
Abstract
Parkinson’s disease (PD) is a prevalent neurodegenerative disease (NDD) affecting millions of individuals. The pathogenesis of PD centers around α-synuclein (α-Syn), a pivotal protein whose aggregation significantly impacts disease progression. Although existing treatments mainly focus on managing motor symptoms by targeting the dopaminergic system, they frequently overlook other non-motor symptoms. The intricate nature of PD pathogenesis contributes to challenges in disease analysis and has hindered the development of effective PD treatments. In recent years, various novel therapies utilizing immunotherapy methods have exhibited promise in preclinical animal models. In NDDs, immunotherapy aims to counteract the detrimental effects of protein accumulation by neutralizing toxic species and aiding their elimination. Numerous active therapy (AI) and passive immunotherapy (PI) strategies have been devised for PD and related synucleinopathies, many of which are currently undergoing clinical trials. Despite demonstrating remarkable success in animal models, immunotherapies encountered substantial setbacks during the late stages of clinical trials, with the exception of lecanemab, which targets amyloid-β (Aβ) in Alzheimer’s disease (AD) and has recently received approval from the Food and Drug Administration (FDA). The lack of translation from experimental investigations to successful clinical outcomes, particularly in terms of cognitive and functional evaluations, highlights the limitations of relying solely on animal studies to comprehend the effects of immunotherapeutic approaches. This comprehensive review focuses on α-Syn-based immunotherapies and delves into their underlying mechanisms of action. Furthermore, Furthermore, the article discusses recent advancements and future prospects concerning the potential of immunotherapeutic strategies for PD. The focus is on highlighting the latest research in this domain to illuminate the challenges and opportunities related to the development of efficacious immunotherapies for individuals with PD.
Keywords
Parkinson’s disease, α-synuclein, immunotherapy, misfolded protein, preclinical, clinical trial, synucleinopathies, neurodegenerative diseaseIntroduction
Neurodegenerative diseases (NDDs) are a group of neurological disorders characterized by the progressive loss of neurons in the central nervous system (CNS) or peripheral nervous system (PNS) [1]. The hallmarks of NDDs are the abnormal misfolding and aggregate (aggre) of proteins [1]. The aggregation of disease pathological protein produces distinct lesions on the course of Alzheimer’s disease (AD), Parkinson’s disease (PD), Dementia with Lewy bodies (DLB), and multiple system atrophy (MSA) [1]. PD is characterized by a progressive loss of dopaminergic neurons in the substantia nigra (SN), resulting in motor symptoms, such as tremors at rest, bradykinesia, stiffening, and gait impairments [2, 3]. Non-motor symptoms of PD include gastrointestinal dysfunction, neuropsychiatric symptoms, neuroinflammation, mitochondrial dysfunction, oxidative stress, and intracellular aggres of misfolded α-synuclein (α-Syn) that appear later in the disease’s progression [1, 2, 4].
PD is the world’s second most common NDD after AD, affecting more than 10 million people globally and is expected to reach 12 million by 2050, with 1 million sufferers in the United States alone [1, 2]. The annual expenses of PD in the United States are approximately $51.9 billion, estimated to surpass $79 billion by 2037 [2, 5]. Despite decades of research, there is no cure for PD; existing treatments aim to manage symptoms rather than address the disease’s underlying cause. Increasing dopamine (DA) levels, brain stimulations, physical therapy, and dietary changes are the mainstays of PD treatments. Other methods include lifestyle changes and brain stimulation [2, 6]. However, treatment for non-motor symptoms has yet to be discovered [7, 8]. Several novel therapies demonstrating promise in preclinical animal models are currently undergoing evaluation in clinical trials, employing immunotherapy approaches.
Immunotherapy uses the body’s immune system to fight diseases such as cancer or autoimmune disorders [9]. Over the past few decades, immunotherapy strategies that target proteinopathy in NDDs have grown gradually, and several active and passive therapies are currently undergoing various preclinical and clinical stages [10]. The initial section of this review article provides an overview of the current understanding of α-Syn physiological function and recent advancements in immunotherapy for PD. Particular emphasis is placed on treatments that aim to inhibit or reduce α-Syn aggregation. Currently, there is no established role of immunotherapy in treating PD. However, these studies are still in the initial stages, and it is unclear whether immunotherapy will be a practical treatment possibility for PD. Immunotherapy treatments have grown in interest among NDDs after the recent Food and Drug Administration (FDA) approval of lecanemab, a monoclonal antibody (mAb) therapy for reducing amyloid-β (Aβ) plaques in AD. Overall, immunotherapy is a fast-evolving therapeutic field, with next-generation technology enabling the design and development of highly specific vaccines targeting NDD-associated proteins. Few immunotherapies for synucleinopathies have been produced, and even fewer are in clinical studies. The objective of this article is to offer a comprehensive understanding of the key principles and challenges associated with the design of immunotherapies. The review encompasses a thorough analysis of experimental and clinical trials in PD immunotherapy, with the aim of identifying potential factors contributing to variations in their outcomes.
α-Syn in PD
Synucleinopathies are among the most devastating neurological diseases of age-related NDDs [6]. PD, DLB, and MSA are all synucleinopathies in which the protein α-Syn plays a critical role in the disease’s progression [11]. The neuropathological hallmark of PD is the accumulation of α-Syn oligomer (oligo) and amyloid fibrils in intracellular inclusions known as Lewy bodies (LB) and Lewy neurites (LN) [12]. The etiology of PD has several recognized causes, including genetics accounting for 5–10% of the cases, while most are sporadic [13, 14]. Most of the genetic causes of PD include mutations in 23 loci [leucine rich repeat kinase 2 (LRRK2; PARK8), SNCA (PARK1), Parkin RBR E3 ubiquitin protein ligase (PRKN; PARK2), PINK-1 (PARK6), and DJ-1 (PARK7)] [13]. Several studies have shown that genetics and environmental factors (pesticides, pollution, heavy metals, etc.) contribute to the etiology of the disease [13].
Initially, a comprehensive examination of α-Syn’s normal structure and function is essential to comprehend its abnormal physiological and biophysical characteristics. α-Syn is an intrinsically disordered (ID) and highly flexible small protein (14 kDa) with 140-amino acid (AA) lacking cysteine and tryptophan residues α-Syn has three domains: (1) an N-terminus lipid-binding α-helix, (2) a non-amyloid component (NAC), and (3) an acidic C-terminus tail (Figure 1) [15]. The N-terminus domain (residues 1–87) is positively charged and rich in lysine [15]. Characterized by a sequence of 11-residue imperfect repeats, each based on a highly conserved KTKEGVXXXX hexametric motif that may adopt the amphipathic α-helical structure [15, 16]. The central region or NAC domain (residues 61–95) develops cross-b-sheet and has a highly hydrophobic sequence that underpins its aggregation propensity and leads to protofibril and fibril formation (Figure 1) [15]. The NAC domain is essential for α-Syn aggregation; several studies have demonstrated that deleting significant portions of this motif reduces α-Syn oligomerization and fibrillization [15, 17, 18]. The highly acidic and proline-rich C-terminus domain (residues 96–140) represents a very flexible ID region [15, 19]. The C-terminus contains negatively charged residues such as tyrosine and serine (Ser) that can be phosphorylated and impact the structure leading to aggregation and toxicity (Figure 1) [20]. α-Syn is hyperphosphorylated at Ser-129 in individuals with PD and other synucleinopathies (Figure 1) [21]. Almost 90% of α-Syn in LB is phosphorylated on Ser-129, whereas only 5% of α-Syn in normal brains is similarly changed [22]. Phosphorylation inhibition appears to be promising for inhibiting protein aggregation [21]. In the cytosol, α-Syn exists in a dynamic equilibrium of unfolded monomers and statistically disfavored helical oligos, i.e., tetramers [23].
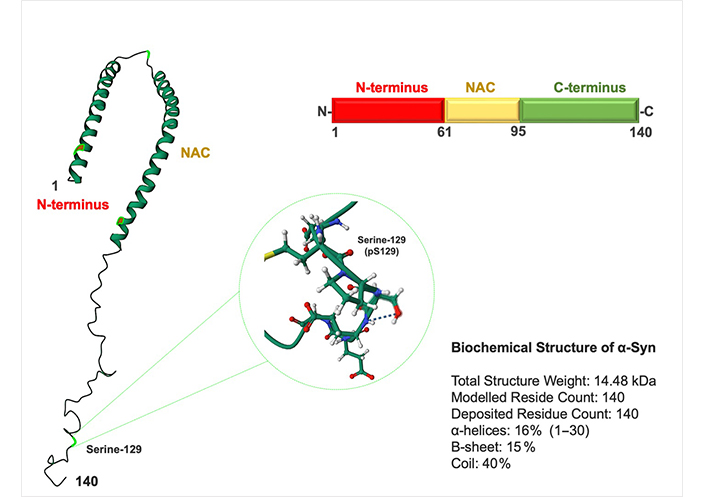
Structure of α-Syn. α-Syn is a 140-AA protein composed of three domains: an N-terminus domain (AA 1–60), an NAC domain (AA 61–95), and a C-terminus domain (AA 96–140). The NAC and N-terminus domains contain an 11-residue sequence repeated seven times. Most disease-causing mutations are identified in the N-terminus domain. NAC domain is responsible for its propensity to misfold into b-sheet–rich amyloid aggres. The C-terminus domain contains one Ser-129 (pS-129) and three tyrosine residues (125, 133, and 135) that can be phosphorylated and alter protein toxicity
Note. Adapted from the protein data bank (http://www.wwpdb.org). CC0 1.0.
The physiological function of native, monomeric α-Syn is unknown [24]. It is thought that α-Syn modulates synaptic transmission by controlling vesicle docking, membrane channels, synaptic plasticity, fusion, and neurotransmitter release at axon terminals and present in neuronal presynaptic terminals [24]. However, it has been observed that α-Syn function in monomeric form during neuron-to-neuron communication is associated with presynaptic homeostasis and neurotransmitter exocytosis on axon terminals [24]. In PD, α-Syn undergoes misfolding and aggregation, leading to the formation of insoluble protein deposits [25–29]. The accumulation of these aggres is believed to contribute to neurodegeneration in PD by disrupting normal cellular processes, impairing protein clearance mechanisms, and inducing cellular toxicity. Additional research is necessary, as the molecular processes and variables governing α-Syn aggregation remain unidentified. A deeper understanding of both the normal and pathological actions of α-Syn is deemed a crucial initial step in the development of novel disease-modulating therapeutic strategies.
α-Syn seeding, aggregation, and propagation
The levels of α-Syn in the CNS are determined by the rate of α-Syn synthesis, aggregation, and clearance [24, 30]. An imbalance between these systems results in abnormal amounts of α-Syn, which may favor the synthesis and accumulation of toxic oligomeric and fibrillar species (Figure 2) [19, 30]. In familial cases of an inherited form of PD, genetic mutations such as A53T/E, A30P, H50Q, E46K, and SNCA lead to higher accumulation of α-Syn due to enhanced protein expression [15, 24, 30]. In a transgenic (tg) mice study, the overexpression of human α-Syn decreased synaptic vesicle exocytosis and neurotransmitter release [31]. PD and other synucleinopathies may be impacted by increased expression and accumulation of α-Syn because of mutations and failure in the clearance. Recent research suggests that the spread and transmission of aggregated forms of α-Syn contribute to the etiology of PD [31].
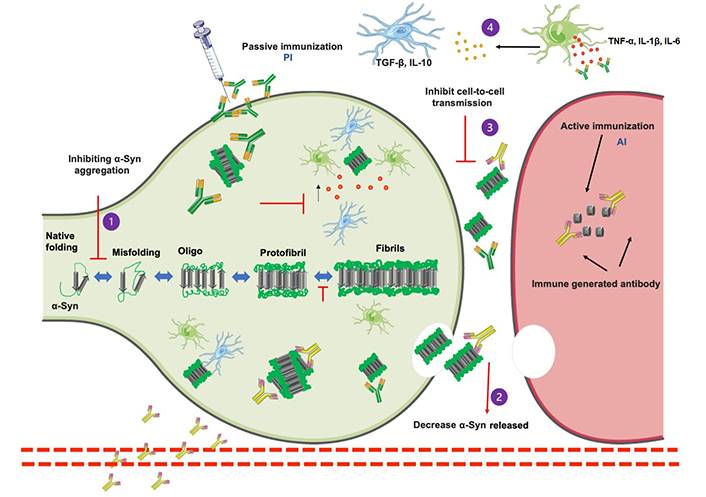
Schematic representation of α-Syn aggregated pathway and immunotherapy approaches. α-Syn monomers include several stages, from intrinsically oligomeric aggres to fibrils and protofibrils that develop into pathogenic inclusions and the activation of microglia and astrocytes by proinflammatory cytokines [tumor necrosis factor α (TNF-α), interleukin 1β (IL-1β), IL-6, and interferon β (IFN-β)]. α-Syn toxicity may be reduced using a variety of treatment approaches. Immunization may be able to (1) prevent α-Syn aggregation, (2) decrease the number of α-Syn released into the extracellular environment, (3) inhibit cell-to-cell transmission, and (4) antibodies against immune system activation being another immunotherapy method that attempts to inhibit pro-inflammatory cytokine release, increase anti-inflammatory microglial activity, and therefore prevent α-Syn disease progression. A small fragment of α-Syn antibody (AI) is presented to the immune system as part of the active immunization pathway, which causes the immune system to recognize the peptide as foreign and create antibodies that promote phagocytosis and protein clearance. By injecting in vitro-produced antibodies (P1) that recognize various structures of α-Syn (P2) and promote phagocytosis of α-Syn. Parts of Figure 2 were generated with the Servier Medical Art Commons Attribution 3.0 Unported License (http://smart.servier.com)
Aggregation of α-Syn is a multistep process that involves the oligomerization of monomeric α-Syn into insoluble fibrils, followed by bigger aggres [16, 30]. α-Syn oligos and fibrils can function as “seeds” for α-Syn propagation in neighboring cells [25]. One of the most significant advances in the study of protein misfolding and aggregation in NDDs has been the discovery of their abilities to transfer pathogenic seeds between affected and healthy cells, referred to as “cell-to-cell or prion-like” transmission (Figure 2) [28]. In a study, tg mice treated with α-Syn protofibrils and fibrils increased α-Syn pathology and propagation, raising the possibility that extracellular α-Syn seeds function in a prion-like manner (Figure 2) [11, 32–34].
Several preclinical studies using human brain tissues, animals, and cell models in recent years have been able to comprehend better and provide insight into the mechanics of this disease transmission model [33–35]. Aggres eventually expand to the brainstem, cortex, and limbic regions [34, 35]. This transmission theory has been demonstrated in cell transplant treatment using brain-derived cells as therapy [32, 36, 37]. α-Syn aggres were found in the transplanted cells, demonstrating that transmission occurred from the host to the grafted cells [38, 39]. These α-Syn aggres play a critical role in the pathogenesis and progression of synucleopathies [25–28]. It will be interesting to examine how damaged seeds impact different brain areas over time and how the patterns may alter depending on the condition, which will aid in the development of new therapies.
α-Syn targeted therapeutics for PD
Mounty of evidence has shown that α-Syn is more than a marker for PD, but plays a significant role in its development [24]. Treatments for synucleinopathies may include reducing intracellular and extracellular α-Syn accumulation levels or enhancing its clearance, which are potential therapeutic methods for PD and related diseases [6, 40]. The discovery of pathogenic α-Syn species and evidence of cell-to-cell transmission has created an avenue for various treatment strategies to halt or decrease the course of the disease [6]. These strategies generally consist of (1) immunization, (2) decreasing α-Syn expression, (3) inhibiting of α-Syn oligomerization, (4) reducing the accumulation of intracellular or extracellular α-Syn aggregation, (5) increasing the clearance of α-Syn, (6) targeting genes/proteins that affect α-Syn aggregation or processing (silencing SNCA), (7) increasing proteolytic breakdown of α-Syn, and (8) reducing the posttranslational modifications such as oxidation, nitration, phosphorylation, and C-terminal cleavage (Figure 2) [6, 15, 41]. It is worth noting that while these therapeutics show promise, many are still in the early stages of development and require further testing in clinical trials to determine their safety and efficacy in treating α-Syn-related diseases. Immunotherapy aimed at modulating extracellular α-Syn transmission is clinically significant [10]. Theoretically, antibodies-based therapies are the best types of treatment to target pathogenic proteins, particularly those found in the extracellular environment [42, 43]. Other targeting approaches in various preclinical and clinical studies have been created to indirectly slow the spread of α-Syn (Tables 1 and 2) [9]. These strategies have long-term clinical applications since inhibiting the extracellular release of the aggregated form of α-Syn and preventing the spread of pathogenic proteins. This might constitute the basis of neuroprotection in PD patients.
Active and passive immunization studies in PD animal models
| Type | Name | Antibody | Model | Route | Time and dose | Target site | Effect on α-Syn | Effect on behavior | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Active | PDO1A AFFiRiS | AFF | PDGF, mThy1, MBP α-Syn mice | s.c. | 8 months; 80 μg/mL, 100 μL | C-terminus | ↓ α-Syn accumulation prevented α-Syn spreading | Motor improvements: ↓ errors on round beam test | [44, 45] |
| PDO3A AFFiRiS | PLP α-Syn mice | Preserved nigral DA neurons ↓ α-Syn olig levels and microglial activation | Improved gait deficits Motor improvements: ↓ errors on round beam test | [46] | |||||
| UB-312 Vaxxinity | UB-312 | Thy1SNCA/15 mice (line 61) | i.m. | 15 months; 40 µg per injection | C-terminus | ↓ Levels of α-Syn olig | Motor, learning, and memory improvements: ↓ errors on round beam ↓ Path and latency to platform in water maze and challenging beam test | [47–51] | |
| Passive | Prasinezumab (PRX002) | 9E4 | PDGF, Thy1 (line D, 61)-α-Syn mice | i.p./i.v. | 6 months; 10 mg/kg | C-terminus | ↓ DA cell loss ↓ Calpain-cleaved α-Syn olig ↓ α-Syn accumulation | Motor performance: ↑ Cylinder, Pole, and Rotarod test | [45, 51, 52] |
| Cinpanemab | BIIB054 (NI-202) | Rat and cynomolgus monkey. A53T (M83), BAC A53T α-Syn PFF | i.p./i.v. | 2–4 months; 10 mg/kg and 30 mg/kg | N-terminus | ↓ α-Syn pathology preserved and improved DAT levels | Motor improvements: ↑ latency in time to fall in wire hang test | [53, 54] | |
| TAK-341 | MEDI1341 | Rat and cynomolgus monkey | i.v. | 13 weeks; from 3 mg/kg to 100 mg/kg; 25, 75, and 150 mg/kg | C-terminus | ↓ Contralateral and ipsilateral α-Syn ↓ Interstitial and CSF fluid α-Syn levels ↓ α-Syn propagation along axons | - | [55] | |
| ABBV-0805, BAN0805 | ABBV-0805, mAb47 | A53T, Thy1-A30P | i.p./i.v. | 2–4 months; 0.1, 1, 10, and 20 mg/kg | C-terminus; protofibrils | ↓ Soluble and insoluble α-Syn ↓ pS129 α-Syn pathology | ↑ Mean survival on A30P mice | [56–58] | |
| Lu AF82422 | Rat and cynomolgus monkey | i.v. | 4 weeks, 1 dose; 0–600, 0–300, and 1–30 mg/kg | C-terminus | ↓ α-Syn in plasma and CSF | - | - | [59] | |
| Ab274 | PDGFb (line M) α-Syn mice | i.p. | 4 weeks; 1 mg/mL | C-terminus | ↓ NeuN cell loss; ↑ Cathepsin-D and α-Syn; ↑ α-Syn clearance by microglial; ↓ TNF-α and IL-6; ↑ Iba-1; ↓ α-Syn (70–80%) | ↓ Latency to turn (Pole test) ↓ Total activity in the open field and Pole test | ↓ Latency to turn (Pole test) ↓ Total activity in the open field and Pole test | [47] | |
| 1H7, 5C1 | Thy1 α-Syn (line 61) mice | i.p. | 6 months; 10 mg/kg | C-terminus | ↓ TH loss in striatum (35%) ↓ α-Syn aggregation ↑ Synaptophysin + MAP2 ↓ Astro and microgliosis | ↓ Memory and learning deficits ↓ Error on transversal beam ↓ Memory and learning deficits | ↓ Memory and learning deficits ↓ Error on transversal beam ↓ Memory and learning deficits | [45] | |
| AB1 | Nigral AAV-CBA α-Syn in wt rats | i.p. | 3 months; 1 mg/rat (2x first), 0.5 mg/mL | N-terminus | ↓ α-Syn in SN ↓ α-Syn brain homogenate ↓ DA and NeuN cells loss | Motor improvement: ↑ on Cylinder test | Motor improvement: ↑ on Cylinder test | [60] | |
| AB2 | C-terminus | ||||||||
| Syn303 | wt mice PFF | i.p. | 180 days; 30 mg/kg | N-terminus | Blocked α-Syn spreading ↓ DA cells loss ↓ Insoluble α-Syn aggregation and pS129-α-Syn | ↑ Latency to hang (wire-hang time) | ↑ Latency to hang (wire-hang time) | [61] | |
| Syn-O1 | mThy1 (line 61) α-Syn mice | i.p | 3 months; 30 mg/kg | Oligos | ↓ α-Syn accumulation | - | - | - | |
| Syn-O4 | Oligos | ↓ NeuN cell loss | - | - | - | ||||
| Syn-F1 | Fibrils | ↓ Oligomeric α-Syn | - | - | - |
↓: decreased; ↑: increased; -: not applicable. MBP: myelin basic protein; NeuN: neuronal nuclei; MAP2: microtubule-associated protein 2; Iba-1: ionized calcium-binding adapter molecule 1
Immunotherapeutic agents on the clinical trial for PD
| Type | Drug | Target | Mechanism of action | Phase of deve-lopment | Clinical trials.gov ID | Statuses | Sponsor | Result | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Active | PD01A AFF | C-terminus | Peptide-based vaccine | Phase I | NCT01568099 | Completed | AFFiRiS AG | Good tolerability and safety of subcutaneous administration | [62, 63] |
| Mimics α-Syn peptide vaccination acts as B cell epitope | Phase IB | NCT02618941 | Completed, result pending | Safe and tolerable | |||||
| PD03A AFF | Phase I | NCT02267434 | Completed | ||||||
| UB-312 | C-terminus | Targets olig and fibrillar α-Syn | Phase IA and B | NCT04075318 | Completed | United Neuroscience Ltd. | Safe, well tolerated, and induced anti-α-Syn antibodies in serum and CSF | [64] | |
| Passive | Prasinezumab (PRX002) | C-terminus | Aggre α-Syn at AA 118–126 | Phase II | NCT03100149 | Active, not recruiting | Hoffman-La Roche. Prothena Biosciences | Safe and well tolerated ↓ Free serum α-Syn | [65–67] |
| Prasinezumab (PRX002) | Phase IIB | NCT04777331 | Recruiting | - | - | ||||
| Cinpanemab (BIIB054) | N-terminus | Aggre, fibrillar α-Syn | Phase I | NCT02459886 | Completed | Biogen | Safe and well tolerated with no TEAEs | [68] | |
| Cinpanemab (BIIB054) | N-terminus | Aggre, fibrillar α-Syn | Phase II | NCT03318523 | Terminated | Biogen | The study did not meet its primary outcome | [69] | |
| MEDI1341/TAK-341 | C-terminus | Directly bind to all from of α-Syn | Phase I | NCT03272165 | Completed | AstraZeneca; Takeda Pharmaceuticals | - | - | |
| MEDI1341/TAK-341 | C-terminus | Monomeric and aggre α-Syn at the AA 103–129 region | Phase I | NCT04449484 | Completed | Result pending | [55] | ||
| Phase III | NCT05526391 | Recruiting | - | - | |||||
| ABBV-0805/BAN0805 | C-terminus | Aggre α-Syn oligo/proto-fibrils at AA 121–127 | Phase I | NCT04127695 | Withdrawn | AbbVie, BioArctic Neuroscience | - | - | |
| Lu AF82422 | C-terminus | Aggre α-Syn at AA 112–117 | Phase II | NCT05104476 | Recruiting | H. Lundbeck | - | - | |
| UCB7853 | C-terminus | Aggre α-Syn | Phase I | NCT04651153 | Active, not recruiting | UCB Biopharma SRL | Ongoing | - |
-: not applicable. TEAEs: treatment-emergent adverse events
Neuroinflammation in PD
Neuroinflammation is a prominent feature of PD, characterized by the activation of immune cells, particularly microglia and astrocytes, in the CNS [70]. The immune response to α-Syn aggres has been linked to disease progression [30, 71]. Mounted evidence suggests that aggregated forms of α-Syn activate microglia and produce pro-inflammatory immune factors, which harm nearby neurons and neuronal connections (Figure 2) [28, 72, 73]. Recent research has identified a strong association between innate and adaptive immunity in PD pathobiology, allowing therapeutic immune regulation to ameliorate disease progression [70, 74]. In PD, microglia are the primary scavengers of extracellular α-Syn aggregated (Figure 2) [72, 75, 76]. Excessive microglial activation has been seen to become phagocytic, express more major histocompatibility complex (MHC) antigens, and secrete both pro- and anti-inflammatory cytokines (Figure 2) [75]. This microglia alteration result in an increase in the inflammatory response mediated by proinflammatory cytokines such as TNF-α, IL-1, IL-6, and IFN-γ in the striatum and cerebral spinal fluid (CSF; Figure 2) [75]. With the existing relationship between inflammation and the pathology of PD and the role of microglia and the immune system, immunotherapies are likely the most viable therapeutic option for PD (Figure 2) [41, 74–77].
Multiple in vitro and in vivo investigations have provided substantial evidence demonstrating that toxic α-Syn is a potent activator of microglia-mediated inflammation [26, 77]. The activation of microglia has been shown to impact their capacity to uptake and eliminate extracellular α-Syn, thus contributing to the propagation of toxicity [27]. Notably, neuron-derived exosomes have been identified as carriers of toxic α-Syn species, facilitating the spread of its toxicity across neural and non-neural cells (Figure 2) [78, 79]. In vivo studies have further substantiated the involvement of microglia in the dissemination of α-Syn through exosome release [27, 73, 79, 80]. Injection of human α-Syn preformed fibrils (PFFs) into the mouse brain’s dorsal striatum resulted in the presence of α-Syn within CD11b+ exosomes [80, 81]. Furthermore, the reduction of α-Syn transmission corresponded to a decline in exosome synthesis by microglia [80, 81]. These findings collectively demonstrate that microglia-released exosomes containing α-Syn can induce protein aggregation in recipient neurons and anatomically related regions [80, 81].
In summary, inflammatory processes are a double-edged sword, assisting in removing toxins and pathogens while increasing cytotoxicity and neurodegeneration. Researchers [72, 76, 82, 83] have identified immune response mechanisms and how they may influence neuronal cell death in PD. TNF, IL-1, IL-6, IFN-plasma levels, and TNF-α in serum have been discovered to be elevated in PD patients’ brains, CSF, and sera. The severity and course of PD may affect the concentration of these indicators in peripheral tissues. Therefore, it is reasonable to consider whether inflammation is merely a byproduct rather than an active component. Immunotherapies can reduce extracellular and endogenous α-Syn levels to prevent pathologic inflammatory activation [43, 61].
The exact mechanisms underlying neuroinflammation in PD are still being elucidated, and it is likely a multifactorial process involving interactions between genetic, environmental, and immune factors. The sustained activation of microglia and astrocytes, coupled with the release of inflammatory mediators, can contribute to the ongoing neurodegenerative processes observed in PD. Understanding the science behind neuroinflammation is crucial for developing targeted therapies that aim to modulate the immune response, reduce inflammation, and potentially slow down disease progression. How to target the aggregated α-Syn and inflammatory response in PD is now an urgent focus of study in the field, with several current possible immunotherapies for PD directly targeting α-Syn in clinical studies.
Immunotherapies in NDDs
Immunotherapies have gained significant attention and have been extensively studied in recent years across various fields of medicine, including NDDs. Immunotherapies in NDDs suppress the activated immune state in the CNS, which reduces neuroinflammation and creates a neuroprotective environment. The administration of vaccines to activate the immune system and confer protection against specific diseases, known as immunization, has emerged as a prospective approach for PD [10]. The underlying hypothesis of PD immunization suggests that targeting α-Syn with a vaccine could potentially impede or diminish the progression of the disease [9, 10].
Following the approval of a new medication for AD by the FDA, there has been significant interest in the development of antibody-based therapies for PD. Clinical trials using antibodies targeting Aβ have demonstrated a reduction in abnormal protein accumulation; however, no substantial cognitive benefits were observed [84]. Numerous clinical trials have been conducted to assess the effectiveness of immunization approaches in PD [9]. One strategy involves utilizing antibodies to target α-Syn and developing vaccines to generate antibodies against α-Syn [43]. These approaches can be categorized as active immunization and passive immunization (Figure 2). In animal models, both active and passive immunization have demonstrated neuroprotective properties [2, 5, 6, 9]. Studies conducted on α-Syn tg mice have shown that anti-α-Syn antibodies with high affinity to the C-terminus can reduce intracellular α-Syn accumulation and improve motor and cognitive impairments [65].
The mechanism of how immunotherapy works in PD is still unclear and many studies are still under early phase trials (I or II) [30]. Immunotherapy relies on the production (active) or the use (passive) of antibodies that directly target antigen, α-Syn in this case, to counteract disease by activating the immune system. A vaccination containing α-Syn-antigen is frequently delivered intramuscularly in active immunizations [74, 85]. However, in passive immunization, mAbs are administered directly by intravenous infusions or subcutaneous injection and must cross the blood-brain barrier (BBB) to reach the brain parenchyma [86]. Only a small fraction of antibodies introduced into the peripheral circulation (0.1–1%) can be detected in the brain or CSF [87]. mAb against α-Syn is supplied directly by intravenous infusions or subcutaneous injections in passive immunization [15, 71, 87]. In PD, antibodies may be responsible for the disassembly of α-Syn deposits in the brain or prevention of reassembly and inhibition of toxicity through direct binding to oligos [47, 87]. By directly preventing the uptake of harmful protein by the cells, antibodies against α-Syn stop the transmission and spread of the disease (Figure 2) [47]. Immunization against α-Syn is still under investigation, and additional study is required to determine the safety and efficacy of this technique. Immunization against α-Syn, if successful, could provide a novel therapeutic option for PD [85]. However, this method is still in the experimental stage and is not currently available as a medical therapy for PD. Finally, while immunotherapies offer enormous promise, it is crucial to recognize that their development and clinical application are dotted with challenges, such as potential adverse effects, optimization of delivery strategies, and variability in immune responses. Nonetheless, the possible disease-modifying effects and specific approach of immunotherapies make them an intriguing field of study with tremendous potential for the treatment of NDDs and other disorders.
Active immunization
Active immunization is a type of immunity that occurs after being exposed to an α-Syn antigen and involves the production of antibodies in the receiver [33, 74, 88]. The immunity response takes days or weeks to develop, but it can be long-lasting, even lifetime [74]. With only a few vaccines, patients should be able to develop a long-lasting antibody response; however, the unpredictability of the induced response can be an issue, especially when working with elderly patients [87, 89]. Adverse side effects from active immunization may occur, such as an aberrant immune response that rises with age, diminishing immune system competency, and increasing the likelihood of developing autoimmune responses [74, 89, 90]. Approximately ten years ago, a group of researchers conducted studies on active immunization against α-Syn, utilizing tg mouse models to evaluate the vaccine’s efficacy and safety; they found that active immunization with the α-Syn vaccine resulted in a significant reduction in α-Syn pathology, including decreased accumulation of aggregated α-Syn and reduced neuronal degeneration [24, 44, 46, 85, 91]. Moreover, the vaccinated mice exhibited improved motor, cognitive performance compared to control groups, and an increase in α-Syn-specific antibodies in the vaccinated mice, which correlated with the reduction in pathology. These antibodies were shown to effectively target and clear α-Syn aggres, preventing their accumulation and subsequent neurotoxicity [44, 85]. Injection of recombinant human α-Syn into tg mice expressing α-Syn under the influence of platelet-derived-growth-factor b (PDGFb), results showed a reduction in accumulated α-Syn in neurons with microglia activation [44, 85]. Today, several clinical trials have been carried out to investigate the efficacy of immunization in PD. One approach involves using antibodies to target α-Syn. Another method is to use a vaccine to stimulate the immune system to produce antibodies against α-Syn. To be effective, active immunization strategies in PD must overcome two rate-limiting obstacles: (1) reprogram the host immune system’s self-tolerance for target α-Syn antigens and (2) guarantee a sufficient supply of antibodies cross the BBB.
Active immunization strategies preclinical and clinical studies
AFFiRis: PD01A and PD03A
Mandler and colleagues [85] were the first to test active immunization paradigms on PD animal models; these studies served as the foundation for developing the AFFITOPE (AFF) vaccines. AFFiRis completed phase I clinical trials for the two peptide vaccines PD01A and PD03A, or AFF. AFFiRis’ AFF vaccines implement short peptide fragments that mimic the C-terminus residues of α-Syn (110–130) [46, 85]. AFF peptide produced high titers of antibodies against aggregated forms of α-Syn that were detectable in CSF, plasma, and brain parenchyma using two different tg mouse models overexpressing α-Syn under the PDGF promoter and the thymus cell antigen 1 (Thy1) promoter [85].
AFF® PD01A on murine Thy1 (mThy1)-α-Syn tg mice reduces α-Syn accumulation and protects against tyrosine hydroxylase (TH) fiber loss and motor impairments (Table 1) [85]. AFF® PD01A immunization reversed motor dysfunction, impaired spatial memory, and reduced striatal degeneration and α-Syn deposition in PDGF-α-Syn tg mice (Table 1) [85]. AFF® PD01A on the MSA mouse model demonstrated comparable protection against α-Syn aggregation and TH fiber loss, reduced demyelination, and motor protection [44]. These animal models demonstrated that these peptides improved motor impairment and cognitive function without affecting physiologic α-Syn levels [44, 85]. Although AFFiRis is the only active peptidic vaccine that has reached the clinical development stage, multiple other peptidic vaccines have demonstrated preclinical efficacy in vivo.
The initial vaccination study targeting recombinant α-Syn was conducted in PDGF, which demonstrated significant preservation of synapses attributed to the affinity of the antibodies for membrane-bound aggre species [91]. DNA-based vaccinations offer the potential for a more robust impact on overall proteinopathy by targeting naive protein substrates before aggre recruitment. However, it is important to consider the lack of selectivity for pathologic α-Syn conformers and the potential risk of excessive reduction of functional monomeric α-Syn when developing nucleic acid-based active immunization strategies [85, 90, 92]. In another study, the therapeutic efficacy of PD03, a novel AFF immunotherapy strategy, alone or in combination with Anle138b, was investigated using a proteolipid protein (PLP) α-Syn mice model (Table 1) [46]. The combined therapy of AFF® PD03 and Anle138b showed improvements in gait deficits and preservation of nigral dopaminergic neurons, accompanied by reduced levels of α-Syn oligos, decreased microglial activation, and lower immunoglobulin G (IgG) binding, resulting in attenuation of neurodegeneration (Table 1) [46]. Both approaches hold promise as potential disease-modifying therapies for α-synucleinopathies [44, 46, 62, 85, 91, 92].
Various phase I clinical trials of AFF® PD01A/PD03A in patients with PD and MSA have been conducted based on preclinical observations (Table 1). The results of clinical trials were obtained from the clinicaltrials.gov database (Table 2). Two phase I trials evaluated the safety and tolerability of the vaccine in patients with MSA or PD [93]. In the initial clinical trials of AFF®, a randomized parallel-group phase I study enrolled 32 early PD patients [94]. The dosing regimens tested included a single dose or four doses of 15 μg or 75 μg administered every 4 weeks (Table 2) [62]. Both doses were well tolerated, and no major adverse events related to the drug were observed [62, 93, 94]. Subsequently, the participants continued in the AFF® 12-month phase Ib follow-up extension study, which included a booster immunization 36 weeks after the initial injection with six doses of AFF® PD01A (Table 2) [93, 94]. All dosing regimens led to a significant increase in antibodies over time [62, 94]. AFF® PD01A was well tolerated locally and systemically, with no severe adverse events or significant adverse responses reported. An immunological response was observed in 19 out of 22 vaccinated individuals (86%), with specific serum antibodies produced in 12 of the 19 responders (63%). Compared to the placebo, AFF® PD01A reduced oligomeric α-Syn in the CSF by 51% and fibrillar α-Syn compared to its monomers [62]. Clinical PD ratings remained stable throughout the trial period, although additional data were not provided due to the lack of a double-blind design. In an open-label evaluation, PD01A patients demonstrated stable clinical ratings compared to the placebo group, leading to the design and initiation of phase II clinical studies (NCT01568099, NCT02618941) [63, 93, 94].
Another AFF® antibody, AFF® PD03A, was evaluated in a similar manner as PD01A vaccination in patients with early PD and MSA in a separate clinical trial (NCT02267434), with a 36-week duration and injection doses ranging from 15 μg to 75 μg [63, 94]. AFF® PD03A induced a sustained IgG antibody response against the PD03 peptide and demonstrated a favorable safety and tolerability profile in PD patients [63]. Both PD03A dosages were tolerated locally and systemically and induced prolonged anti-αSyn titers, with a 58% responder rate [63]. Over 98 patients were treated with either AFF® PD01A or PD03A and followed for up to 48 months (AFF PD01A) or 12 months (AFF PD03A) [62, 63, 94]. Both treatments elicited an antibody response, with PD01A showing a more significant response than PD03A. AFF PD01A demonstrated safety and tolerability in early-stage PD patients and appeared to reduce α-Syn levels in the blood. However, the larger phase II trial did not show a significant difference in disease progression between the vaccine and placebo groups [63, 93].
United neuroscience: UB-312
UB‐312 is a synthetic peptide vaccine developed to overcome immune tolerance and generate a humoral antibody response to target oligomeric and fibrillary α-Syn. As an active vaccination, UB-312 induces an immune response against the aggregated form of α-Syn [48, 64, 95]. UB-312 targets the 10-AA fragment of the protein’s C-terminus fused to a short peptide that stimulates T-helper cells, which is then mixed with adjuvant for intramuscular injection [42, 49]. The United Biomedical, Inc. and United Neuroscience Ltd. compounds UB-312 in PD were designed based on UBITh technology on the related AD vaccine UB-311 [41, 64, 96]. The so-called UBITh® approach produced the United Biomedical, Inc. and United Neuroscience Ltd. drugs UB-312 for PD and MSA disorders [43, 97]. Vaccines developed using this technology are made up of heterologous T-helper cell epitopes linked to a short B-cell epitope as an antigenic target site either directly or indirectly via a heterologous spacer [42, 48, 49]. The synthetic T-helper cell determinant stimulates an immunological response that produces many antibodies directed against the target [9, 42, 48, 49]. In the first preclinical study, researchers used guinea pigs and a 12-AA precursor of UB-312-induced antibodies against the toxic form of α-Syn fibrils and oligos but not monomers [42, 49, 50].
Immunohistochemical staining was performed on human cases of PD, DLB, and MSA using guinea pigs immunized with the UB-312 vaccine and IgG fractions from the resulting immune sera (IgG-1, IgG-2, or IgG-3). The immunoreactivity results were compared to a commercially available α-Syn antibody from Novacastra (NOV), commonly used for diagnostic purposes [42, 49]. The vaccine-generated antibodies showed higher detection of α-Syn pathology compared to NOV, with IgG-3 exhibiting the highest levels of α-Syn recognition. These antibodies were able to detect α-Syn inclusions in the SN and basal ganglia of patients with PD, DLB, or MSA. Slot blot analysis confirmed the antibodies’ specificity for native oligos and fibrillar α-Syn [42, 49].
In a study involving young tg (Thy1SNCA/15) and wildtype (wt) mice, a 6-week immunization regimen followed by a 9-week observation period was conducted [48]. Behavioral assessment was performed before immunization and 15 weeks after the first dose. Injection of UB-312 in 10-week-old line 61 tg mice resulted in reduced α-Syn oligos in the brain, hippocampus, and striatum (Table 1) [50]. Vaccinated mice exhibited improved performance in motor tests, such as crossing a beam and hanging from a wire (Table 1) [48, 50]. Therefore, UB-312 was found to bind α-Syn oligos and fibrils in post-mortem brain tissue from PD, MSA, and DLB patients. However, the efficacy of UB-312 in animal models of these disorders was not assessed in preclinical research studies [42, 48–50].
The main goal of phase I clinical trial was to assess the safety, tolerability, and immunogenicity of UB-312 vaccination in healthy participants [64]. Fifty healthy participants were enrolled in a 44-week, randomized, placebo-controlled, double-blind study (NCT04075318). Participants in seven cohorts were assigned to three intramuscular UB312 or placebo injections (40–2,000 μg) [64]. Safety and tolerability were assessed by adverse events, clinical laboratory, vital signs, electrocardiograms (ECGs), and neurological and physical examinations [64]. Immunogenicity was assessed by measuring serum and CSF anti-α-Syn antibody concentrations [64]. This phase I study in healthy volunteers revealed that UB-312 was generally safe, well tolerated, with no concerning safety findings, and immunogenic, with a high responder rate, antibodies pass the BBB, and induced anti-β-Syn antibodies in serum and CSF of all participants receiving the 300/300/300 μg dose regimen (Table 2) [64]. The company has reported that all patients have completed the vaccination series, and full results are expected by mid-2023. Part A of the UB-312 investigation was a dosage escalation period in which the optimum dose was determined, followed by a safety and tolerability review period in part B. Based on clinicaltrials.gov, in December 2022, researchers at NYU Langone Health registered a phase IB experiment in which four participants with MSA and four with PD will get three priming and five booster injections of 300 g UB-312 over two years [95]. Approximately 6 months following the previous boost, serum and CSF levels of anti-α-Syn antibodies are measured. The project’s estimated start date is April 2023, and the estimated completion date is scheduled for April 2025 [95].
There was no discernible difference between the treatment and placebo groups while using AFF PD01A, and the reason behind this is still unresolved. The organization has not indicated whether it would continue with a more modified therapy. On the other hand, in the phase IA study, which examined safety and tolerability, the UB-312 demonstrated good results. The company plans to begin this year with phase IB, which will treat four people with MSA and four patients with PD. Although the precise future of these active therapies is unknown, it is crucial to be aware that a therapy candidate’s success or failure in clinical trials can be impacted by a number of variables, including effectiveness, safety, tolerability, and the particular endpoints being evaluated. The decision to discontinue or change the development of these candidates can be influenced by a variety of factors, including insufficient effectiveness, unforeseen safety concerns, or strategic decisions made by the sponsor companies.
Passive immunization
Passive immunity refers to the production of IgG antibodies that offer protection against infections [74]. It can be categorized as natural or acquired, with natural passive immunity lasting for weeks or months until the antibodies degrade [86]. Acquired passive immunity involves collecting serum from immune individuals, pooling, and concentrating it, and injecting it into the patient to provide protection [74]. In the context of passive immunization for NDDs, engineered antibodies targeting specific α-Syn domains are produced externally and then administered through injection [33]. These antibodies disperse in the bloodstream and function similarly to endogenous antibodies [33]. In the treatment of PD, patients receive anti-α-Syn antibodies to bind with extracellular α-Syn and inhibit its spread [86].
Passive immunization offers several advantages, including the ability to precisely target disease-related proteins or pathology markers in neurodegenerative disorders. It has a rapid therapeutic effect upon administration, potentially leading to immediate symptom reduction or halting disease progression. Additionally, certain PI approaches involve engineered antibodies or immunological molecules designed to penetrate the BBB, allowing for direct therapeutic action within the brain. By targeting and reducing disease-associated proteins, PI can modify the underlying disease process in neurodegenerative disorders. Today, preclinical, and clinical studies have demonstrated favorable safety and tolerability profiles for PI [45, 51–60, 66–69, 86, 98–100].
Passive immunization strategies preclinical and clinical studies
Prothena-roche: PRX002
Masliah and colleagues [45, 51, 52] published the first study on passive α-Syn immunization in 2011. Several passive antibody therapies are being studied in clinical trials. Studies on prasinezumab (PRX002/RG7935), Lu AF82422, MEDI1341, ABBV-0805, and BIIB054/cinpanemab are now being studied in phase I–III clinical studies for passive immunization against PD and MSA [51, 86]. In the preclinical study, Masliah et al. [51] used a tg mouse model mThy1 (line 61) of PD overexpressing human wt α-Syn was injected with the mAb 9E4 (Table 1) [51]. The antibody binds to the C-terminus and prevents morphological changes that might lead to α-Syn oligomerization [51]. Over 6 months, 10 mg/kg of 9E4 was administered intravenously once a week (Table 1) [51]. There was a reduction in the buildup of calpain-cleaved α-Syn aggres in cortical and hippocampal neurons’ axons and synapses. Additionally, treated mice showed motor improvement on the pole and rotarod and improved cognitive impairment in the water maze when compared with IgG1-treated control animals [51]. In this study, 9E4 antibody was designed to target the C-terminus region of α-Syn since there is scientific evidence showing that α-Syn cleavage at the C-terminus is involved in the generation of toxic α-Syn oligos (Table 1) [51].
Prasinezumab (PRX002, RO7046015, RG7935, NEOD002), a humanized IgG1 mAb, targets aggregated α-Syn in the pathogenesis of PD and DLB. Following the promising result of the PRX002 mouse model with 9E4 antibody. Prothena began a single-center study in 40 healthy adults to compare the safety and pharmacokinetics (PK) of a single infusion of 0.3, 1, 3, 10, or 30 mg/kg to placebo [51, 52]. In March 2014, a phase I single ascending-dose (SAD) study of prasinezumab demonstrated that the antibody was safe, well-tolerated, and dose-dependently reduced the concentration of free α-Syn in the blood down to 4 percent with a maximal effect at a dose of 30 mg/kg (Table 2; NCT02157714) [65, 98].
Following the success of PRX002 in phase II clinical trials, Roche launched PASADENA. A two-year phase II trial evaluating the effectiveness of 1,500 mg or 4,500 mg prasinezumab infusions versus placebo was done in early PD patients (~316) [66]. The primary outcomes of this study included a decrease in the active arm’s part III motor scale Movement Disorder Society-Unified PD Rating Scale (MDS-UPDRS) on the second endpoint [66]. However, the most recent report on the 52-week phase II study found that therapy with PRX002/prasinezumab had no meaningful influence on key end measures compared to placebo expected to be completed in 2026 (NCT03100149) [66]. In May 2021, scientists from Roche began a phase IIB trial (PADOVA) in patients with more advanced PD symptoms than in PASADA (NCT04777331; Table 2). In the PADOVA study, 575 participants taking stable DA agonists received monthly injections of prasinezumab or a placebo for 18 months [67]. The primary outcome was significant advancement on the part III (MDS-UPDRS), defined as a drop of more than 5 points. The study, initiated in late 2022, underwent modifications to align with a confirmed motor advancement event [67]. It encompasses various secondary outcomes, including assessments of motor function, clinical changes, adverse events, PK, and anti-drug antibodies (ADAs). Spanning across 120 locations in North America and Europe, the trial is projected to extend until 2024 [67].
Biogen: BIIB054
Biogen’s mAb, BIIB054 (cinpanemab), binds to N-terminus (residues 1–10) of α-Syn without cross-reactivity to β- or γ-Syn [53, 54]. It is a recombinant human IgG1 antibody derived from the memory B cells of older adults without neurological disorders [54]. Previously, the same technological platform was utilized to create aducanumab, an antibody targeting aggres studied in AD therapy [54]. BIIB054 has an 800-fold greater affinity for fibrillar α-Syn than monomers [54]. Rats and cynomolgus monkeys were given a dose of 10 mg/kg to assess the PK properties (Table 1) [53]. It was shown that antibodies reached the CNS according to dose, peaking between 24 h and 72 h after injection [53]. In the study, wt, tg, and bacterial artificial chromosome (BAC) A53T mice were given injections of PFFs (2 L) [54]. BIIB054 (30 mg/kg) resulted in a 30% reduction of α-Syn spreading, a 20% reduction in the loss of DA transporter (DAT) density in dopaminergic terminals, and a reduced behavioral impairment in the wire hanging test by 50% (Table 1) [54]. Treatment was found to postpone the development of paralysis by 7 days (Table 1) [54]. Preclinical findings provided a strong argument for the clinical development of BIIB054 for the treatment and prevention of PD [54]. Targeting α-Syn by passive vaccination seems to be a promising disease-modifying treatment in animal PD models (Table 1). As a result, this method has been applied in human clinical studies.
A randomized, double-blind, placebo-controlled study tested the safety and PK of BIIB054/cinpanemab (NCT02459886) [68]. Initial cohort of BIIB054 was tested in healthy and mild to moderate PD participants using a single dosage of BIIB054 (15 mg/kg or 45 mg/kg) or a placebo [68]. For 16 weeks, participants underwent clinical, neuroimaging, ECG, and laboratory evaluations [68]. Serum and CSF ratios of BIIB054 concentrations were measured. It was similar to the result obtained in the preclinical studies (0.4%), with a favorable safety profile and the ability to pass the BBB [68]. According to Biogen, cinpanemab is more selective for aggregated than soluble α-Syn but binds soluble protein at high dosages. Overall, BIIB054 was found to be well tolerated, with no significant adverse effects reported (Table 2) [68]. Biogen started SPARK (NCT03318523) to examine the safety and PK of BIIB054/cinpanemab, a two-year phase II multicenter, randomized, double-blind, placebo-controlled trial [69]. Over two years, PD patients were given monthly doses of BIIB054 ranging from 250 mg to 3,500 mg and placebo [99]. The primary objectives of the SPARK research were changes and improvements in the MDS-UPDRS total score and DaT-SPECT changes from baseline to week 52 [69]. However, neither the primary nor secondary endpoints were met, and the study was therefore discontinued from further development [69]. Since BIIB054/cinpanemab binds to the N-terminus and prasinezumab binds to the C-terminus of α-Syn, cinpanemanb results possess less favorable outcomes than prasinezumab.
The clinical trials followed substantial preclinical research in rodent models employing cinpanemab and prasinezumab, which revealed significant reductions in PD-like symptoms. Both studies enrolled patients in the early stages of PD, recognizing the limited potential for reversing downstream pathological processes in late-stage PD, even with the stoppage of basic pathogenic mechanisms [67–69]. In conclusion, while the studies involving α-Syn mAbs (cinpanemab and prasinezumab) did not show efficacy in early PD, the findings highlight the need to reconsider the potential limitations of preclinical studies and clinical trials, particularly the translation from experimental models to endpoint evaluations [67–69]. Incorporating developing technology may aid in overcoming the problems of α-Syn-based immunotherapy. Despite the detrimental results, it would be premature to terminate the investigation of these therapeutic techniques.
AstraZeneca: MEDI1341
MEDI1341 is an α-Syn mAb distinguished by its high affinity and specificity [55]. MEDI1341 possesses therapeutic properties by binding to the C-terminus of monomeric and aggregated α-Syn, most likely in the epitope at AA 103–129 [55]. In in vitro model (SH-SY5Y cells), the antibody inhibited cell-to-cell transmission and spreading of human α-Syn PFF [55]. Preclinical testing of MEDI1341 demonstrated rapid infiltration in the brain and reduced free α-Syn unblocked α-Syn propagation [55]. Intravenous administration of MEDI1341 to rats at 100 mg/kg showed a decrease in CSF α-Syn levels by 81.5% at peak, with the drop remaining at 45% after 4 days, and decreased brain interstitial fluid (ISF) levels by 75% after intravenous administration (Table 1) [55]. In a series of target engagement tests, MEDI1341 was examined for its ability to enter and capture extracellular α-Syn in the CNS [55]. MEDI1341 was also administered intravenously to cynomolgus monkeys and resulted in measurable amounts in the CNS, although at 0.2% of serum levels, up to 88% less free α-Syn was present in the CSF (Table 1) [55]. The combination of characteristics and properties of MEDI1341 mentioned above are novel and different from other α-Syn antibodies currently in human clinical trials. AstraZeneca’s MEDI1341/TAK-341 is currently in early clinical development. Recently, two phase I clinical research is conducted to examine the safety, tolerability, and PK properties of MEDI1341 and its capacity to sequester α-Syn in the CNS [55].
In the study, SAD was administrated in healthy volunteers and multiple ascending-dose in patients with PD (NCT04449484, NCT03272165; Table 2) [55]. The aim of phase I study was to evaluate safety and tolerability, PK, and pharmacodynamics of SAD of MEDI1341. In October 2017, AstraZeneca initiated phase I with 36–48 healthy volunteers (Table 2). In August 2019, AstraZeneca changed qualifying criteria to exclude participants with severe eye disease and added vision and eye health assessment to the critical objectives. From August 2020 to January 2022, AstraZeneca conducted a second phase I study in patients with PD, evaluating several ascending dosages. Takeda took over development in November 2022, with a phase II study in 138 patients with MSA. After one year, the study outcome is a change in the Unified MSA Rating Scale part 1. Secondary results include PK, other clinical indicators, survival time, adverse events, and ADAs. No data disclosed has been released.
At the time of this review, no data from the study had been released. These data imply that MEDI1341 offers promise as a potential therapeutic option for α-Syn-related illnesses, and further clinical research will provide more information about its safety, effectiveness, and disease-modifying effects.
AbbVie: ABBV-0805
AN0805/ABBV-0805, also known as BAN0805, is a humanized mAb targeting α-Syn oligos and protofibrils at the C-terminus [58]. Preclinical investigations studied the effects of ABBV-0805 on two mouse models of PD, one expressing human α-Syn and the other expressing A53T mutant human α-Syn (Table 1) [58]. Mice were treated with ABBV-0805 at a range of 0.1, 1, 10, and 20 mg/kg or control for 2–4 months; treatment was assessed by measuring α-Syn levels, motor function, and survival (Table 1) [58]. ABBV-0805 decreased the level of α-Syn fibrils, prevented motor impairment, and extended longevity; the antibody generally protected against α-Syn toxicity [58]. These findings suggest that ABBV-0805, a novel antibody selective for soluble aggregated α-Syn, has therapeutic potential for PD [58]. Further research is needed to determine the long-term effects and safety of ABBV-0805 in humans [57, 58]. Lindström and colleagues [56] created a novel α-Syn protofibril-specific mAb47 similar to ABBV-0805.
The antibody mAb47 was evaluated in 14-month-old A30P tg mutant mice under the Thy1 promoter, resulting in the formation of α-Syn fibrils (Table 1) [56]. Mice were treated for 14 weeks with a weekly intraperitoneal injection. mAb47 decreased soluble α-Syn protofibril levels in the spinal cord without changing the level of monomeric forms and decreased motor dysfunction compared with untreated mice [56, 57]. Although levels of α-Syn protofibril, monomeric, and fibrillar were decreased, there was no difference in levels across groups in the brain and spinal cord tissue [56, 57]. In a similar study, Thy1 α-Syn mice were weekly immunized with specific antibodies (Syn-O1, Syn-O4, and Syn-F1) that exclusively recognized oligos and fibrils but not monomeric α-Syn to prevent accumulation and synaptic alterations, resulting in reduced astrogliosis and microglia activation in the hippocampus [100]. In 2019 AbbVie, in partnership with BioArtic, began a phase I trial of ABBV-0805 in healthy volunteers (Table 2). Phase I data from AbbVie on PK excellent tolerance and safety supported moving forward to phase II. During placebo-controlled phase IB with 32 idiopathic, mild to moderate PD patients, ABBV-0805 was withdrawn by the firm in July 2020, citing strategic considerations. At the time of this review, no results from the trial have been published yet. It was planned to randomly assign them to receive three increasing doses of the antibody, or a placebo administered intravenously every 28 days, with a fourth dosage added pending preliminary findings (Table 2; NCT04127695). Although ABBV-0805 showed promising preclinical results, further clinical investigations and trials are necessary to fully understand its efficacy, safety, and potential as a therapeutic option for PD.
H. Lundbeck: Lu AF82422
The humanized mAb IgG1 Lu AF82422 targets the C and N-terminus of all forms of α-Syn [59]. Blood cells present α-Syn, yet most human blood cell types do not bind to the antibody [58]. A nonclinical study was completed in rats and cynomolgus monkeys at dosages up to 600 mg/kg delivered intravenously every 10 days to support a first-in-human (FIH) SAD trial with Lu AF82422 (Table 1) [59]. No-observed-adverse-effect met the criteria at the highest dosage tested, and there were no treatment-related side effects. A single dose showed PK parameters in the predicted range for mAb with a soluble target and target engagement in plasma and CSF [59]. A preclinical study showed enough safety margins between documented safe doses in animals and the intended dosage levels in the FIH SAD study [59]. The study concluded that Lu AF82422 represents a promising therapeutic approach for synucleinopathies and further clinical investigation [59]. In July 2018, Lundbeck initiated a phase I SAD trial with Lu AF82422 in healthy non-Japanese and Japanese subjects and patients with PD (NCT03611569; Table 2). Participants were given a single dose of 50 mg/mL or a placebo by infusion, followed by a 12-week monitoring period. Although no findings were provided, the manufacturer classified this antibody as well tolerated. Lundbeck announced the AMULET trial, which will confirm the efficacy of Lu AF82422 as a therapy to slow the rate of disease development in 64 patients with MSA. Lu AF82422 will be administered intravenously once every four weeks for a maximum of 72 weeks (NCT05104476). Finally, the humanized mAb Lu AF82422 targets multiple forms of α-Syn and has demonstrated encouraging preclinical outcomes. Nonclinical trials revealed safety and efficacy, paving the way for additional clinical research. A phase I experiment with Lu AF82422 in healthy persons and PD patients showed satisfactory tolerability. The AMULET trial appears to be the next step in evaluating the efficacy of Lu AF82422 in reducing disease progression in patients with MSA.
Numerous preclinical investigations have revealed passive immunization mechanisms with significant results that have yet to be tested in clinical trials. PDGFb, a tg mouse, was used to test the C-terminus antibody (Ab274; Table 1) [47]. The passively immunized treated group improved behavioral impairments and neurodegeneration [47]. After 4 weeks of therapy, Ab274 successfully reduce aggregated α-Syn transmission (Table 1) [47]. Similarly, preincubating recombinant α-Syn with 1H7 and 5C1 reduced C-terminus cleavage in the mThy1 α-Syn tg mice model [45]. mAbs 1H7, 5C1, and 5D12 were also particularly effective in reducing C-terminus-truncated α-Syn and higher-molecular-weight aggre (Table 1) [45, 52, 91]. The effect of passive immunization against aggregated α-Syn was studied in a mouse model of axonal transit and accumulation of α-Syn. Non-tg, α-Syn knockout, and mThy1 α-Syn tg mice were given unilateral intracerebral injections of a lentiviral (LV)-α-Syn vector construct, followed by systemic treatment with the mAb 1H7 (which recognizes AA 91–99) or control IgG, for 3 months (Table 1) [45, 52, 91]. 1H7 antibody decreased axonal accumulation and improved behavioral abnormalities, suggesting immunotherapy may help synucleinopathy models with behavioral impairment [52]. Overall, Games et al. [45], Masliah et al. [51], and Spencer et al. [52] discovered antibody variants, such as 1H7 and 5C1, specifically targeting the C-terminus of α-Syn, blocking calpain-mediated cleavage and hindered the spread of aggre α-Syn species from cell-to-cell [45, 51, 52].
Anti-α-Syn antibodies that are N-terminally directed have also been created and tested in a PD rat model. The model was created by injecting a recombinant adeno-associated viral (rAAV) vector expressing human wt α-Syn under the control of the chicken β-actin (CBA) promoter into the skin (Table 1) [60]. Control IgG or antibodies against the N-terminus (AB1) or central region (AB2) of α-Syn were administrated intraperitoneally to rats every 2 weeks for 3 months. AB1 and AB2 resulted in a substantial but not statistically significant improvement in behavioral measures and strong protection against the death of nigral dopaminergic neurons [60]. Overall, α-Syn expression and microglial activity in the nigral area were reduced (Table 1) [60]. Syn303 and Syn211, two N-terminus antibodies, were designed to target misfolded α-Syn in a mouse model after being injected with PFFs α-Syn (Table 1) [61]. Weekly intraperitoneal treatment for up to 26 weeks prevented the spread of α-Syn aggres and alleviated behavioral impairments and dopaminergic cell loss (Table 1) [61]. These mAbs could bind to aggregated α-Syn at the N-terminus to promote clearance of extracellular α-Syn and mitigate its pathological consequences (Table 1) [60, 61]. Additional research on PI for synucleinopathies is being conducted, and phase I clinical studies assessing safety and acceptability have been completed. Phase I study on people living with PD and healthy males began in December 2020 by UCB Biopharma/Novartis (Table 2). The trial will enlist 64 patients in the UK and evaluate the safety, tolerability, and PK of single- and multiple-ascending doses of UCB7853 infusion. The trial’s completion date was moved from September 2022 to April 2023 after UCB suspended enrollment in June 2021 (NCT04651153). Early clinical trials suggest that immunotherapy is often well tolerated. The CSF contains much fewer mAbs than the plasma, which may limit their efficacy (Table 2). Overall, PI targeting α-Syn, such as the one stated above, have shown promise in preclinical and early clinical research. More research and clinical trials are required to determine their long-term efficacy and safety in the treatment of synucleinopathies including PD and MSA.
Discussion
The accumulation of α-Syn aggres in PD is known to contribute to neurodegeneration by disrupting normal cellular processes, impairing protein clearance mechanisms, and inducing cellular toxicity. This accumulation leads to the dysfunction and degeneration of dopaminergic neurons in the SN region of the brain, resulting in the motor symptoms characteristic of PD, such as tremors, rigidity, and bradykinesia. Additionally, α-Syn pathology can spread throughout the brain, affecting other regions, and contributing to non-motor symptoms as the disease progresses.
Ongoing immunotherapy research and improvements strive to refine and integrate these techniques to maximize therapeutic efficacy in various diseases and patient populations. The methods of action and roles in influencing the immune response differ between active and PI. The perception that active immunotherapies (AIs) are less potent than PIs can be influenced by various factors, including the mode of stimulation compared to direct delivery, timing and persistence of the immune response, target specificity, and outcomes observed in clinical trials. It is important to acknowledge that both AI and PI have their advantages and limitations. AIs have the potential for long-term effects, immunological memory formation, and broader targeting capabilities. On the other hand, PIs offer immediate and potent therapeutic effects. In preclinical trials, both treatments showed promising results in decreasing α-Syn aggres and enhancing motor functions and cognitive performance in animal models [44, 45, 47, 52, 61, 91]. However, it is important to emphasize that clinical trials of several immunotherapeutic drugs targeting α-Syn have shown disparate results in clinical trials. For example, BIIB054/cinpanemab, which targets the N-terminus of α-Syn, showed negative results in a significant phase II study, while PRX002/prasinezumab, targeting the C-terminus, demonstrated positive results in secondary endpoints, leading to the initiation of a phase IIB study [54, 66–68]. Several factors, including differences in binding sites and individual antibody properties and responses, may account for these disparities and underscore the importance of careful evaluation in determining successful clinical therapies.
Several antibodies, including PRX002/prasinezumab, BIIB054/cinpanemab, MEDI1341, ABBV-0805, and Lu AF82422, have been developed with higher affinities for aggregated forms of α-Syn [54, 55, 58, 59, 67, 68]. A practical immunotherapeutic approach may involve inhibiting cellular uptake, preventing cell-to-cell transmission, and sequestering α-Syn to reduce oligomerization. For example, the antibody Lu AF82422 targets various forms of α-Syn, including monomeric and oligomeric species, as well as N- and C-terminal truncated forms [59]. Immunotherapies for α-Syn are also being investigated in terms of their ability to block the synthesis of toxic α-Syn species by binding to the exact cleavage sites as proteases. However, further research is needed to understand the reasons behind the varying outcomes observed in clinical trials. Additionally, it remains uncertain whether conventional vaccination or other inactive immunotherapies effectively promote cellular immunity or have intracellular effects. Exploring alternative methods beyond antibody-based therapies may be crucial in achieving intracellular immunity against aggregated forms of α-Syn.
While significant progress has been made in the development of immunotherapies targeting α-Syn, it is important to recognize that many of these treatments are still in the early stages of development and require further evaluation in clinical trials to determine their safety and effectiveness in treating α-Syn-related diseases. As the field of immunotherapy continues to evolve, new candidate therapies may emerge. Ongoing research efforts aim to optimize and integrate these approaches to enhance therapeutic efficacy across different diseases and patient populations. Comprehensive investigations and careful considerations are necessary to address the complexities and challenges associated with antibody-based therapy in PD. In conclusion, substantial strides have been made in the development of immunotherapies targeting α-Syn in PD. However, it is crucial to conduct further research and clinical trials to validate the safety and efficacy of these treatments. The potential of immunotherapy to effectively reduce the burden of α-Syn pathology holds promise for the future of PD treatment.
Challenges and limitations in α-Syn immunotherapies
Despite extensive research on α-Syn, the optimal molecular structure to target in PD remains uncertain [101]. This ambiguity arises from the ability of α-Syn to assume diverse conformations across different individuals. Additionally, the absence of reliable biomarkers poses a significant challenge in enabling early detection and intervention strategies. What is known is that treating α-Syn in PD patients is an appealing target mechanism for interfering with aggre formation, reducing the level of seeds or substrate for fibrillization. PD preclinical studies have demonstrated that α-Syn inclines to prion-like behavior, even when compared to other amyloidogenic proteins. Despite repeated failures with immunotherapeutic initiatives against AD, antibody-based therapeutics may yet be a therapy focus for treating synucleinopathies.
Several active and PI approaches targeting α-Syn are currently in clinical trials. However, no strategy has yet been submitted to clinical phase III as those for AD. One of the noted concerns is that the genesis of PD is complex and not entirely understood. Considerations for immunotherapy in synucleinopathies are confounded by various aspects, and these limitations must be addressed.
The most significant challenges to α-Syn immunotherapy drugs are target specificity. Achieving specific targeting of pathological α-Syn aggres while sparing normal, physiological forms of the protein is crucial. Additionally, the limited knowledge of intracellular α-Syn aggregation and how the seed propagates in the brain is a significant challenge in the field. There is a lack of reliable biomarkers to evaluate the α-Syn proteopathic in synucleopathies. Establishing defined disease biomarkers, particularly pathologic α-Syn deposition will considerably improve our capacity to identify risk patients in the primary stage of the disease and treat with efficient and effective treatments to prevent progression to clinically apparent disease. α-Syn biomarkers have little clinical validation and may not be applicable to all PD patients, negatively affecting effective target engagement. Ensuring that immunotherapies selectively bind to pathological species and do not interfere with the normal function of α-Syn is a challenge that requires careful design and characterization of therapeutic antibodies.
Another factor to consider is the limited BBB penetration of antibodies, including those delivered passively and those produced in the body following AI. Developing strategies to enhance the delivery of therapeutic antibodies across the BBB is a significant challenge in the field. The accessibility of less permeable regions might be circumvented by lowering the molecular weight of the antibodies, preparing smaller antibody fragments, or engineering antibodies to enhance brain penetration. Passive antibody treatments should be tested in numerous models of synucleinopathy to validate target engagement across assembly conformations or aggregation paradigms because mutation-driven variability in α-Syn assemblies can manifest differently in clinical outcomes. Additionally, contrary to positron emission tomography (PET) imaging of Aβ and tau, clinical imaging of synucleinopathy has not produced any clinically meaningful structures, even though some approaches have indicated possible clinical implications. Effectively monitoring the α-Syn disease situation and prospective target engagement for anti-α-Syn therapeutic strategies may depend on non-invasive imaging methods such as an α-Syn PET tracer. The resolution of current imaging techniques for patients at the late stage of the disease with α-Syn burdens is probably insufficient to extrapolate clinical achievements.
Antibodies’ inability to distinguish between monomeric and aggregated fibril forms and the possibility of inflammatory autoimmunity are significant pitfalls that might occur. The most crucial aspects of PD immunotherapy should be reducing the inflammatory response and, more effectively, bridging the BBB. To successfully target the intracellular α-Syn of neurons, nanoantibodies should be further researched, and antibodies should be produced to pass this barrier more readily. Moreover, PD patients in the early stages of the disease must be chosen for clinical trials to assess the effect of immunotherapy. These insights can also be drawn from research on immunotherapy for AD. For a long time, α-Syn aggregation has been observed in PD, complicating diagnosis. It is crucial to consider the effect on the patient if targeted, as it may not be adequate to prevent or slow the progression of PD when combined with other proteinopathies.
Furthermore, aggregated α-Syn can vary depending on the condition, implying that special immunotherapy for PD may not respond as well for other synucleinopathies. Further research is necessary to clarify these discrepancies. Antibodies must be selective and efficient for the targeting of aggregated α-Syn, and if these precautions are implemented, there is a probability that there will be a breakthrough soon.
Other challenges and limitations include the potential immunogenicity of immunotherapeutic agents is an important consideration. Repeated administration of therapeutic antibodies may lead to the production of ADAs in some patients, which can reduce the effectiveness and safety of the treatment. Managing and minimizing immunogenicity is a key challenge in the development of α-Syn immunotherapies. Multifunctional immunotherapy designs targeting several proteins may substantially lower the proteostatic burden and diminish reciprocal interactions between toxic aggregation forms. Non-α-Syn proteinopathy treatment may be sufficient to resist Lewy pathology. Combinatorial therapies may include anti-inflammatory and immunomodulatory therapy as well as anti-α-Syn immunotherapeutic. Clinical approval for such strategies will need sufficient safety data, which may only be possible with better foundational principles in disease progress assessment. Some other commenters on this thread have noted that the reason for therapeutic failures in AD is the timeframe of interventions concerning the moving target of misfolded proteins and propagation. Identifying the optimal timing and stage of the disease for initiating immunotherapy is a challenge. The effectiveness of immunotherapies may vary depending on the disease progression and the extent of α-Syn pathology. Determining the appropriate patient population and disease stage for intervention is critical to maximize therapeutic benefits. It is worth noting the long-term safety and efficacy of therapies. Long-term safety and efficacy data for α-Syn immunotherapies are still limited. Understanding the potential side effects and monitoring the durability of treatment effects over extended periods is essential. Long-term studies and post-marketing surveillance are necessary to assess the sustained benefits and safety profiles of these therapies. Preclinical models, erroneous study population, improper study design, aggregation hypothesis, inadequate disease pathways, and the complexity of NDDs all contribute to the lack of efficacy of present treatments.
Besides the aforementioned challenges and limitations, species differences are topics of consideration and have been an area of active research. PD is a complex disorder with diverse mechanisms and clinical presentations, which animal models only partially replicate. This makes accurate evaluation of immunotherapy efficacy and safety challenging. Additionally, significant species differences in genetics, immune system function, and disease progression can affect the response to immunotherapies and limit the applicability of results from animal studies to humans. Immunological variations between animals and humans further complicate interpretation, as immune responses may differ. Most of the preclinical study is based on mouse disease models, which are influenced by the neuropathological traits of NDDs. Specific genes are typically altered to establish relationships between neuropathological characteristics or protein species and functional abnormalities in animal models. It is important to notice that studies on mice have consistently and accurately predicted immunotherapy’s neuropathological, PK, and dynamic effects. However, the important outcome measures of clinical trials, based on cognitive scores, are not reflected in neuropathological findings. Several preclinical studies and behavioral and functional studies of immunotherapy in mice have been disregarded. However, with relatively moderate effects seen in clinical studies, better cognition in mice has not been directly translated to people. Additionally, ethical considerations also impose constraints on animal research. Researchers are actively working to develop more representative animal models and exploring alternative techniques like in vitro and computational modeling to enhance PD immunotherapy research.
In the past decade, significant advancements have been made in understanding how NDDs profoundly alter the immune system’s response and behavioral patterns. Designing robust clinical trials for α-Syn immunotherapies presents challenges due to the complexities of PD, the heterogeneity of patients, and the need for appropriate biomarkers to assess treatment response. Defining meaningful clinical endpoints, optimizing trial duration, and selecting suitable outcome measures are critical aspects that require careful consideration. Addressing these challenges and limitations is essential for the successful development and deployment of α-Syn immunotherapies. Continued research, collaboration, and innovation in the field will contribute to overcoming these hurdles and advancing the potential of immunotherapeutic approaches for PD.
Overall, extensive research on α-Syn in PD has yet to determine the optimal molecular structure to target, as α-Syn can assume diverse conformations across individuals. The absence of reliable biomarkers poses a challenge for early detection and intervention. However, targeting α-Syn in PD patients shows promise for interfering with aggre formation and reducing fibrillization substrates. Despite failures in immunotherapeutic initiatives against AD, antibody-based therapeutics remain a focus for synucleinopathies. Several α-Syn immunotherapy approaches are in clinical trials, but no phase III strategy has been submitted yet. Challenges include target specificity, limited understanding of intracellular aggregation and propagation, and the lack of reliable biomarkers. Ensuring immunotherapies selectively bind to pathological species, enhancing BBB penetration, and validating target engagement are key challenges. Considerations for managing immunogenicity, identifying optimal timing and patient population for intervention, and assessing long-term safety and efficacy are important. Animal models have limitations in replicating PD complexity, and ethical considerations impact animal research. Robust clinical trial design, meaningful endpoints, and collaboration are necessary to overcome these challenges and advance α-Syn immunotherapies for PD.
Conclusions
Different immunotherapies (active and passive) have been shown to benefit neuropathological and behavioral indicators using experimental models of PD and AD. According to earlier studies in Tables 1 and 2 [44–48, 50–61, 63, 64], active immunization reduces aggregation, improves motor deficits, modifies neuroimmune responses, and encourages neuroprotection. At the same time, passive immunization against pathogenic α-Syn species may improve pathological protein clearance and prevent neurodegeneration. Most research on α-Syn aggregation and transmission has been conducted in animal models. It is still debatable whether a similar process exists in humans. Creating animal models that closely resemble most neuropathological traits identified in patients is crucial to extrapolate human results from immunotherapy more accurately. Thus, further research is needed to understand if and how antibodies work in the human brain to provide neuroprotection. Due to this, mouse models are being developed, including humanized mice, double- and triple-tg mice to simulate the various proteinopathies that exist in humans, and double- and triple-tg mice to mimic the human state of disease. From our perspective, a greater understanding of disease courses would make pinpointing therapeutic windows much more effective for NDD intervention.
Another factor to consider in future research is the inability of antibodies to cross the BBB. Furthermore, the delivery of real-time immunotherapy will be necessary. Immunotherapy should begin considerably earlier than now while the brain adapts to abnormal conditions. Diagnostic biomarkers are needed to identify people at risk of developing the disease before the neurodegenerative process begins. PD becomes clinically apparent when 80 percent of dopaminergic neurons in the SN are lost owing to neurodegeneration. Like AD trials, selecting appropriate patients and timing to begin treatment will be critical. As our understanding of PD causation and treatment grows, it will become clear whether immunization targeting α-Syn is viable. Some of the research mentioned in this review, for example, did not show the efficacy of α-Syn antibodies in PD, although upcoming technology may help address the issues.
Abbreviations
| AA: |
amino acid |
| AD: |
Alzheimer’s disease |
| ADAs: |
anti-drug antibodies |
| AFF: |
AFFITOPE |
| aggre: |
aggregate |
| AI: |
active immunotherapy |
| Aβ: |
amyloid-β |
| BBB: |
blood-brain barrier |
| CNS: |
central nervous system |
| CSF: |
cerebral spinal fluid |
| DA: |
dopamine |
| DLB: |
Dementia with Lewy bodies |
| IFN-β: |
interferon β |
| IgG: |
immunoglobulin G |
| IL-1β: |
interleukin 1β |
| LB: |
Lewy bodies |
| mAb47: |
monoclonal antibody 47 |
| MDS-UPDRS: |
Movement Disorder Society-Unified Parkinson’s Disease Rating Scale |
| MSA: |
multiple system atrophy |
| mThy1: |
murine thymus cell antigen 1 |
| NAC: |
non-amyloid component |
| NDDs: |
neurodegenerative diseases |
| oligo: |
oligomer |
| PD: |
Parkinson’s disease |
| PDGF: |
platelet-derived-growth-factor |
| PFFs: |
preformed fibrils |
| PI: |
passive immunotherapy |
| PK: |
pharmacokinetics |
| SAD: |
single ascending-dose |
| Ser: |
serine |
| SN: |
substantia nigra |
| tg: |
transgenic |
| TH: |
tyrosine hydroxylase |
| Thy1: |
thymus cell antigen 1 |
| TNF-α: |
tumor necrosis factor α |
| wt: |
wildtype |
| α-Syn: |
α-synuclein |
Declarations
Author contributions
GH: Writing—original draft. GH and MN: Writing—review & editing.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2023.