both HVA and LVA ICa in vitro conditions [71], and genetic deletion of the Cavα2δ1 also blocks the effects of TSP4 on cytoplasmic Ca2+ [82]. Briefly, the TSP4/Cavα2δ1 interaction is responsible for the effect of PNI on the ICa in the DRG [71].
Changes in the DRGs are very important for also central sensitization. Various changes in gene transcription and protein synthesis in DRGs also contribute to the facilitation of synaptic transmission by altering synaptic functions [66, 73]. Exposure of the sensory neuron somata in the DRG to high levels of TSP4 may provide a suitable environment for the post-injury dorsal horn (DH) synaptogenesis, which also plays a key role in the conversion of acute pain to chronic pain [41, 71, 73].
These mechanisms that develop following PNI cause abnormal signal transmissions from primary sensory afferent neurons to secondary neurons [23] and the development of spontaneous and paroxysmal shooting pain without external stimuli [6].
TSP4 and mechanisms contributing to central sensitization
Under physiological conditions, spinal and supraspinal antinociceptive systems continuously suppress the transmission of pain inputs in the CNS. The spinal antinociceptive system includes A-β fibers, inhibitory interneurons, and endogenous opioid systems, while the supraspinal antinociceptive system consists of pathways with extensions in the mesencephalon, pons, medulla oblongata, and locus coeruleus. These pathways directly inhibit synaptic transmission by releasing serotonin and noradrenaline into the synaptic space or indirectly activating enkephalinergic neurons in the spinal DH.
Central sensitization, which is responsible for most of the temporal, spatial, and threshold changes in acute and chronic pain states [83], is accepted as the basic mechanism underlying many chronic pain states [67, 84]. It involves certain neurochemical changes in the CNS regions that mediate the processing of the pain signal and a process in which the balance between the pronociceptive and antinociceptive systems is disturbed [23, 85].
The International Association for the Study of Pain defines central sensitization as “the increased response of nociceptive neurons in the CNS to normal or sub-threshold afferent impulse inputs” [86, 87]. Repetitive ectopic firings that occur during the peripheral sensitization process are probably responsible for also the initiation of central sensitization of spinal DH neurons [2, 67, 84]. In addition, expressions of ion channels are altered in spinal DH and thalamus neurons after both peripheral and spinal cord lesions, similarly to peripheral sensitization [88].
PNI also causes activation of glial cells in the spinal cord and inflammatory reactions in the injury site [89]. Both astrocytes and microglia, glial cells in the CNS, have contributed to the development of behavioral hypersensitivity, including hyperalgesia and allodynia associated with NP [89–91]. In active microglia, various Ca2+-sensitive intracellular signaling cascades such as extracellular signal-regulated kinase (also known as mitogen-activated protein kinase 1) and cyclic adenosine monophosphate-response element binding protein that lead to the production of pro-inflammatory cytokines are stimulated [89, 90] and increased neuronal excitability [83]. Astrocytes are activated more slowly and longer than microglia after PNI and may play a greater role than microglia in maintaining hypersensitivity in NP [83]. Injury-induced astrocyte activation in the spinal cord contributes to chronic pain by inducing abnormal excitatory glutamatergic synaptogenesis [92]. Glutamate is the major excitatory neurotransmitter of the CNS. The expression of glutamate receptors increases in the superficial DH rapidly in response to repetitive ectopic firings [65, 83]. As a result, membrane excitability and synaptic activity and hence transmission of excitatory inputs increase [83]. It has been reported that spinal cord astrocyte activation contributes to the development of behavioral hypersensitivity, and suppression of this increase in activation alleviates the development of allodynia due to nerve injury [92]. Expression of TSP4 also is increased in the dorsal spinal cord (DSC) following PNI, which is accompanied by an increase in the number and/or activation of astrocytes. Both injury-induced TSP4 expression and exogenous administration of TSP4 by intrathecal injection cause the development of behavioral hypersensitivity and increase the frequency of miniature excitatory postsynaptic currents (mEPSCs) in the L5 superficial spinal DH in rodents [93]. Therefore, TSP4 secreted from astrocytes is thought to mediate the effects of astrocytes on abnormal synaptogenesis and central sensitization [29, 41, 53, 94].
Dysfunction of endogenous pain control systems, including decreased synthesis, release, or activity of inhibitory transmitters due to apoptosis of GABAergic interneurons [66, 83], and/or loss of GABAergic inhibitory currents and/or decreased expression of inhibitory receptors in primary afferent terminals and postsynaptic neurons [72] may also contribute to this condition [87]. In conclusion, central sensitization represents a major functional shift in the somatosensory system from high-threshold nociception to low-threshold pain hypersensitivity. This hypersensitivity may persist even after the main pathology has been eliminated and in the absence of noxious stimuli [83].
Measurement of NP in animals
Rats and mice are most commonly used in experimental animal models of NP. To evaluate the presence of the NP in animals, it is often determined whether allodynia (pain response to a non-painful stimulus) and hyperalgesia (exaggerated pain response to a stimulus that normally causes pain), which are also known as behavioral hypersensitivity, develop or not. These phenotypes accepted as pain behavior are assumed to resemble the sensation responses that occur against stimuli such as brushing, touch, pressure, or hot or cold, in humans with NP.
Evidence of the relationship between TSP4 and NP
The results from animal studies to reveal the relationship between TSP4 expression and behavioral hypersensitivity show that:
In various NP models, it was determined that the increase in TSP4 expression in the spinal DH [30, 41, 95–98] and TSP4 level in DRGs [73] correlated with the development of behavioral hypersensitivity.
Intrathecal administration of TSP4 protein to naive rodents resulted in the development of behavioral hypersensitivity [30, 41, 93, 99, 100].
Intrathecal TSP4-antisense oligodeoxynucleotides (AONs) or TSP4 antibodies reduced the increased TSP4 expression in the spinal DH and reversed the behavioral hypersensitivity [41, 97, 98].
Blocking the injury-induced TSP4 expression in the spinal DH by intrathecal TSP4-AONs prevented the development of behavioral hypersensitivity [30, 41].
SNL did not lead to the development of behavioral hypersensitivity in THBS4 knockout mice [41, 93].
Intrathecal TSP4 protein which was administered before C6/7 facet joint distraction (FJD), exacerbated injury-induced tactile allodynia [30].
The effects of TSP4 on behavioral hypersensitivity were independent of gender in mice [100].
Detailed information has also been presented in Tables 1 and 2.
In addition:
Both intrathecal administration of TSP4 to naive animals [93, 100] and, FJD [30, 96] increased excitatory synapses in the DSC.
Blocking the injury-induced TSP4 expression in the spinal DH by intrathecal TSP4-AON reduced synaptogenesis [30].
Animal studies documented a correlation between increased TSP4 expression and behavioral hypersensitivity development in the spinal cord/DRG
| Study | Animals (species/types) | Procedures | Results |
|---|---|---|---|
| Kim et al. [41], 2012 | Male young adult Harlan Sprague-Dawley rats | L5/6 SNL | TSP4 expression increased in L5/6 DSC on the 2nd and 10th postoperative days. Tactile allodynia and thermal and mechanical hyperalgesia peaked on the 30th, 20th, and 10th postoperative days, respectively |
| TSP4 protein, 45 μg/rat, intrathecal | Tactile allodynia and thermal and mechanical hyperalgesia reached peak levels around the 3rd–4th post-injection days | ||
| Adult male THBS4 knockout mice | L5 SNL | Tactile allodynia did not develop after SNL | |
| Zeng et al. [98], 2013 | Adult female Sprague-Dawley rats | SCI | TSP4 expression in the L4–6 DSC significantly increased 30–40 days after SCI. Mechanical and thermal hyperalgesia reached peak levels approximately four weeks post-SCI |
| Li et al. [97], 2014 | Adult male Sprague-Dawley rats | CCI of the infraorbital nerve | TSP4 expression in the C1/2 spinal cord and orofacial tactile allodynia peaked on the 3rd postoperative week |
| Crosby et al. [30], 2015 | Adult male Holtzman rats | C6/7 FJD | TSP4 expression increased in the spinal cord on the 7th postoperative day. Tactile allodynia developed one day after surgery and persisted for seven days. Excitatory synapse density increased in the superficial DH |
| TSP4 protein, at different doses, intrathecal | Tactile allodynia developed at 30 μg/rat and higher doses of TSP4 protein and peaked three days after TSP4, 60 μg/rat, intrathecal | ||
| TSP4 protein, 60 μg/rat, intrathecal (3 days before FJD) | A single dose (60 μg/rat) of TSP4 protein, administered three days before FJD, exacerbated the FJD-induced tactile allodynia | ||
| Pan et al. [73], 2015 | Male Sprague-Dawley rats | L5/6 SNL or SNI | Tactile allodynia developed following SNL and SNI. TSP4 expression increased in neurons, the extracellular space, and DRG on the 2nd and 24th days after SNL and SNI surgery |
| Park et al. [93], 2016 | Adult male Harlan Sprague-Dawley rats | TSP4 protein, 45 μg/rat, intrathecal | Tactile allodynia that peaked on the 3rd day developed. mEPSCs increased in the L5 superficial spinal DH neurons |
| THBS4 knockout mice | L4 SNL | Behavioral hypersensitivity did not detect | |
| Park et al. [99], 2018 | Male adult Harlan Sprague-Dawley rats | The EGF-like domain of TSP4 protein, 10 μg/rat, intrathecal | Tactile allodynia and thermal hyperalgesia developed within two days and lasted for about one week (similar to that induced by full-length TSP4 proteins) |
| Yu et al. [100], 2018 | 129sv adult male and female mice | TSP4 protein, 5 μg/mouse, intrathecal | Tactile allodynia peaked on day 4 post-TSP4 injection, and excitatory synapses in DSC increased by approximately 37% |
| Düzenli et al. [95], 2022 | Wistar rats | CCI of the sciatic nerve | On the 4th postoperative day, TSP4 expression increased in the L4–6 spinal cord and tactile allodynia and mechanical and thermal hyperalgesia developed. |
CCI: chronic construction injury; SCI: spinal cord injury; SNI: spared nerve injury
Inhibition or blockage of TSP4 expression through TSP4-AON or antibody reversed or prevented the development of behavioral hypersensitivity
| Study | Animals (species/types) | Procedures | Results |
|---|---|---|---|
| Kim et al. [41], 2012 | Male young adult Harlan Sprague-Dawley rats | TSP4 antibody 80 μg/rat, intrathecal, two weeks after L5/6 SNL | Both tactile allodynia and mechanic and thermal hyperalgesia reversedAll effects peaked at 6 h after TSP4 antibody injection |
| TSP4 antibody, 80 μg/rat per day, intrathecal immediately before, for eight days, and after SNL | Pre-emptive TSP4 antibody treatment prevented allodynia onset | ||
| TSP4-AON, 50 μg/rat per day, intrathecal, for four days after SNL | Tactile allodynia and thermal and mechanical hyperalgesia reversed time-dependently | ||
| Zeng et al. [98], 2013 | Adult female Sprague-Dawley rats | TSP4-AON, 50 μg/rat per day, intrathecal, post-SCI for four days | The antiallodynic effect developed after the last injection and lasted more than two daysSCI-induced TSP4 expression was reduced in the DSC by 40% |
| Li et al. [97], 2014 | Adult male Sprague-Dawley rats | TSP4-AON, 50 μg/10 μL per rat per day, intrathecal, for four consecutive days, starting at least three weeks post-CCI of the infraorbital nerve | After the last injection, the antiallodynic effect peaked at approximately one day and lasted over two days. Simultaneously, injury-induced TSP4 expression diminished in the C1/2 DSC |
| Crosby et al. [30], 2015 | Adult male Holtzman rats | TSP4-AON, 75 μg/rat per day, intrathecal, for three days before FJD and seven days after FJD | Pre-emptive TSP4-AON treatment prevented the development of tactile allodynia and the increase in TSP4 expression in the C6/7 spinal cord following FJDFJD-induced excitatory synapse density is reduced in the superficial DH |
Effect mechanism of TSP4 in the NP
The results of experimental studies performed to determine the effect mechanism of TSP4 in NP show that:
Cavα2δ1 gene ablation (by anti-Cavα2δ1 small hairpin RNA, intrathecal) prevented both injuries- and intrathecal TSP4-induced behavioral hypersensitivity and the intrathecal TSP4-induced the frequency of mEPSCs in the DSC [93].
Intrathecal gabapentin diminished both injury- [30] and intrathecal TSP4-induced [93, 99, 100] behavioral hypersensitivity. It also reduced both the injury- [30] and intrathecal TSP4-induced [93, 99] excitatory synaptogenesis in the spinal DH.
Intrathecal gabapentin quickly (within 2 h) but briefly (< 4 h) reversed intrathecal TSP4-induced behavioral hypersensitivity [99] and also blocked intrathecal TSP4-induced excitatory synaptogenesis in the DSC if administered early [100].
In THBS4 knockout mice, SNL induced the expression of Cavα2δ1 in the DSC without increasing excitatory synapses and developing behavioral hypersensitivity [93].
In the Cavα2δ1 over-expressing transgenic mice, TSP4 ablation (by TSP4 antibody, intrathecal) reversed the behavioral hypersensitivity [93, 99] and reduced the increased frequency of mEPSC in the superficial spinal DH neurons [93].
EGF-like, a domain of TSP4, is a functional determinant in promoting aberrant excitatory synaptogenesis and sensory hypersensitivity resulting from activation of a TSP4/Cavα2δ1 pathway. Intrathecal administration of EGFD355–369, a peptide within EGF-like as highly conserved in rodents and having a high binding affinity to Cavα2δ1, reversed the intrathecal TSP4-induced behavioral hypersensitivity in Cavα2δ1 over-expressing transgenic mice [99].
All these results indicate that PNI or intrathecal TSP4-induced behavioral hypersensitivity and excitatory synaptogenesis require the interaction of TSP4/Cavα2δ1. Results of increasing the interaction of TSP4/Cavα2δ1 in DRG and DSC have been presented in more detail in Figure 3.
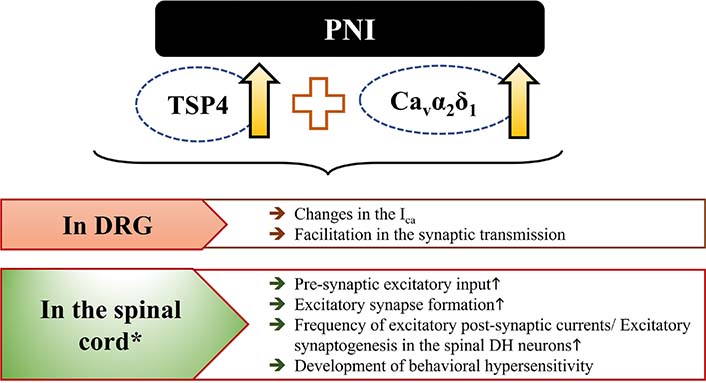
Events mediated by increasing the interaction of TSP4/Cavα2δ1 in DRG and DSC following PNI. It has been inspired by [93]. *: intrathecal administration also leads to similar results in DSC. The upward arrow (↑) represents “increase”
Conclusions
Expression of TSP4 secreted by astrocytes increases in the spinal DH following PNI. Both injury-induced TSP4 expression and intrathecal administration of TSP4 cause the development of behavioral hypersensitivity and trigger abnormal synaptogenesis in rodents. Blockage of spinal TSP4 expression reverses behavioral hypersensitivity and reduces the development of aberrant excitatory synaptogenesis, a fundamental mechanism in central sensitization responsible for the chronicity of pain. In addition, increased TSP4 level in the DRG following PNI leads to the transmission of abnormal signals to the CNS by disrupting ICa. All these results support that TSP4 has an important role in both peripheral and central sensitization and may also be a key mechanism in both initiation and maintenance of NP. These effects of the TSP4 mentioned above require the TSP4/Cavα2δ1 interaction. Therefore, blocking the TSP4/Cavα2δ1 interaction may be an effective strategy in the targeted therapy of NP. Indeed, gabapentin, an α2δ1 ligand, has been shown to reduce or reverse these changes by preventing [101] the TSP4/Cavα2δ1 interaction. However, these effects of gabapentin are short-term and diminish as treatment initiation is delayed. Therefore, it has been thought that gabapentin cannot permanently block the TSP4 signal and may be more likely to prevent the initial increase of TSP4/Cavα2δ1 interaction [30, 100]. The development of combinations that will potentiate the analgesic potency and/or duration of action of gabapentinoids without an increase in its adverse effects and/or the need for dose escalation and also the development of pharmacological agents that may inhibit TSP4 expression or prevent the interaction of TSP4/Cavα2δ1 for a long-time may be a promising strategy in the treatment of NP.
Abbreviations
| AONs: | antisense oligodeoxynucleotides |
| Ca2+: | calcium |
| Cavα2δ1: | α2δ1 subunits of the voltage-gated calcium channel receptor |
| CNS: | central nervous system |
| DH: | dorsal horn |
| DRG: | dorsal root ganglion |
| DSC: | dorsal spinal cord |
| EGF: | epidermal growth factor |
| FJD: | facet joint distraction |
| GABA: | gamma-aminobutyric acid |
| HVA: | high-voltage-activated |
| ICa: | calcium current |
| LVA: | low-voltage-activated |
| mEPSCs: | miniature excitatory postsynaptic currents |
| Na+: | sodium |
| NP: | neuropathic pain |
| PNI: | peripheral nerve injury |
| PNS: | peripheral nervous system |
| SNL: | spinal nerve ligation |
| THBS4: | the gene encoding the thrombospondin 4 protein |
| TSP4: | thrombospondin 4 |
| vWF_C: | von Willebrand type C domain |
Declarations
Acknowledgments
We want to thank Mr. Taylan Erekli for helping prepare the figures.
Author contributions
ND contributed to conceptualization, investigation, visualization, and project administration and wrote the original draft. AÖ and CC contributed the validation of the investigation, reviewed, and edited the article. All authors contributed to manuscript revision and read and approved the submitted version.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2022.