 Review
Review

Affiliation:
1PeQuiM - Laboratory of research in Medicinal Chemistry, Institute of Chemistry, Federal University of Alfenas, Alfenas 37133-840, Brazil
2Post-Graduate Program in Pharmaceutical Sciences, Federal University of Alfenas, Alfenas 37130-001, Brazil
ORCID: https://orcid.org/0000-0003-4978-0944
Affiliation:
1PeQuiM - Laboratory of research in Medicinal Chemistry, Institute of Chemistry, Federal University of Alfenas, Alfenas 37133-840, Brazil
ORCID: https://orcid.org/0000-0001-9354-0708
Affiliation:
1PeQuiM - Laboratory of research in Medicinal Chemistry, Institute of Chemistry, Federal University of Alfenas, Alfenas 37133-840, Brazil
3Post-Graduate Program in Chemistry, Federal University of Alfenas, Alfenas 37130-001, Brazil
ORCID: https://orcid.org/0000-0003-1674-818X
Affiliation:
4Institute of Biomedical Sciences, Federal University of Alfenas, Alfenas 37130-001, Brazil
ORCID: https://orcid.org/0000-0003-2545-7288
Affiliation:
1PeQuiM - Laboratory of research in Medicinal Chemistry, Institute of Chemistry, Federal University of Alfenas, Alfenas 37133-840, Brazil
2Post-Graduate Program in Pharmaceutical Sciences, Federal University of Alfenas, Alfenas 37130-001, Brazil
3Post-Graduate Program in Chemistry, Federal University of Alfenas, Alfenas 37130-001, Brazil
Email: cvjviegas@gmail.com
ORCID: https://orcid.org/0000-0002-7799-992X
Explor Neuroprot Ther. 2023;3:71–89 DOI: https://doi.org/10.37349/ent.2023.00038
Received: June 28, 2022 Accepted: December 29, 2022 Published: March 29, 2023
Academic Editor: Shile Huang, Louisiana State University Health Science Center, USA
The enteric nervous system (ENS) is considered by some authors as the second human brain, given its fundamental role in the regulation process of the central nervous system (CNS). Recent data from scientific literature have shown the existence of close bidirectional communication between the gut microbiota and the CNS, influencing physiological and biochemical changes related to cognition, emotion, behavior, anxiety, depressive symptoms, and stress. Furthermore, the existence of mediators in the connection between intestinal microorganisms and the CNS is evident, which includes neural networks, signaling, immune, and endocrine responses. However, the mechanisms underlying the effects of gut microbiota on brain processes still need to be determined. Therefore, understanding the relationship between the gut and neurodegenerative diseases (NDs) is essential for developing effective prophylactic alternatives and disease-modifying drugs that can prevent or slow the progression of such diseases. Herein, this short review aimed to present the most recent data from the scientific literature associated with the physiological, biochemical, and cellular aspects involved in the interrelationship between the gut-brain axis and NDs, discussing the role of the intestinal microbiota, and its relationship with CNS disorders.
The gut is a complex ecosystem that houses a dense and diverse microbial community called the gut microbiota, which co-evolves with the host and establishes a mutualistic relationship [1–3]. However, despite its significant influence on the state of human health and its relationship to the development or progression of diseases, it has only been studied in more detail in the recent years, coming to be highlighted by several researchers in the health field [1–3]. The most recent scientific advances have shown a close connection between the gastrointestinal tract (GIT), through the vagus nerve, with the central nervous system (CNS), contributing to the regulation of secretion, permeability, motility, and immunity of the digestive tract, exerting its effect on the enteric nervous system (ENS) [1, 4–6]. Due to this prominent role in the regulation and functionality of the CNS, the ENS has been considered a “second brain” since the structure of enteric neurons is a key factor in the control of the physiological response to the environment, muscle tissue, and the intestinal mucosa via the efferent autonomic nerve pathways [5, 7–9].
ENS influences brain functions through afferent signaling pathways and the secretion of biologically active substances in an intrinsic and bilateral relationship, providing a functional interaction and contributing to the coordination of the body’s metabolic and homeostatic functions [4, 6, 7, 10]. Thus, ENS has been suggested as an essential agent in the pathophysiology of chronic diseases, such as diabetes, metabolic syndromes, obesity, several autoimmune diseases, stress-induced psychosomatic disorders, schizophrenia, and some neurodegenerative diseases (NDs), as well as in the search for therapeutic targets and new drugs [7–9].
The human gut consists of approximately 1,000 species and 7,000 strains of bacteria, mainly belonging to the phyla Firmicutes (51%) and Bacteroidetes (48%). The remaining species (1%) belong to other divisions such as Proteobacteria, Actinobacteria, Fusobacteria, Spirochaetes, Verrucomicrobia, and Lentispherae [6]. The microorganisms consortia, which constitute the intestinal microbiota in healthy human beings, undergo important changes in quantity and quality among individuals and in pathological situations, as is the case of patients with NDs [11–13].
Dementias are some main causes of disability in the elderly, accompanied by physiological changes that include alteration in the intestinal microbiota, which seems to form a consensus regarding the relationship in the reduction of the number of species and their quantitative composition of Bifidobacterium and Lactobacillus in aging, with an increased incidence of chronic diseases [14–17]. One possible explanation is the reduction of symbionts that control mucus production. Once disturbances in mucus production occur, the possibility of damage to the intestinal epithelial barrier increases, leading to gut functionality restriction and increased susceptibility to gastrointestinal infections [10, 15, 18]. Thus, significant changes in the gut microbiome with aging constitute a significant risk factor for NDs [1, 10, 16, 18].
In this perspective, unhealthy diet, poor sleep quality, circadian rhythm disorders, sedentary behavior, drug abuse, and environmental factors may contribute to changes in the gut microbiota and, in turn, trigger NDs [1, 19–21]. Therefore, in this narrative review, the role of the gut microbiota, its relationship with CNS disorders, and the known mechanisms that drive dysbiosis, including its role in the pathophysiology of NDs and potential therapeutic aspects were discussed.
The gut microbiota plays a fundamental role in the biosynthesis of vitamins and cofactors, cleaving the structure of complex lipids and polysaccharides, and promoting detoxification by residual particles [2, 15, 21, 22]. The gut microbiota also contributes to the fermentation of non-digestible substrates, such as dietary fibers and endogenous intestinal mucus, supporting the growth of microorganisms specialized in the production of short-chain fatty acids (SCFAs), such as acetate, propionate, and butyrate, and certain gases [2, 22–24]. Regarding SCFAs, another of their functions is to contribute to the improvement of intestinal health through a series of effects that include maintaining the integrity of the intestinal barrier, mucus production, and protection against inflammation [23–25]. Besides, evidence supports a potential key role of SCFAs in gut-brain axis signaling [23].
Preclinical and clinical studies have shown that SCFAs activate a series of receptors involved in the regulation of satiety and hunger by stimulating L cells, which are located in the intestine’s distal ileum of enteroendocrine cells (EECs) [23, 25]. They are responsible for the secretion of peptide YY (PYY) and glucagon-like peptide-1 (GLP-1), which induce satiety and behavioral changes [23, 26]. Alterations in this signaling process may favor CNS disorders that range from neurodevelopmental alterations to the NDs onset [23, 25]. Furthermore, the signaling role could be seen in intestinal microbial enzymes that contribute to the metabolism of bile acids, generating non-conjugated and secondary bile acid derivatives that act as signaling and regulatory molecules in different metabolic pathways [2].
The gut microbiota harbors a wide variety of microorganisms that have a central role in the fermentation of complex indigestible substances, contributing to the regulation of the immune system, and in the synthesis of vitamins. Taking into account, bacteria of the Bifidobacterium and Lactobacillus genera, have a prominent role for their active contribution in the production of γ-aminobutyric acid (GABA), one of the main inhibitory mediators of the CNS in humans and other mammals, involved in the modulation of neurotransmitters and metabolic processes in the brain [10, 14, 26–28]. Additionally, experimental evidence has revealed that GABA levels in the gut are related to its concentration in the CNS. However, a decreased population of Bifidobacterium and Lactobacillus could lead to brain dysfunction associated with synaptogenesis disorders, depression, and cognitive impairment [14, 26–28].
The imbalance in the gut microbiome triggers the overproduction of harmful molecules, which becomes a serious problem because these metabolites inform the ENS of the content of the gut lumen. Once the ENS translates the signal generated by the unbalanced microbiota, this information is transmitted to the CNS via the vagus nerve in milliseconds. In principle, the correlation between ENS and NDs can be established by the metabolism of essential amino acids, as they constitute one of the primary sources of neuromodulators, neurohormones, and neurotransmitters. Thus, changes in the production of these compounds impact the CNS with consequent effects on the stimulation of central receptors, peripheral neural stimulation, and endocrine and immunological mediators [6, 27–30].
Therefore, the gut microbiota contributes to the processes of fermentation of complex food [22, 31], including amino acids, such as L-tryptophan (compound 1, Figure 1), which is metabolized by the main serotonergic and kynurenic pathways. In turn, the products of its metabolism play an indispensable role in brain functioning and CNS physiology [3, 6, 32, 33].
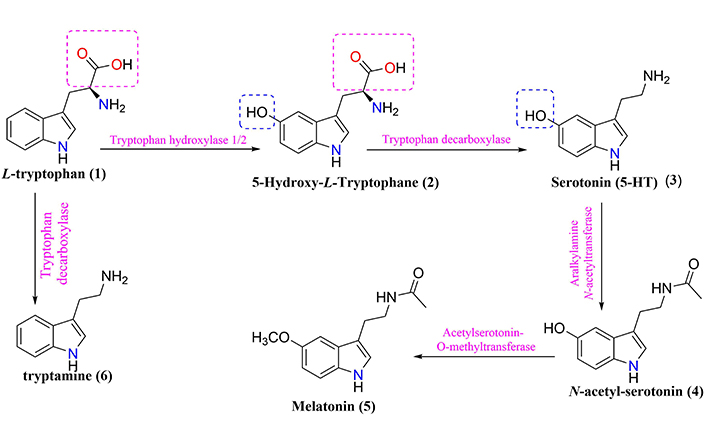
General scheme of L-tryptophan metabolism generating serotonin (3) and melatonin (5), tryptamine (6), and other metabolites
Intestinal microbiota in a state of eubiosis/homeostasis has an important impact on several functions of the host’s ENS, including essential physiological processes like maintenance of metabolic and nutritional homeostasis, maturation and stimulation of the immune system, regulation of neurotransmitters, and activation of antioxidant enzymes [3, 5, 8, 9, 34]. The role of SCFAs (e.g., butyrate) can also be highlighted, whose concentrations are related to the production of mucin, which has anti-inflammatory effects and increases protein levels at the tight junction, promoting the maintenance of the intestinal barrier and reducing the permeability of the intestinal mucosa [35–38]. Changes resulting from dysbiosis in the elderly lead to increased intestinal permeability, which leads to systemic inflammatory processes, affecting a series of vital physiological cascades that favor depression, anxiety, and NDs [9, 18, 29, 35].
In sporadic forms of NDs, which are generally recorded in the elderly, the gut microbiome shows a reduction in diversity and quantity of essential microorganisms, such as Bifidobacteria, Lactobacillus, and Bacteroides, which guarantee the tolerance of the intestinal immune system [15, 18, 25, 39–41]. Changes in the gut microbiota composition in aging, seem to constitute an important risk factor for NDs, psychological disturbs, and other chronic pathologies due to cumulative systemic exposure to toxins [1, 18, 29, 42]. Thus, the maintenance and modulation of the homeostasis of the intestinal microbiota are currently seen as a beneficial path in the prevention of chronic diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) [18, 29, 42]. In this regard, under homeostatic conditions, epithelial cells produce antimicrobial peptides (AMPs) in response to interleukins (ILs, e.g., IL-22), and also express Toll-like receptor (TLR), a class of pattern recognition receptors. Besides regulation of mucosal secretion and AMPs production, intestinal microbiota also regulates/improves intestinal barrier integrity through the production of SCFAs. Goblet cells or caliciform cells produce mucus to restrict the invasion of pathobionts, and lymphoid cells [T helper 17, (TH17)] play a role in host defense by producing controlled arrays of IL-22. Moreover, dendritic cells (DCs) induce activation and differentiation of B cells to produce plasma cells that, in turn, produce commensal specific immunoglobulin A (IgA) in the lamina propria. IgA is transported into the intestinal lumen as secreted IgA (sIgA) via polymeric immunoglobulin receptors (pIgR), where sIgA then binds to commensal microbes soluble antigens, thereby restricting their adherence to the host epithelium and leakage across the intestinal barrier. However, under a dysbiosis condition, the altered microbiota composition can weaken the intestinal epithelium leading to increased permeability and the leakage of opportunistic pathobionts and their by-products across the intestinal barrier, which, in turn, instigates hyperinflammatory responses, and eventually increases host susceptibility to neurodegeneration [6, 8, 15].
L-tryptophan (compound 1, Figure 1), one of the 20 essential amino acids for humans, is metabolized in the intestine and used for protein synthesis, being the only biosynthesis precursor at the level of enterochromaffin cells (EC), which modulates neuronal signaling in the ENS [32, 43]. The potential role of L-tryptophan metabolism in the gut-brain axis serves as a sole precursor for the biosynthesis of the neurotransmitter serotonin (compound 3, Figure 1) and, subsequently, a pineal hormone called melatonin (compound 5, Figure 1) [32]. Notwithstanding, the manipulation of gut microbiota composition (e.g., antibiotics and probiotics) could contribute to changes in central L-tryptophan metabolism between serotonin synthesis and L-tryptophan degradation pathways, which thus influence individual functionality and behavior [32].
The signaling pathway occurs through the conversion of the biosynthetic precursor 5-hydroxy-L-tryptophan (compound 2, Figure 1) into the neurotransmitter serotonin, whose secretion in CNS plays a fundamental role in the modulation of emotional control, appetite, sleep, sex, temperature, and pain processing [3, 6, 43]. Therefore, L-tryptophan metabolism products, such as serotonin, melatonin, and tryptamine (compound 6, Figure 1) have profound effects on the interaction between gut microbiota and neuroendocrine system, and gut immune responses [3, 7, 32]. However, evidence supports those changes in gut microbiota composition may have implications for modulating L-tryptophan metabolism in the axis-gut-brain interaction [6, 32, 43]. Consequently, messages sent to the brain can propagate harmful signals, which are manifested as inflammatory processes, oxidative stress, imbalance of energy homeostasis, and a general increase in cellular degeneration [3, 32, 43].
It is worth mentioning that the L-tryptophan metabolism comprises two main pathways, both influenced by the intestinal microbiota, namely: (A) The serotonin production pathway in EC via L-tryptophan hydroxylase (Figure 1), and (B) The kynurenine and QUIN pathway (Figure 2), which plays a critical role in inflammatory mechanisms, neurobiological functions, and immune responses in epithelial cells via indoleamine 2,3-dioxygenase 1 [6, 32, 44, 45].
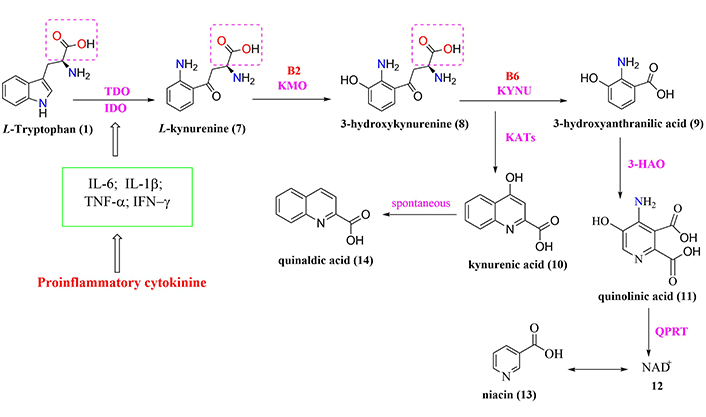
General scheme of tryptophan-degradation pathway metabolism. 3-HAO: 3-hydroxyanthranilic-3,4-dioxygenase; B2: riboflavin; B6: pyridoxine; IDO: indolamine-2,3-dioxygenase; IFN-γ: interferon-gamma; KATs: kynurenine aminotransferase; KMO: kynurenine-3-monooxygenase; KYNU: kynureninase; NAD+: nicotinamide adenine dinucleotide; QPRT: quinolinate phosphoribosyltransferase; TDO: tryptophan-2,3-dioxygenase; TNF-α: tumor necrosis factor-alpha
Regarding serotonin metabolism, about 95% is produced and stored in EC, and enteric neurons, while only 5% is stored in the CNS [46, 47]. Thus, serotonin can be presented into two distinct forms, central and peripheral serotonin. The serotonergic pathway (peripheral and central) of L-tryptophan contributes to average cell growth, protein synthesis, and insulin secretion, as well as regulating essential physiological mechanisms, such as serotonin, melatonin, and vitamin B3 (niacin) [6, 32, 48–50]. Therefore, to meet the body’s basic needs, it is estimated that 4–5 mg/kg of L-tryptophan daily is needed [51, 52]. However, the dysfunction of the axis-gut-brain may reflect changes in L-tryptophan metabolism and, in turn, in the serotonin availability, which is the neurotransmitter responsible for the homeostasis of mood and well-being sense in individuals [51, 52]. Under physiological conditions, peripheral serotonin cannot cross the blood-brain barrier (BBB), and its distribution to the brain is still poorly understood [3, 46, 47]. However, most of the known evidence suggests the involvement of vagal receptors that detect intestinal regulatory peptides, inflammatory molecules, dietary components, and bacterial metabolites to transmit signals to the CNS [3, 17, 33, 47, 53]. In this sense, it is possible to assume that the hormones and metabolites secreted by the microbiota and the intestinal EC influence CNS physiology [7, 8, 49]. Furthermore, dysregulation of the enzyme L-tryptophan hydroxylase, which is one of the central enzymes in the biosynthesis of serotonin, is a common disorder of psychiatric and gastrointestinal functions, such as anxiety and irritable bowel syndrome, suggesting the importance of this enzyme in the serotonin bioavailability [3, 47, 53]. Therefore, serotonin produced in the intestine exerts several local effects, including stimulation of intestinal motility [47]. On the other hand, even without crossing the BBB, the intestinal microbiota indirectly affects the central serotonergic pathways, modulating the bioavailability of L-tryptophan and tryptamine [3, 47, 53].
L-tryptophan is the unique precursor of the biosynthesis of serotonin and melatonin and plays an important regulatory role in the sleep-wake cycle [50]. During the day, melatonin occurs at low levels. However, a healthy individual has optimal or ideal levels during the night that confer high power to scavenge free radicals and fight against oxidative stress, improving cognitive performance, vigilance, and psychological conditions. Thus, it explains the importance of sleeping in maintaining inflammatory homeostatic functions and why its loss is linked to changes in the immune response [33, 43, 50, 54]. Due to sleep deprivation, through the hypothalamus-pituitary-adrenal (HPA) pathway, L-tryptophan catabolism occurs through the activation of the TDO enzyme through an increase in corticosterone, a regulator of lipid, protein, glucose, hormone, and stress response metabolism, via IDO1 inflammatory signaling enzyme (TNF-α, IFN-γ) [43, 50, 54–56]. However, the serotonergic system is susceptible to inadequate or fragmented sleep, favoring the catabolism of L-tryptophan by the kynurenine and quinolinic acid (QUIN) route due to its association with higher levels of inflammation promoted by changes in immune responses, induction of apoptosis, and resulting in oxidative stress. It is important to note that L-tryptophan can be directly converted into various metabolites, including aryl hydrocarbon receptor (AhR) ligands, which have been suggested to impact the inflammatory process [6, 33]. These cellular and chemical changes increase the levels of inflammatory mediators, such as IL-6, TNF-α, and IFN-γ, which activate the IDO1 enzyme and initiate the catabolism of L-tryptophan by the QUIN.
The QUIN is responsible for metabolizing more than 95% of L-tryptophan in the GIT. QUIN allows the entry of L-tryptophan into the bloodstream and its passage through the BBB, and this mechanism is a critical factor for central serotonergic signaling [7, 44, 54, 57]. Thus, the QUIN pathway is related to neurodegenerative processes due to its indispensable role in the biosynthesis of several important neuroactive intermediates, such as kynurenic acid, niacin, and the cofactor NAD+. In addition, QUIN is also involved in the production of other neurotoxic intermediates, such as QUIN, which promotes excessive stimulation of the NR2A and NR2B subunits of the glutamate N-methyl-D-aspartate (NMDA) receptor [48, 54, 57]. Furthermore, excessive stimulation of QUIN promotes calcium influx into neurons, contributing to the generation of reactive oxygen species (ROS) and free radicals [48, 54, 57]. In turn, even in this context of excessive stimulation of the NMDA receptor by the agonist binding of QUIN, there is an increase in glutamate release and inhibition of its reuptake. As a consequence, the depolarization of the postsynaptic membrane occurs with the removal of the Mg2+ blockade of the channel ionic acid bound to the NMDA receptor, stimulating lipid peroxidation, thus compromising its fluidity and permeability, which results in neuronal damage and its respective death [48, 54, 57].
Experimental data support that QUIN can impair BBB function by inducing nitric oxide production, which can cause hyperphosphorylation of cytoskeletal intermediate protein filaments in astrocytes and neurons [48]. At a low QUIN concentration, it can induce stem cell proliferation, besides being an intermediate metabolite of QUIN that synthesizes NAD+ in human brain cells [48, 57]. However, QUIN-induced damage may vary depending on the brain area due to the degree of neuronal susceptibility, with cortical, striatal, and hippocampal neurons being the most sensitive. These changes could explain, at least in part, the increased degree of neurodegeneration in these brain regions of AD patients, which may be resultant of increased levels of QUIN and the consequent installation of an inflammatory process [57, 58].
Another neurotoxic intermediate is 3-hydroxykynurenine acid (compound 9, Figure 2), responsible for inducing oxidative stress and neuronal apoptosis through its interaction with xanthine oxidase (XO) and ROS production, such as the superoxide radical (O2–) and hydroxyl radical (OH–), among other free radicals. All these chemical species are capable of cleaving DNA and promoting apoptosis, with consequences for neuronal damage and promotion of cognitive and motor dysfunctions [48, 57]. Both intermediates L-kynurenine (compound 7, Figure 2) and 3-hydroxy-kynurenine (compound 8, Figure 2) have physiologically similar characteristics and distribution in the CNS, with higher concentrations in the cerebral cortex, striatum, and hippocampus [48, 57, 58].
To date, due to the relatively low number of in vivo studies, the role of 3-hydroxy-kynurenine in neuropathological processes remains poorly understood. However, high levels of 3-hydroxy-kynurenine are associated with neuroinflammation and may exhibit antioxidant and pro-oxidant properties depending on its concentration. Conversely, low concentrations of this metabolite are associated with strong pro-oxidant activity and, in turn, with neuronal toxicity, while resistance is related to increased oxidation at its higher concentrations [48]. On the other hand, this biosynthetic intermediate can stimulate glutathione S-transferase, superoxide dismutase, and activation of nuclear factor erythroid 2-related factor 2 (Nrf2), an important nuclear transcription factor and antioxidant regulator [8, 48].
The physiological balance between neurotoxic kynurenines (compound 8, compound 9, and compound 11, Figure 2), and neuroprotective kynurenines (compound 10, compound 12, and compound 13, Figure 2) plays a pivotal role in CNS homeostasis, once it contributes to neuroprotection against oxidative stress and ROS production. In this regard, the effects of all these metabolites are associated with an established interaction between astrocytes, microglia, and neurons since, under physiological conditions, astrocytic QUIN serves as a source of production of the neuroprotective agent kynurenic acid (compound 10, Figure 2), while the neuronal QUIN produces NAD+, improving cellular energy status [56].
By contrast, in pathological conditions, inflammatory signals stimulate the QUIN pathway in macrophages, microglia, and DCs to produce high amounts of quinolinic acid (compound 11, Figure 2) [8, 48, 51, 54, 57]. This fact requires a balance regulator in the action of neuroactive QUIN. Therefore, IDO establishes the control of this balance that activates L-tryptophan catabolism via the QUIN pathway as opposed to alternative pathways of production of serotonin [3, 36]. However, the conversion of L-tryptophan via the QUIN pathway occurs in the liver by the action of TDO, which is activated in response to corticosteroid stress hormones. In contrast, IDO is activated by inflammatory stimuli mediated by IFN-γ, TNF-α, and amyloid-β (Aβ) in different regions, such as brain, lung, heart, kidneys, and intestine [48, 54].
Inflammation triggers the production of the significant neurotoxic metabolites 3-hydroxykynurenine and QUIN, affecting cognitive function and promoting the development and progression of neurodegeneration [54, 56]. This evidence was corroborated by Baratta and co-workers [43, 46], which demonstrated that L-tryptophan and kynurenic acid were increased in the plasma of rats subjected to chronic sleep deprivation.
It is well-known that increased kynurenic acid affects cognition in the brain and may even establish a molecular link between sleep loss and cognitive impairment. By contrast, lower levels of kynurenic metabolites from L-tryptophan metabolism (Figure 3) improve learning and memory by affecting glutamatergic and cholinergic transmission [50, 52, 56, 59]. Therefore, this imbalance among neurotoxic, neuroprotective, and immunomodulatory QUIN metabolites has been reported in NDs such as AD, PD, multiple sclerosis, ALS, and HD [23, 54, 60–62].
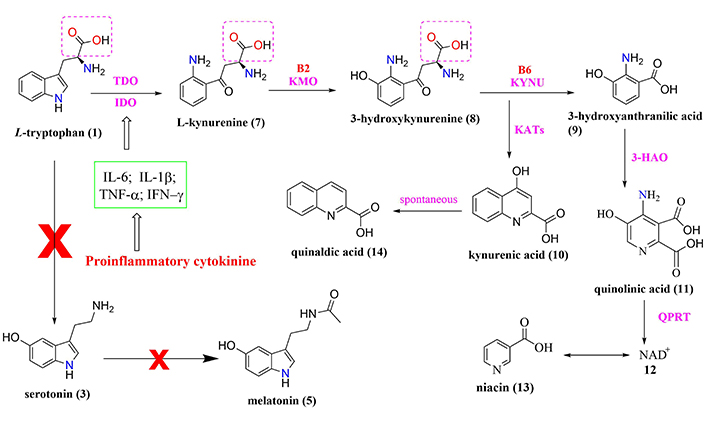
Biochemical cascade involved in the relationship between sleep deprivation, HPA axis, and L-tryptophan metabolism with increased production of kynurenine metabolites altering various neurotransmitter systems in the cortical and striatal regions
Note. Adapted from “Effects of sleep deprivation on the tryptophan metabolism,” by Bhat A, Pires AS, Tan V, Babu Chidambaram S, Guillemin GJ. Int J Tryptophan Res. 2020;13 (https://doi.org/10.1177/1178646920970902). CC BY.
Abnormalities in the functional communication of the gut-brain axis can usually reflect a disruption of the integrated network of neurotransmitters in the endocrine, intestinal, and brain systems [8, 51, 57, 63, 64]. Three basic mechanisms mediate the communication between the gut and the CNS: i) direct neuronal communication, ii) endocrine signaling mediators, and iii) through the immune system. Therefore, the combination of these three systems establishes a highly integrated molecular communication network, that links systemic imbalances to the development of neurodegeneration, including insulin regulation, fat metabolism, oxidative markers, and immune signaling [7, 28]. Since gut-brain axis communication is efficient, disturbances in the gut microbial balance may be associated with AD and PD.
The functioning of the microbiota in AD patients was evaluated, considering a group of patients with mild cognitive and amnestic impairment and healthy individuals, based on the functional pathway of the Kyoto Encyclopedia of Genes and Genomes (KEGG) [13, 65]. The results demonstrated changes in the taxonomic and functional composition of the intestinal microbiota, capable of influencing brain functions, suggesting the effect of the intestinal microbiota on the development of amyloid pathology [10, 13, 65]. In this context, a study carried out by Kennedy et al. [64] using feces from transgenic mice that express the human amyloid precursor protein (APP) and Presenilin-1 (PS1) gene (CONVR-APPPS1), that is accepted as an animal model of AD, showed differences in the composition of the intestinal microbiota compared to the control group.
Given the complexity of the pathogenesis and pathophysiology of AD, which imposes challenges in finding therapeutic targets and effective drugs, recent scientific advances have suggested an intrinsic and bilateral relationship among the gut microbiota, ENS, and the CNS with a decisive influence on brain functions. In this context, a critical reflexive analysis of the biosynthesis of methionine (Met) and acetylcholine (ACh) modulated by gut microbiota was carried out.
Met is a biosynthetic precursor of choline, cysteine, and other neuroactive biogenic amines and plays a pivotal role in the synthesis of ACh and the consequent implications in neurodegeneration. Choline, in turn, is an essential nutrient that functions at the intersection of multiple metabolic pathways, such as the synthesis of glycerol phospholipids, the main lipid components of cell membranes, which include very-low-density lipoprotein (VLDL), phosphatidylcholine, and sphingolipids (Figure 4). These lipid components are all produced in the liver and are released into the bloodstream to reach different body tissues where they are important in phospholipid metabolism and biliary metabolism, regulation of whole-body lipids, lipoprotein, and energy metabolism [34, 36, 66, 67].
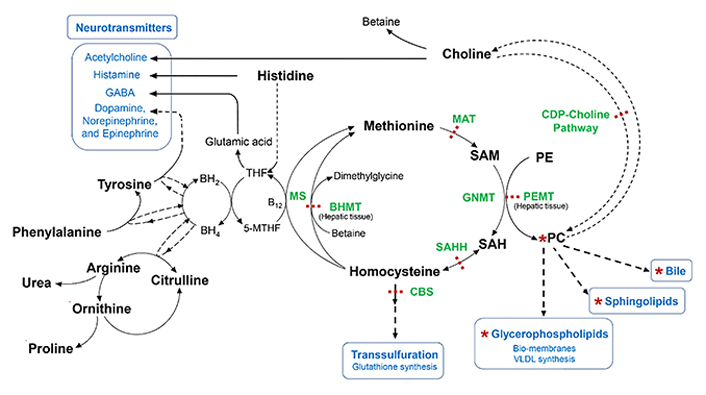
Metabolism of Met, choline precursor, cysteine, neuroactive biogenic amines, and urea. 5-MTHF: 5-methyltetrahydrofolate; B12: Vitamin B-12 or cobalamin; BH2: dihydrobiopterin; BH4: tetrahydrobiopterin; MS: methionine synthase; BHMT: betaine-homocysteine methyltransferase; CBS: cystathionine β-synthase; CDP-choline: cytidine-diphosphate-choline; GNMT: glycine N-methyltransferase; MAT: Met-adenosyl transferase; PC: phosphatidylcholine; PE: phosphatidylethanolamine; PEMT: phosphatidylethanolamine N-methyltransferase; SAH: S-adenosylhomocysteine; SAHH: S-adenosylhomocysteine hydrolase; SAM: S-adenosylmethionine; THF: tetrahydrofolate
Note. Adapted from “Dysregulated choline, methionine, and aromatic amino acid metabolism in patients with wilson disease: exploratory metabolomic profiling and implications for hepatic and neurologic phenotypes,” by Mazi TA, Sarode GV, Czlonkowska A, Litwin T, Kim K, Shibata NM, et al. Int J Mol Sci. 2019;20:5937 (https://doi.org/10.3390/ijms20235937). CC BY.
The biosynthesis of Met is preceded by a methylation reaction of homocysteine through betaine, the oxidized form of choline, which is catalyzed by betaine-homocysteine methyltransferase to generate dimethylglycine derivative, an antioxidant and lymphocyte proliferation stimulator and B12 vitamin involved in folate metabolism and aromatic amino acids to generate neurotransmitters (Figure 4). Furthermore, Met is an excellent and universal donor of S-adenosylmethionine, whose biosynthesis involves the enzymatic participation of MAT. This methylating donor contributes to the conversion of phosphatidylethanolamine into phosphatidylcholine to generate choline that will culminate in the production of ACh (Figure 4) [41, 58, 67].
Therefore, if choline is the precursor of ACh and other neuroactive biogenic amines are derived from the essential amino acids phenylalanine, tryptophan, and histidine, except for tyrosine, which is not essential, it further supports the idea of an intrinsic ENS-CNS relationship.
In addition, Met is an essential precursor in the biosynthesis of histamine, serotonin, epinephrine, norepinephrine, and dopamine. These neurotransmitters are important endogenous chemical messengers responsible for neural transmission and regulation of locomotion, muscle tone, mood, attention, and behavior [29, 37, 38, 40]. It is important to note that changes in the microbiota, driven by exogenous or endogenous factors, could effectively affect the metabolism and production of several neurotransmitters, which are, in turn, essential for the proper brain functions, and the organism as a whole, as well as to contribute to the progression of neurodegenerative processes [34, 35, 42, 68].
In the mid-1970s, post-mortem studies of AD brains showed a significant reduction in choline acetyltransferase (ChAT) levels. This enzyme is responsible for the synthesis of ACh, an important neurotransmitter in both central and peripheral nervous systems [61, 64, 66]. ACh is one of the main brain neurotransmitters and is especially involved in the neuromodulation of learning, memory, attention, sensory information, and cognitive functions [61, 64, 66].
During the last decades, findings related to the decreased concentration of ChAT in AD brains were recognized as a major milestone in the first discoveries related to the pathophysiology of AD and, above all, the prominent role of biosynthesis and modulation of acetylcholinesterase (AChE) in the complex cascade of multiple events that characterize the progression of AD remains under deep discussion, despite all data pointing out its key role in cholinergic deficit [41, 61, 64, 66]. Indeed, ChAT concentrations have been most drastically reduced in the hippocampus and frontal cortex, the brain regions where neurodegeneration is most pronounced during AD progression [61, 64, 66]. ACh production occurs in the cytoplasm of cholinergic neurons, from choline and acetyl-coenzyme A, catalyzed by ChAT and transported to the vesicular ACh transporter (VAChT, Figure 5). However, it was realized that disturbances in ACh levels could contribute to NDs, as well as depressive disorders, and this led to the cholinergic hypothesis. According to this hypothesis, the cholinergic function could be improved through AChE inhibitors or nicotinic or even muscarinic receptor agonists, which could enhance the release or the availability of ACh, and this was the main strategy used for the development of almost all currently available drugs for AD treatment [61, 64, 66].
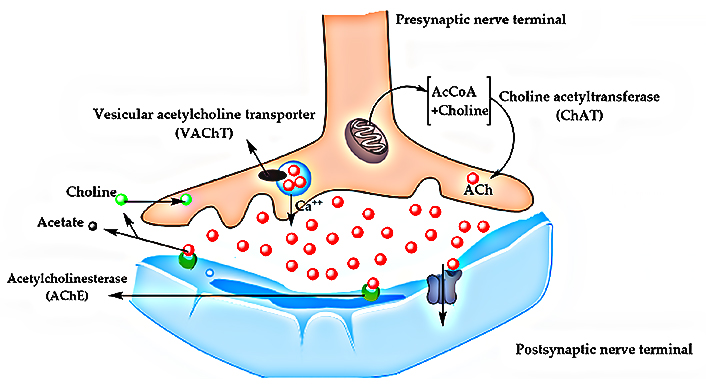
Biochemical pathway of ACh synthesis and transport between presynaptic and postsynaptic nerve terminals. AcCoA: acetyl Conzyme-A
Note. Adapted from “Comprehensive review on Alzheimer’s disease: causes and treatment,” by Breijyeh Z, Karaman R. Molecules. 2020; 25:5789 (https://doi.org/10.3390/molecules25245789). CC BY.
Indeed, cholinergic hypothesis was supported by the fact that AChE is the main enzyme responsible for AChE degradation, and that inhibition of AChE would increase ACh levels in the synaptic cleft, delaying neurodegeneration. In addition, current experimental data suggest that AChE may also be involved in the brain inflammatory response, neuronal apoptosis, oxidative stress, pathological aggregation of Aβ oligomers, and the formation of tau tangles, which are all hallmarks of AD pathogenesis [41, 61, 64]. The fact that AChE has several important functions related to NDs has opened the field of development of AChE inhibitors, and some of them are currently available on the market. Despite this class of drugs only acts symptomatically and does not effectively offer a cure for AD, during the last three decades, medicinal chemists and pharma industry have dedicated great efforts to the discovery and development of new AChE inhibitors, which is attested by the thousands of publications in the literature and several ongoing clinical studies. All these investments have led to the discovery of a number of new promising bioactive chemical entities, with optimized pharmacological properties about the approved AChE inhibitors, but no advance has been achieved to slow the progression of AD effectively [35, 41, 66].
Therefore, a close link between the metabolism of Met by the intestinal microbiota, ENS, and AD pathogenesis can be established considering that Met is the biosynthetic precursor of choline, which is, the precursor of ACh, that occurs at low levels in the brain of AD patients. Considering that, the ACh deficit in AD, may also be associated with dysbiosis, since this process leads to decreased production of choline and, in turn, in the availability of ACh [41, 64].
The main neuropathological characteristics of PD are the decrease of dopaminergic neurons in the pars compacta region of the substantia nigra (SN) up to the caudate nucleus and in the putamen of the dorsal corpus striatum (nigrostriatal axis). Moreover, the presence of cytoplasmic inclusions of insoluble polymers of α-synuclein is evidenced as toxic aggregates called Lewy bodies [68], which represent the neuropathological signature of PD [69].
The early stage of PD is characterized by a set of non-motor signs and symptoms that foreshadow physiological changes in the individual (prodromal period), such as loss of smell, sleep dysfunction, mood disturbances, and constipation [70]. When there is a decrease of about 50% in dopaminergic neurons, classic motor symptoms appear, such as bradykinesia, tremors, and muscle stiffness at rest [71]. Several pieces of evidence have shown that PD can be originated in the gut [69, 72–74]. According to the topographic distribution of Lewy bodies identified after the autopsy of the brains of PD patients, it was possible to identify that α-synuclein can originate in the ENS and be transported to the brain, in a slow and progressive process that could take up to 20 years, first reaching the olfactory bulb and gradually reaching the dopaminergic region [73, 75, 76].
The hypothesis of Braak et al. [74] postulates that aberrant α-synuclein accumulation starts in the gut and spreads via the vagus nerve to the brain in a prion-like manner. This hypothesis is supported by pathophysiological evidence, where α-synuclein inclusions appear early in the ENS and in the glossopharyngeal and vagal nerves [73, 75, 77, 78]. Corroborating with Braak et al. [74], an epidemiological cohort study of patients undergoing truncal or selective vagotomy has demonstrated that patients undergoing complete truncal vagotomy showed a reduced risk of developing PD, suggesting the critical involvement of the vagus nerve in the PD pathogenesis [78].
Several evidence from the literature suggests that the neurodegenerative cascade may indeed be initiated in the gut with subsequent dissemination to the brain, as a reflex of the gut-brain axis, and that the gut microbiota may be involved in this process [79, 80]. Changes in the composition of the gut microbiota can cause changes in the intestinal barrier, affecting intestinal permeability, affecting not only the cells of the epithelium and the immune system, but also modulating brain biochemistry [81, 82]. The composition and diversity of the microbiota of individuals with PD are significantly different from the microbiota of healthy individuals [79]. Gastrointestinal dysfunction is highly prevalent in PD and can precede motor symptoms by more than a decade [79]. Thus, it may be directly related to the process of α-synuclein misfolding, since it is already well documented that PD patients have α-synuclein aggregates in their colon [77, 80, 83–85].
Understanding the greater influence of the intestinal microbiota on brain functions poses a great challenge to the research related to NDs, such as AD and PD, especially in the search for new therapeutic targets that could effectively prevent, or delay the NDs. It seems a consensus that the current strategies, which are focused only on the CNS and specific targets, such as AChE, and led to all available drugs worldwide, do not seem to reduce symptoms or modify the progress of the diseases [14, 18, 42]. Emerging evidence suggests that modulation of the gut microbiota by the use of probiotics, prebiotics, symbiotics, and antibiotics, or dietary change, could exert effective neurological and psychological changes [18, 86, 87]. For example, the supplementation with probiotics have shown to improve performance in cognitive tests and to increase the concentration of substances capable of delaying the neurodegenerative AD process in experimental models [14, 87–89]. In general, probiotics applications are considered a safe alternative treatment for several diseases and disorders because they can positively alter the host’s gut microbiome and improve the health status [87–90].
According to AD mice models studies, the supplementation with Lactobacillus and Bifidobacterium can restore GABA and glutamate levels in the frontal cortex and hippocampus, besides improving memory and learning functions [37, 38]. Bonfili and co-workers [87] used an AD model based on transgenic 3xTgDA mice, treated with the probiotic formulation SLAB51, which consists of a mixture of lactic acid and Bifidobacterium. As a result, significant inhibition of Aβ aggregation and reduction of oxidative stress were observed in the animal brains with the involvement of the sirtuin1 (SIRT1).
In another approach, this perspective was further enriched by Abdelhamid and co-workers [91], that evaluated the effects of oral supplementation with Bifidobacterium breve MCC1274 on cognitive function and AD-like pathologies in mice. Their results reinforced that the probiotic supplementation was effective in the prevention of memory impairment in mice AppNL−G−F, as well as led to decreased hippocampal Aꞵ levels by increasing levels of α-disintegrin and metalloproteinase 10 (ADAM10). Moreover, administration of the probiotic activated the extracellular signal-regulated kinase (ERK)/hypoxia-inducible factor-α (HIF-1α) signaling pathway responsible for increasing ADAM10 levels. Furthermore, Bifidobacterium breve MCC1274 supplementation led to attenuation of microglia activation, reducing pro-inflammatory cytokine, messenger ribonucleic acid (mRNA) expression levels in the brain, as well as increasing the levels of synaptic proteins in the hippocampus [91].
Based on the above-mentioned corroborative studies, the effect of Bifidobacterium and Lactobacillus probiotics was evidenced to be effective in improving memory, learning functions, inhibition of Aβ aggregation, and reduction of oxidative stress related to AD.
A new approach with AD patients was recently carried out by Leblhuber and co-workers [92]. A group of twenty AD outpatients (9 females, 11 males) aged 76.7 ± 9.6 years, were submitted to routine laboratory tests, as well as a mini-mental state examination with a score of 18.5 ± 7.7. The study was carried out for four weeks with 18 patients and was analyzed before and after supplementation using biomarkers of immune activation, serum neopterin, L-tryptophan breakdown, and markers of intestinal inflammation and microbiota composition in fecal samples. After treatment, a decline in fecal zonulin concentrations and an increase in Faecalibacterium prausnitzii compared to baseline were observed, with a concomitant increase in the concentrations of serum kynurenine. Experimental data showed that the multispecies probiotic supplementation in AD patients influenced the gut bacteria composition, as well as L-tryptophan metabolism in serum. In addition, it was evidenced that the correlation between kynurenine/L-tryptophan and neopterin concentrations points to the activation of macrophages and/or dendritic cells [92].
Some groups worldwide have been studying a therapeutic strategy of faecal microbiota transplantation from healthy individuals to patients with NDs. These studies have aimed to restore the intestinal microbiota to treat these diseases. However, so far, it’s still too early, and there is no evidence of its efficacy in the progression of AD and PD [21, 93, 94].
The benefits of probiotics supplementation can induce immune modulation, changes in the endocrine pathway, and increase neurotrophic factors, mainly by the production of high levels of SCFAs [21, 92–94]. As stated before, SCFAs regulate immune pathway, block pro-inflammatory mediators, while up-regulating anti-inflammatory mediators (Figure 6). Through the endocrine pathway, probiotics activate the HPA axis, stimulating the adrenal release of cortisol, which is the most potent anti-inflammatory hormone. In addition, probiotics stimulate the production of GLP-1 and PYY hormones by L cells in EECs via the endocrine pathway. By way of neuronal factors, probiotics stimulate the secretion of certain neurotransmitters, such as glutamate, or modulate the secretion of neurotransmitters through EC, such as serotonin. These neurotransmitters and neuroactive metabolites, released together, seem to exert neuroprotective effects, preventing neuronal apoptosis [95–97].
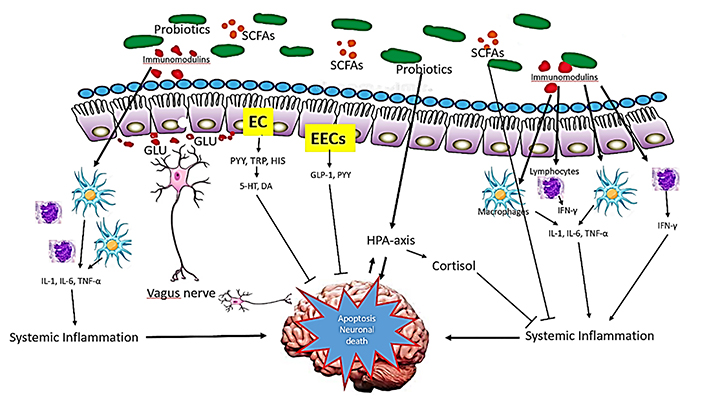
Mechanism of action of probiotics in NDs. Probiotics influence brain function through three main functions: immune modulation, endocrine pathways, and neuronal regulation exerting neuroprotective effects and preventing neuronal apoptosis. 5-HT: hydroxy L-tryptophan or serotonin; DA: dopamine; GLU: glutamate; HIS: histamine; TRP: tryptophan
Note. Adapted from “Probiotics for Alzheimer’s disease: a systematic review,” by Naomi R, Embong H, Othman F, Ghazi HF, Maruthey N, Bahari H. Nutrients. 2022;14:20 (https://doi.org/10.3390/nu14010020). CC BY.
The intestinal microbiota plays a key role in the metabolism of essential amino acids as precursors of several neuromodulators. The resulting metabolic products have shown an important impact on the neuromodulation involving microbiota-ENS-CNS crosstalk, influencing brain functions through signaling pathways. However, disruption of the gut microbiota homeostasis by dysbiosis may be a determinant factor in the pathophysiology of NDs, such as AD. Therefore, treatments non-CNS-directed have been shown as promising alternatives aimed at restoring the microbiota by applying probiotics, prebiotics, and symbionts.
Nevertheless, to the best of our knowledge, and based on the sparse data in literature, despite the demonstrated relevance of the microbiota, ENS and its close relationship and influence on CNS physiology, research on natural products or the design of new bioactive molecules targeting the ENS-CNS is still very shy among medicinal chemists. Perhaps, in the coming years, the exploration of this alternative may indeed lead to the discovery of new prototype drug candidates with different mechanisms of action from those currently known, opening new ways for the development of more effective and safer disease-modifying medicines capable to modulate the progress and severity of CNS-related diseases.
The microbiota-targeted interventions, such as probiotics supplementation and fecal microbiota transplantation, might represent a potential therapeutic option for ND in the near future. However, before that, it is fundamental to understand the interactions between host and gut microbiota, and elucidate how these interactions are affecting CNS, to find new effective strategies in the treatment and prevention of NDs.
Ach: acetylcholine
AChE: acetylcholinesterase
AD: Alzheimer’s disease
Aβ: amyloid-β
BBB: blood-brain barrier
ChAT: choline acetyltransferase
CNS: central nervous system
EC: enterochromaffin cells
EECs: enteroendocrine cells
ENS: enteric nervous system
GABA: γ-aminobutyric acid
GLP-1: glucagon-like peptide-1
HPA: hypothalamus-pituitary-adrenal
IDO: indolamine-2,3-dioxygenase
IFN-γ: interferon-gamma
IgA: immunoglobulin A
ILs: interleukins
Met: methionine
NAD+: nicotinamide adenine dinucleotide
NDs: neurodegenerative diseases
NMDA: N-methyl-D-aspartate
PD: Parkinson’s disease
PYY: peptide YY
QUIN: quinolinic acid
ROS: reactive oxygen species
SCFAs: short-chain fatty acids
TDO: tryptophan-2,3-dioxygenase
TNF-α: tumor necrosis factor-alpha
JTE: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. CMD: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. VSG: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. JG: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. CV: Validation, Writing—review & editing, Supervision.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable
Not applicable.
This study was funded by the Brazilian Agencies CNPq [#454088/2014-0, #400271/2014-1, #310082/2016-1], FAPEMIG [#CEX-APQ-00241-15], FINEP, INCT-INOFAR [#465.249/2014-0], and CAPES [#23038.008071/2018-66]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

