
Abstract
Late-onset Alzheimer’s disease (LOAD) is the most common form of Alzheimer’s disease (AD) and its risk increases exponentially with aging. The incidence of LOAD is reported to increase from 1 in every 1,000 people aged 37 to 65 in every 100 people aged 80 years and older. LOAD is extensively associated with aging and cognition decline. Several risk factors, including lifestyle choices, environmental factors, and medical ailments, affect cellular stress. The cellular stress can bring upon epigenetic alterations that affect cellular aging making the individual more susceptible to LOAD development. In due course the cellular stress resulting into epigenetic deregulation, oxidative burden, and genomic mutations leads to increased disease risk. Role of epigenetic and non-epigenetic mechanisms in accelerated cellular aging that are reported to increase the risk of LOAD development are summarized in this review. The underlying biological mechanism of cellular aging and the risk factors that could predispose cellular aging and LOAD development are also discussed in the upcoming sections.
Keywords
Late-onset Alzheimer’s disease, epigenetics, cellular aging, telomere length, immunosensecenceIntroduction
Alzheimer’s disease (AD; named after the German psychiatric Alois Alzheimer) is a type of dementia which can be defined as a gradual progressive neurodegenerative disease characterized by neuritic plaques and neurofibrillary tangles (NFTs) which results in the accumulation of amyloid-beta peptide’s (Aβ’s) in the most affected area of the brain, the medial temporal lobe and neocortical structures [1]. It is a neurodegenerative condition that arises in two forms: an early-onset familial form and a sporadic late-onset form called late-onset AD (LOAD). In the familial early-onset form of the disease, patients at first display minor memory loss but then progress to major cognitive dysfunction, which is prevalent among adults aged 45–64. The late-onset form includes motor difficulties, language problems, behavioral disorders, severe memory loss, and paranoia and occurs in elderly more than 65 years of age [2–6]. Genetic influence plays an important role in the early-onset form of AD and rarely occurs (5% of cases). The most common form is the late-onset form which has been identified in more than 95% of all AD cases, and its genetic determination remains questionable, however, the research is going on, unraveling the genetic networks underlying LOAD. It has a multifactorial etiology with no obvious simple (mendelian) genetic determinants, yet, the risk of developing LOAD could be, in some cases, significantly influenced by genetics [7]. The well-accepted risk factor for disease development in the LOAD is the possession of the ɛ4 allele at the apolipoprotein E (APOE) locus on human chromosome 19. Not all individuals possessing the ɛ4 allele at the APOE locus will develop LOAD, but its presence increases the risk for disease development, promotes the earlier onset of symptoms, and facilitates rapid progression. In particular, individuals homozygous for the ɛ4 allele type are at high risk for disease development [8, 9]. As AD is characterized by neuritic senile plaques (NSP) and NFT, the diagnosis of LOAD comes from post-mortem examination of the density of NSP and NFT, and the overall neuronal death in the affected brain regions including the frontal and temporal lobes [10].
Advanced age and female sex are the two major risk factors for LOAD [11]. Aging comes with natural cellular senescence, decreasing the neuronal and glial cell populations, and leading to the neurodegenerative condition. Aging-associated cellular senescence is the consequence of the activation of apoptotic molecular machinery due to oxidative stress or mitochondrial dysfunction [12]. Oxidative stress generates reactive oxygen species from the oxidation of biomolecules and is considered the major cause of progressive aging, including both physical and mental aging [13]. Mitochondrial dysfunction is associated with the disruption in ATP production and apoptosis regulation, and these defects in mitochondrial dynamics can lead to irreversible damage to neuronal cells [14]. Hence, multiple pathways are involved in cell death during natural aging, and another phenomenon that impacts natural aging is epigenetic alterations while aging. Very few studies have been conducted considering how natural aging shapes the epigenome and vice versa. The epigenetic changes associated with aging have gained much attention in the aging biomarker research field [15]. Alterations of the epigenetic mechanisms (including DNA methylation, histone modifications, and non-coding RNAs) affect most nuclear processes, such as gene transcription and silencing, DNA replication and repair, and cell cycle progression, affecting natural aging. Recent studies have shown rapid, dynamic, and persistent epigenetic modifications in neurons and in neurodegenerative diseases [16–18]. DNA methylation patterns are largely remodeled during aging [16], where a trend toward global loss of DNA methylation with hypermethylation at specific loci is observed [17]. Although with some differences among brain regions, age-associated epigenetic changes interest the brain also, likely contributing to the structural and functional alterations that can result in progressive cognitive decline and increased susceptibility to neurodegenerative disorders including LOAD [18]. Non-epigenetic mechanisms including telomere shortening and immunosenescence are also involved in accelerated cellular aging and contribute to the LOAD development in the elderly [19].
Biological mechanism of cellular aging
Aging is the irreversible physiological process linked to the lifespan of cells and the body and is the primary cause of age-related diseases. Several biological mechanisms mediate aging and its primary feature is the accumulation of cellular senescence driven on by harmful stimuli from both inside and outside the cell [20, 21]. When determining the aging phenotype, nine criteria and associated potential markers are typically taken into account, including genomic instability, reduced telomere length, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, cellular senescence, stem cell exhaustion, mitochondrial dysfunction, and altered intercellular communication. These factors are based on the research of many different types of species, particularly mammals [21]. One of the most accepted factors of aging is the genomic instability that occurs due to the accumulation of genetic damage from time to time and causes interference with cellular homeostasis [22]. Another factor i.e. shortening of telomere length observed in naturally aging cells, however, due to detrimental external or internal stimulus, cells might undergo accelerated telomere shortening, which gives rise to cellular senescence [23–25]. Cellular senescence is a critical aging factor and is characterized by cell cycle arrest. The senescent cells are presented with senescence-related secreted phenotype and result from DNA damage [20, 26]. Another factor i.e. epigenetic mechanisms are the key regulators of cellular functioning; however, with aging the epigenetic machinery might lose its control giving rise to age-related defects [27]. The disturbance in protein homeostasis is linked to the onset of aging and the majority of aging-related illnesses. Aging is characterized by altered post-translational modification, and protein aggregation [28]. The disruption in the endoplasmic reticulum (ER) homeostasis leads to protein aggregation and causes ER stress. The ER stress response can be seen to increase with aging due to several factors like hyperhomocysteinemia, lack of glucose or energy, redox changes, disruption of calcium homeostasis, and others [29]. The protein degradation machinery in the cells i.e. autophagy and proteasome mediated degradation is widely hampered with aging [30, 31]. Nutrient sensing is the cell’s ability to identify and respond to the nutrient in the body, insulin/insulin growth factor-1 (IGF-1) signaling is one of the pathways involved in this process and is known to regulate aging and aging-related disease [32]. Another crucial aspect of aging is the decline in the capacity of tissues and organs to regenerate [33]. A number of age-related disorders are caused by inflammation, that occurs due to mitochondrial dysfunction, which also increases the formation of reactive oxygen species, lowers ATP levels, promotes apoptosis, and causes defects in the respiratory chain [34, 35]. The regulation of neuroendocrine, endocrine, and neuronal levels via cellular communication is another significant factor in the aging process [36]. Renin-angiotensin, adrenergic, and insulin/IGF1 signaling are the neurohormonal signaling that mediates aging when inflammatory responses rise and the immunosurveillance environment changes in the cell [37]. The molecular mechanisms can be targeted to reduce the risk of age related diseases including LOAD.
Cellular aging in LOAD brain vs. healthy brain
A naturally aging brain undergoes significant changes in activity, however, the evident structural changes are near to negligible, and the LOAD and AD brain shows significant atrophy in different brain regions with aging [38, 39]. Neuronal loss is significant in AD, while the number of neurons does not change in healthy aging brains [40]. Brain atrophy in LOAD is positively correlated with cognitive impairment in LOAD patients, which is attributed to neuronal cell death, shrinkage of neurons, and axonal damage [38, 39]. Another factor that is commonly observed even in the natural aging and cognitive fit brain is NFTs [41], however, in LOAD patients the abundance and localization of NFTs are strongly correlated with neurodegeneration [42, 43]. Although significant structural changes like NFTs deposition in the brain with aging could cause LOAD’s development, other factors might also play a significant role in initiating the neurodegenerative process. One such factor is declining neurotransmission, which is affected by altered dendritic morphology, reduced spine density, and spine volume with natural aging and in LOAD. These complications are associated with age-related deficits in learning and memory formation commonly seen in elderly people [42, 43]. The cholinergic system is most commonly studied in classical AD and cognition impairment is also associated with cholinergic degeneration with aging. LOAD is also associated with cholinergic degeneration presented with reduced choline acetyltransferase (CHAT) activity, total acetylcholine levels, and reduced receptor binding [42, 43]. CHAT activity can be influenced by gene polymorphisms and is majorly associated with AD etiopathogenesis [44]. Hence the neurotransmission is vividly affected in natural aging but intensely deteriorated in LOAD.
Oxidative damage in cells is another driving force of aging. The free radical theory of aging states that the constant buildup of reactive oxygen species for an organism’s life results in cellular senescence and aging of the organism [45]. Increased oxidative stress in the brain is another factor that may act as the critical threshold for LOAD development in the naturally aging brain. Increased oxidative stress and lipid peroxidation is observed in LOAD patients [46, 47]. Oxidative damage can also give rise to DNA damage leading to mutations. Mitochondrial mutations are well reported with aging and aging-related diseases [46, 47], but an increased number of nuclear mutations and mitochondrial mutations have been reported in LOAD [48]. One such mutation identified is CD36 gene mutations that are linked with delaying the onset of AD by eight years. CD36 is involved in Aβ clearance by phagocytosing microglia and is involved in reactive oxygen species production and cytokine production [49]. Therefore, alterations in brain morphology, functioning, and oxidative damage are increased extensively in LOAD brain giving rise to mutations and epimutations.
Epigenetic mechanisms in cellular aging
Epigenetics is the study of heritable changes in the genome environment leading to variations in gene expression without altering the DNA sequence, hence inducing a phenotypic change in organisms [50]. Three major epigenetic mechanisms involved in cellular regulation include DNA methylation, histone modifications, and non-coding RNA-mediated regulation. These epigenetic mechanisms following random occurrences and environmental influence have been identified as contributors to the aging phenotype. Numerous studies have suggested that due to the higher occurrence of epigenetic alterations compared to genetic mutations, epigenetic alterations might be predominantly associated with age-related phenotypes [51, 52]. Epigenetic alterations might be tolerated to a certain degree, however, after a certain threshold these might lead to organ dysfunction and accelerated cellular aging, as illustrated in Figure 1. It is suggested by Wang et al. [53] suggested that every individual is predisposed to have AD at a certain time point after crossing the threshold of epigenetic deregulation and that LOAD can be presented as the extreme form of aging with epigenetic alterations in the brain. Epigenetic markers, including DNA methylation, histone modifications, and non-coding RNAs served as the critical factors in regulating brain physiology and pathology of several neurodegenerative diseases, including AD as well as aging [54].
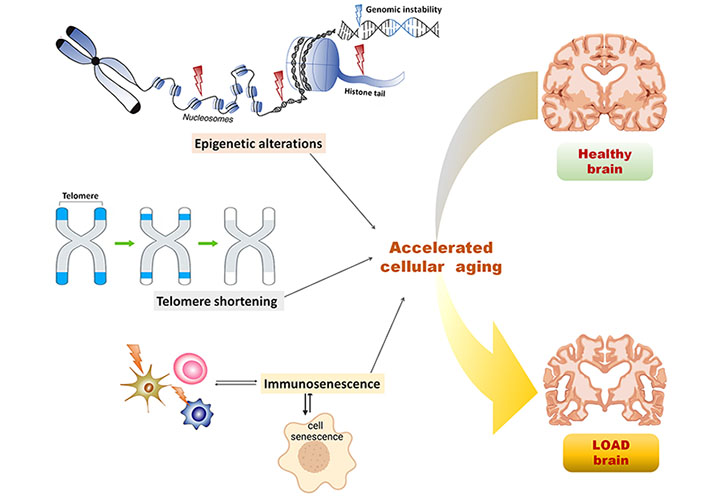
Epigenetic and non-epigenetic alterations in the cellular aging predisposing LOAD. Epigenetic alterations including nucleosome remodeling, histone modifications, and DNA methylation leads to genomic instability, thereby causing accelerated cellular aging and crossing the threshold for LOAD predisposition in the healthy brain; non-epigenetic alterations including telomere shortening and immunosenescence influenced by epigenetic alterations and aging, in turn, leads to accelerated cellular aging in the brain and predispose LOAD symptoms
DNA methylation
DNA methylation involves the addition of methyl group at the 5′ position of cytosine residue of DNA, leading to the formation of methylated cytosine base [5-methylcytosine (5mC)] which is further involved in gene expression regulation. Various enzymes including the family of DNA methyl transferases (DNMT), and ten-eleven translocation methylcytosine dioxygenase (TET) are involved in the addition and removal of methyl groups from the cytosine-phosphate-guanine (CpG) islands in DNA, thereby altering the gene expression at the transcriptional level. DNA methylation at the promoter region leads to the increased hindrance and reduced binding of specific transcription factors at their recognition sites on the promoter, thereby reducing gene expression while demethylation at the promoter region leads to overexpression of the gene because of the increased binding of the specific transcription factors at the recognition site on the promoter and disrupting the regulatory mechanism of gene expression. Another mechanism of DNA methylation-mediated transcriptional inhibition involves binding of the specific transcriptional repressors and methylated cytosine binding proteins at the site of transcription and altering the chromatin structure [55]. A study suggested that the site-specific methylation pattern mediates differential regulation of gene expression, i.e. methylation at gene bodies leads to increased transcription while methylation at promoter region leads to reduced transcription [56]. Several other studies have suggested that the conversion of 5mC to 5-hydroxymethyl cytosine causes more hindrance to the DNA binding protein, resists their binding at the recognition sites, and leads to transcriptional repression [57–59]. DNA methylation also influences gene transcription by regulating histone modifications. Methylated DNA provides a hindrance for histone acetylation and hence methylated DNA is associated with deacetylated histone leading to reduced transcription and unmethylated DNA is associated with acetylated histone leading to increased transcription [60]. Given the significance of DNA methylation in various biological mechanisms, aberrant DNA methylation is widely associated with several diseases, including cancers and neurological diseases, and heritability to the next generation [57–59].
Data on DNA methylation have lately been utilized to estimate cellular age and accelerated age. A number of studies have shown a substantial correlation between chronological age and DNA methylation patterns across the epigenome [61]. Two algorithms are most prominently used to predict the chronological age based on the DNA methylation age, given by Hannum et al. [62] and Horvath [63]. The “epigenetic clock” is assumed to be indexed by these DNA methylation age estimations. Although both algorithms exhibit high correlations with chronological age, there is still enough variation within each estimate at the individual level. Several diseases and health consequences, including Alzheimer’s-related neurocognitive impairment [64], cancer [65], Parkinson’s disease [66], and others, have been linked to the accelerated DNA methylation age estimated using the Horvath algorithm. Several studies have reported global hypomethylation and hypermethylation with age and the age-related progressive reduction in the levels of DNMT1, which is involved in maintaining DNA methylation across the genome [67–69]. Several loci were found hypermethylated with age which was otherwise in a hypomethylated state due to the overexpression of the DNMT3a/b (enzymes involved in de novo DNA methylation), hence reducing transcription of several genes with progressive aging [68].
Several studies in AD patients and animal models have shown impaired DNA methylation levels of several genes, impacting the global methylation status and transcription of the respective genes. Previous studies in LOAD on DNA methylation status in brain biopsies and autopsy material [70–76], and peripheral blood lymphocytes [77, 78] showed differential cytosine methylation at CpG dinucleotides, suggesting the impaired epigenetics in LOAD. In another study depicting the impaired epigenetics in AD, the analysis of postmortem prefrontal brain tissue and peripheral lymphocytes from AD patients concluded the hypermethylation at the promoter region of the methylene tetrahydrofolate reductase (MTHFR) gene [53]. MTHFR is involved in the formation of 5-methyltetrahydrofolate which is a co-substrate for methionine formation, which is further involved in DNA or histone methylation processes. Hypermethylation of MTHFR gene may lead to gene expression suppression, resulting in MTHFR deficiency and hyperhomocysteinemia resulting in an imbalance in DNA methylation, as observed in AD patients [53]. In another study hypermethylation of genes involved in synaptic transmission responsible for compromised synaptic plasticity in LOAD was reported in cortical pyramidal layers of brain samples of 32 LOAD patients [79]. Another gene, ANKYRIN1, which is involved in protein tyrosine kinase 2β induced calcium-induced signaling cascade responsible for modulating the activation of microglia and macrophages, was found to be hypermethylated in the prefrontal cortex and superior temporal gyrus region in LOAD patients [80]. Additionally, animal models of AD have also demonstrated age-related epigenetic alterations in DNA methylation [81].
Histone modifications
Nucleosomes are fundamental units of chromatin, consisting of DNA wrapped around histones and forming a highly stable chromatin structure. There could be two types of modifications on nucleosomes: (1) ATP dependent chromatin remodeling complexes that use energy to restructure nucleosomes and allow the access of transcriptional factors to the transcription site; and (2) amino acid modifications on the histone tails such as histone acetylation, deacetylation, methylation and so on [82]. ATP-dependent chromatin remodeling complexes can recruit the transcriptional activator as well as the repressors hence activating and suppressing the transcription of genes respectively. Histone tail modifications are also associated with transcriptional activation and suppression depending upon the type of modification, for instance, histone acetylation leads to the loosening of chromatin structure, resulting in transcriptional activation, while histone deacetylation leads to closed chromatin structure resulting in transcriptional repression. Various enzymes like histone acetyltransferases (HAT) and histone deacetylase (HDAC) are involved in the acetylation and deacetylation of the histone tails at specific amino acids. Histone tail methylation is involved in the transcriptional regulation in a site-specific manner and the type of methylation. For instance, monomethylation at a specific site is associated with transcriptional activation while trimethylation at the same site can be associated with transcriptional suppression, also methylation at specific sites like histone 3 at lysine 4 (H3K4), H3K36, and H3K79 is associated with transcriptional activation while methylation at H3K9 and H3K20 is associated with transcriptional suppression and hence gene silencing. When two or more modifications are present at a time, the crosstalk among the histone modifications also plays a major role in regulating gene expression. One such regulation is presented by the co-occurrence of H3K4 dimethylation and H3K27 trimethylation, where these two modifications simultaneously lead to transcriptional activation of the gene despite H3K27 trimethylation being a transcriptional suppressor [50].
Aging is also known to have an impact on histone tail modifications. Several changes in histones occur with the progression of aging like the reduction in the histone levels and accumulation of histone variants resulting in aberrant nucleosomes, the differential histone modifications including methylation, acetylation, phosphorylation, and ubiquitylation of specific amino acid residue on histone tails, and the global increase or decrease in active or repressive marks of histone modifications [83]. Despite the overall knowledge of the aging impact on nucleosome remodeling, the lack of studies, particularly in LOAD has made it difficult to analyze the histones status in LOAD patients.
Non-coding RNAs
Non-coding RNAs are the transcribed genes that are not destined to undergo translation and comprise a large percentage of the human genome. Only 2% of the genes carry information for protein synthesis and undergo translation. Depending upon the size, these can be long non-coding RNAs (> 200 nucleotides) and small non-coding RNAs (< 200 nucleotides). These play significant roles in the epigenetic regulation of gene expression. Small non-coding RNAs can be involved in protein syntheses, such as ribosomal RNAs, transfer RNAs, and small nuclear RNAs. These can be regulatory RNAs like microRNAs (miRNAs), silencing RNAs, and others involved in regulating messenger RNAs (mRNA; coding RNAs). miRNAs are non-specific and play a dual role in mRNA regulation. These could bind to the untranslated region of target mRNA and induce translational suppression by interfering in protein synthesis. At the same time, it can also enhance the translation of some of the target mRNAs by binding to the promoter of the corresponding target genes. Additionally, some non-coding RNAs can also be involved in DNA methylation and histone tail modifications, hence regulating epigenetics in several biological mechanisms [50]. Numerous studies have shown the imbalance in several miRNA levels in the brain with aging and neurodegeneration [84, 85]. miRNA-mediated RNA interference is crucial for neuronal homeostasis, and disruption of this regulation might result in increased sensitivity of the aging brain to internal and external stress. Several miRNAs were elevated with increasing age [86] while some were found significantly lower in older populations than in younger individuals [87, 88]. Some of the miRNAs including miR-34 which increases with aging [86], miR-107 which reduces with aging [88], and miR-181 which increases with aging [89] were found consistently deregulated in the brain tissue of LOAD patients according to the systematic review by Herrera-Espejo et al. [90].
Non-epigenetic mechanisms in cellular aging
Telomere length shortening
Telomeres are the non-coding repeated DNA sequences present at the terminals of chromosomes. These maintain the integrity of the information coded in the coding DNA sequences in chromosomes by preventing them from being truncated after every cell division [91]. The incomplete DNA replication leads to the telomere’s shortening, resulting in permanent cell cycle arrest, also known as replicative senescence [19]. Various biological processes, including inflammation, and oxidative stress have been associated with telomere shortening. Various environmental factors have been linked to the aging-associated telomere shortening and are often inferred as an index of accelerated aging [92, 93], illustrated in Figure 1. Several studies have shown that telomere shortening is directly linked to the progression of aging and aging-associated diseases like AD [94–96]. Studies have reported the direct correlation of telomere shortening with cognitive impairment and AD, estimated in terms of telomere shortening rate and telomere length [97]. One study reported that age-dependent telomere shortening predominantly in females than males [98]. In another study, AD patients with a homozygous allele for the APOE gene had shown significantly shorter telomere length than the patients with a heterozygous allele for APOE [99]. Telomere shortening had been established as the risk factor for AD and is reportedly associated with rapid cognitive decline giving rise to long-term dementia [95].
Telomere shortening in AD has been associated with low serum folate levels and increased serum homocysteine (Hcy) levels [100]. Reduced folate levels lead to DNA damage due to uracil incorporation in DNA, resulting in telomere shortening. Apart from the folate levels, telomere length is also regulated by epigenetic mechanism i.e. DNA methylation [101]. Another factor that plays a role in telomere-mediated regulation of aging is telomerase, an enzyme involved in the restoration of telomere length. The mutations in the telomerase gene and the polymorphism of the telomerase reverse transcriptase gene is strongly associated with an increased risk of AD and partly associated with telomere shortening in AD [102].
Immunosenescence
Immunosenescence is characterized by reduced functioning and efficiency of the immune system with advancing age, indicated by loss of acquired immunity to pathogens and poor response to vaccinations. The immune system becomes less adaptive with aging, as illustrated in Figure 1, and it can be estimated using the ratio of CD4+ and CD8+ T cells [103]. The lower ratio (less than 1) indicates increased matured memory T cells which lead to shortened telomeres, T cell death, and increased inflammation [104]. The inflamm-aging is the term coined to label aging-associated increased chronic inflammation characterized by increased proinflammatory cytokines and reduced efficacy of monocytes and natural killer cells. The resulting decrease in innate and adaptive immune response with aging has been related to various age-associated diseases [105] including AD [106]. Innate immune cells, including microglia, monocytes, and natural killer cells undergo immunosenescence with advancing aging in AD. Studies have shown that the aging brain is deficient in the microglia’s phagocytic capacity, which leads to deposition of Aβ, and due to continuous microglial activation, chronic inflammation, mitochondrial damage, and ultimately neuronal death [107]. Monocytes which undergo differentiation to form macrophages are the major regulator of inflamm-aging. Under the influence of Aβ, the macrophages release proinflammatory cytokines leading to neuronal death [108]. Natural killer cells do not show changes in age-dependent distribution in AD patients, however, these undergo phenotypic changes in the aging brain and become more tenacious for cytokine secretion [109]. The role of adaptive immunity is less prominent in AD pathophysiology as compared to innate immune reactions. The adaptive immune cells including B and T lymphocytes act against toxic molecule deposition like misfolded tau and Aβ proteins. A subtype of T cells is responsible for creating memory against pathogens, however, the age-dependent senescent T cells lead to dysfunctional virtual memory cell phenotypes with reduced reactivity to T cell stimulation [110]. The participation of B cells in AD pathophysiology has been limited, however, these have been involved vividly in the generation of autoantibodies against Aβ. These autoantibodies are involved in clearing amyloid plaques however, the levels of these autoantibodies fall with aging, aggravating AD pathophysiology [111]. Altogether immunosenescence-induced chronic inflammation has been associated with the AD pathophysiology that progresses with aging.
Factors predisposing epigenetic alterations and cellular aging in LOAD
The impact of epigenetic alterations can become more prominent with aging due to the progressive weakening of epigenetic control on several physiological processes. Despite the old age, the epigenetic machinery can also be affected from as early as an embryonic stage or from the transgenerational effects i.e. the epigenetic events experienced by the parents. One study reported that the lead exposure to rat pups and cynomolgus monkeys during the early postnatal period resulted in the overexpression of Aβ precursor protein (APP) and overproduction of amyloidogenic Aβ in older life, proving that the environmental conditions during the early age might impact the disease pathology later in the life [112]. Another factor that significantly influences epigenetics is the lifestyle and dietary habits of the individual. The diet composition and inclusion of essential vitamins and omega-3 fatty acid helps regulate epigenetic mechanisms, while the sedentary lifestyle hampers the same [51].
Another common epigenetic alteration seen in LOAD patients is an aberrant one-carbon methylation metabolism, which is indicated by increased total Hcy and low folate concentrations. Studies have demonstrated that an increase in plasma Hcy levels occurs before dementia develops and that there is an inverse linear relationship between plasma Hcy concentrations and cognitive function in elderly individuals [113]. The reduction in the levels of S-adenosylmethionine, which plays a critical role in the methylation of DNA and histones, was also reported in AD patients [114]. Altogether these findings suggest a disruption in epigenetic pathways before the manifestation of major LOAD phenotypes. Another factor i.e. MTHFR polymorphism has been reported in a large number of AD patients, including LOAD cases and ultimately resulting in severe MTHFR deficiency and hyperhomocysteinemia. Elevated total Hcy levels (> 14 μmol/L) in elderly individuals is a well-established risk factor of LOAD and cognitive deficit [115]. Low folate levels and vitamin B12 deficiency throughout life reportedly have a direct correlation with increasing incidence of cognitive impairment in the elderly population and have been considered as the major contributors to the development of LOAD [113].
The occurrence of aberrant chromatin and altered histone regulation in AD is a further poorly understood phenomenon. AD patients reportedly had reduced condensation of constitutive heterochromatin region of chromosomes 1, 9, and 16 compared to the healthy subjects [116]. This study also suggested age-dependent chromosomal anomalies selectively in the region of APOE in AD patients. In addition to the genetic and environmental effects, the epigenetic variability among individuals is also a strong determinant or the risk factor in the occurrence of LOAD as suggested by Wang et al. [53]. An array of epigenetic markers and interindividual variations can affect LOAD predisposition and disease severity.
Conclusions
LOAD is extensively associated with aging-related dementia, and the lack of biomarkers makes it challenging to find selective targets to reduce the disease burden in the elderly. However, the advanced technologies in the field of genetics and bioinformatics in the past decade have made it easier to linearize the pathophysiology of LOAD. Epigenome being affected by a sedentary lifestyle is associated with cellular stress and accelerated cellular aging, a well-established risk factor in LOAD development. Research findings confirm the significant disruption in epigenetic regulation with age progression and can certainly be used as early biomarkers to identify individuals at higher risk of having LOAD. As discussed in earlier sections, every individual can develop LOAD, as it is the aggravated form of aging associated with cognition deficit and epigenomic alterations, however maintaining a proper lifestyle with healthy dietary habit can make one less susceptible to LOAD and associated comorbidities. The lack of animal models selectively for LOAD to study the molecular pathways and the link of aging with cognition decline is another major setback to tackle, however, the advancements in three-dimensional (3D) organoid models can easily overcome this research gap.
Abbreviations
| AD: | Alzheimer’s disease |
| APOE: | apolipoprotein E |
| DNMT: | DNA methyl transferases |
| ER: | endoplasmic reticulum |
| H3K4: | histone 3 at lysine 4 |
| Hcy: | homocysteine |
| LOAD: | late-onset Alzheimer’s disease |
| miRNAs: | microRNAs |
| MTHFR: | methylene tetrahydrofolate reductase |
| NFTs: | neurofibrillary tangles |
Declarations
Author contributions
KR and PG equally contributed to: Writing—original draft, Writing—review & editing.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2023.