 Mini Review
Mini Review

Affiliation:
College of Medicine, California Northstate University, Elk Grove, CA 95757, United States
†These authors share the first authorship
ORCID: https://orcid.org/0009-0007-2551-3821
Affiliation:
College of Medicine, California Northstate University, Elk Grove, CA 95757, United States
†These authors share the first authorship
ORCID: https://orcid.org/0009-0006-2073-3764
Affiliation:
College of Medicine, California Northstate University, Elk Grove, CA 95757, United States
ORCID: https://orcid.org/0000-0002-8252-9092
Affiliation:
College of Medicine, California Northstate University, Elk Grove, CA 95757, United States
ORCID: https://orcid.org/0000-0002-0101-7037
Affiliation:
College of Medicine, California Northstate University, Elk Grove, CA 95757, United States
Email: forshing.lui@cnsu.edu
ORCID: https://orcid.org/0000-0003-4439-5414
Explor Neuroprot Ther. 2024;4:401–410 DOI: https://doi.org/10.37349/ent.2024.00091
Received: August 02, 2024 Accepted: September 25, 2024 Published: October 24, 2024
Academic Editor: Rafael Franco, Universidad de Barcelona, Spain
The article belongs to the special issue The Urgent Need for New Hypotheses to Develop Effective Therapeutic Tools Against Alzheimer's Disease
Alzheimer’s disease (AD) is the leading cause of dementia worldwide. The disease is characterized by the abnormal accumulation of amyloid β (Aβ) protein creating neuritic plaques, hyperphosphorylated tau (p-tau) protein forming intracellular tangles, and neuronal degeneration. Pathological changes related to abnormal Aβ and p-tau accumulation may begin more than fifteen years before the clinical diagnosis of AD is made. The glymphatic system is the brain’s waste clearance pathway that prevents the accumulation of these abnormal proteins and macromolecules. Glymphatic clearance is negatively affected by physiological conditions such as sleep deprivation, and pathological conditions such as traumatic brain injury and hemorrhagic strokes. These physiological and pathological conditions are strong risk factors for AD. In conclusion, impaired glymphatic clearance is an important pathogenetic mechanism for AD.
Alzheimer’s disease (AD) is the leading cause of dementia worldwide [1]. However, dementia’s pathophysiology involving impaired glymphatic clearance is a relatively novel concept. Sleep deprivation [2], advanced age [3], amyloid angiopathy [4], and genetic predispositions such as apolipoprotein E ε4 (APOE ε4) [5], are associated risk factors for impaired glymphatic clearance. Although more attention has been dedicated to glymphatics since its discovery in 2012, few studies provide an in-depth discussion of impaired glymphatic clearance as a potential risk factor for developing AD. In this mini-review, we aimed to summarize the current diagnostic criteria of AD and provided a brief overview of the glymphatic system, highlighting the significance of disrupted glymphatic flow as a critical mechanism leading to AD.
AD, the most common form of dementia, affects an estimated 416 million worldwide [1]. The diagnosis of AD is typically categorized into several stages based on the patient’s cognitive impairment and disability status. In the preclinical stage, individuals are typically asymptomatic but have laboratory evidence of AD pathology [6]. Leading to the mild cognitive impairment (MCI) stage, AD patients commonly experience a progressive decline in episodic memory and cognitive function [7]. As dementia advances, it often leads to language dysfunction, visuospatial difficulties, loss of insight, and personality changes such as withdrawal, decreased initiative, and occasionally depression [8].
AD pathology is characterized by an accumulation of abnormal neuritic plaques composed of amyloid β (Aβ) and neurofibrillary tangles containing hyperphosphorylated tau (p-tau) [6, 9]. Aβ is produced from a proteolytic breakdown of amyloid precursor protein (APP) [10], resulting in the formation of insoluble Aβ42, which contributes to the pathogenic development of amyloid plaques [11]. Meanwhile, p-tau is due to an imbalance in the function of several kinases and phosphates, such as reduced activity of protein phosphatase-2A (PP-2A) [12]. Aβ and p-tau operate in a feedback loop, where Aβ initiates the transformation of tau from a normal to a toxic state, and toxic tau can subsequently amplify Aβ toxicity [13]. The accumulation of abnormal neuritic plaques and neurofibrillary tangles leads to the damage of neurons and synapses, ultimately resulting in AD [14, 15].
Current diagnostic criteria for AD include both qualitative evaluation of clinical symptoms and quantitative assays. These include neurocognitive examinations, patient history, genetic testing, cerebrospinal fluid (CSF) biomarkers, and imaging. Changes in cognition and daily activities can be evaluated by the Mini-Mental State Examination (MMSE) [16], Clinical Dementia Rating-Sum of Boxes (CDR-SB) [16], and the AD cooperative study-activities of daily living MCI (ADCS-ALD-MCI) [17] which all prove to be accurate assessments to streamline the evaluation of neurocognitive deficits associated with AD across clinicians.
Significant reduction in CSF Aβ1–42 in AD patients [18], the elevation of CSF total tau (t-tau) and p-tau protein [19], and although less specific, elevation of p-tau181 and p-tau231 in blood plasma [20] compared to controls, have all been associated with accurately diagnosing AD quantitatively. Lastly, in addition to noting degenerative changes via magnetic resonance imaging (MRI), other imaging techniques using anti-amyloid dyes and positron emission tomography (PET) have been used to accurately and safely quantify the amount of Aβ in AD patients antemortem [21].
One of the most important non-modifiable risk factors for developing AD is age. It is estimated that 19% of adults aged 75 and older have been diagnosed with AD, increasing to up to 50% by age 85. Some studies have suggested a ‘continuum’ of progression from the normal aging brain to early and eventually advanced AD [22]. Genetics also play a key role, as the increased risk of developing AD has been consistently linked with the diagnosis of AD in a first-degree relative [22]. Certain variations in the APOE gene are considered the strongest genetic risk factors predisposing patients to sporadic late-onset AD, as the APOE protein is critical in the clearance of Aβ [23], and APOE directly interacts with soluble and fibrillar Aβ [24]. While the APOE ε2 allele is considered to be the most protective genetic factor against sporadic AD in both homozygotes and carriers versus the most common allele, APOE ε3 [25], the APOE ε4 allele confers a markedly increased risk of developing AD [26]. Specifically, APOE ε4 promotes Aβ fibril and oligomer aggregation and seeding [27], and its overexpression leads to increased APOE-Aβ complex formation, causing impaired Aβ clearance [23]. Autosomal dominant forms of AD usually present at a younger age (< 65 years) and are strongly associated with the mutated forms of presenilin 1 and 2 due to improper cleavage of APP [5]. As previously stated, in AD, APP is cleaved leading to overproduction of insoluble Aβ42 [11] and Aβ aggregation. Patients with Down syndrome, or trisomy 21, also have a high risk for developing AD as the APP gene is found on chromosome 21. By age 50, approximately 55% of patients with trisomy 21 will have dementia, and nearly all will have AD pathological changes in the brain [28]. Three of these four critical genetic risk factors, presenilin mutations 1, 2, and Down syndrome, cause increased production of Aβ.
Modifiable risk factors of AD include traumatic brain injury (TBI) and hemorrhagic strokes. There is a pathological link between TBI and dementia-related illnesses, such as AD. Previous studies have shown Aβ plaque deposition in postmortem brain tissue from patients who received fatal head injuries [29, 30]. Additionally, significantly increased p-tau can be found in the brain parenchyma following a single head injury during a patient’s lifetime, despite recovering from the incident [31]. It is postulated that both Aβ and p-tau are deposited around cerebral vasculature, disrupting the blood-brain barrier (BBB) and leading to neurocognitive deficits and the development of dementia [32]. These findings suggested that TBI leads to Aβ plaque and p-tau accumulation, compromising cerebral vasculature and leading to AD.
Another notable modifiable risk factor is sleep disturbance. Recent studies have implicated sleep disturbance not only as a marker of AD but also as a potential cause, as those with a history of poor sleep have a higher association with developing AD [33]. Additionally, patients with AD often have microhemorrhages in their brains, which can be detected using susceptibility-weighted (SWI) MRI neuroimaging. These microhemorrhages are the result of Aβ deposits in the cerebral vessels, resulting in cerebral amyloid angiopathy (CAA), a condition linked to AD [34]. Furthermore, other modifiable risk factors include diabetes, hypertension, dyslipidemia, obesity, and smoking [5]. Pathological changes of AD with Aβ deposition can be detected biochemically or on imaging years before an observable decline in cognition occurs [5]. Unlike genetic disorders, these modifiable risk factors appear to have little connection to increased Aβ production, suggesting a different physiological process is involved which we believe to be impaired glymphatic clearance.
Similar in function to the peripheral lymphatic system, the glymphatic system comprises a network of paravascular channels that facilitate the movement of CSF into brain parenchyma to deliver nutrients and neuroactive substances [35]. There are multiple anatomical barriers within the central nervous system (CNS) that prevent and allow the passage of various molecules and metabolites. One of these barriers is the well-established BBB which separates the blood from the cerebral parenchyma using tight junctions on endothelial cells. Recent literature highlighted another barrier known as the blood-CSF barrier (BCSFB) which separates blood from CSF [36]. The BCSFB is composed of epithelial cells that are strongly sealed together via tight junctions creating the choroid plexus [36]. This highly vascularized choroid plexus lining the ventricles of the brain is primarily accountable for creating CSF [37].
Once the CSF is produced by the choroid plexus, pulsatile para-arterial influx generated by cerebral blood flow from the pial artery, penetrating artery, to the Virchow-Robin space (VRS), propels CSF to enter the brain parenchyma [38]. CSF exchanges with interstitial fluid (ISF) through water channels, known as aquaporin-4 (AQP4) on astrocytic end feet. AQP4 channels are necessary for the interchanging of CSF with ISF and maintaining the polarity of glymphatic flow. After the fluid penetrates the brain interstitium, toxic metabolic byproducts and interstitial waste products, such as carbon dioxide, lactate, and proteins such as Aβ, are removed from the ISF in the cerebral parenchyma through para-venous efflux [39] (Figure 1) where it then connects and flows through the conventional peripheral lymphatic system via the cervical lymph nodes [40]. These AQP4 channels are critical proteins involved in maintaining glymphatic flow and are strongly associated with the pathogenesis of AD.
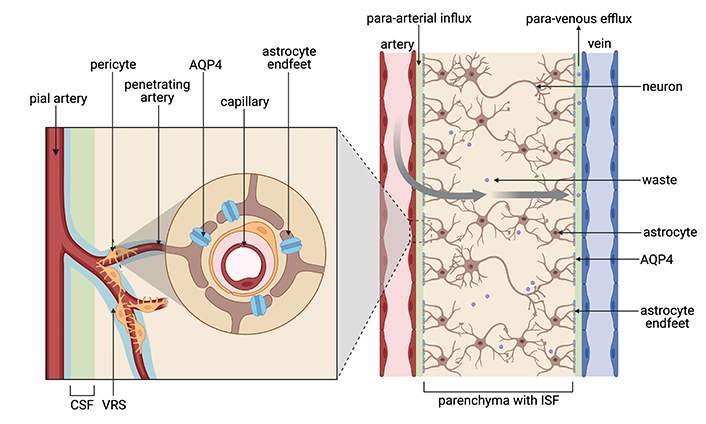
Diagram of the glymphatic system. Blood flow from the pial to the penetrating artery generates a para-arterial influx of CSF into the VRS to exchange with ISF in the cerebral parenchyma by route of AQP4 channels on the astrocyte end feet. Metabolic waste is then cleared from the mixed CSF-ISF fluid via the para-venous efflux. CSF: cerebrospinal fluid; VRS: Virchow-Robin space; ISF: interstitial fluid; AQP4: aquaporin-4. Created in BioRender. Hazzard, I. (2024) BioRender.com/o60i495
However, clearance of a substantial amount of neural metabolite requires several waste clearance mechanisms [39]. Aside from the glymphatic system, another CNS waste clearance mechanism is through meningeal lymphatics in which waste products enter the CSF through compartments rather than crossing the BBB to enter parenchymal circulation [39]. Meningeal lymphatic vessels lining the dura mater of the CNS are present along blood vasculature in mouse models, suggesting that CSF-ISF fluid and waste products enter meningeal lymphatic vessels [39]. This system is proposed to be the anatomical mediator between the glymphatic system and the peripheral lymphatic system such that the CSF-ISF filtrate from the glymphatic system drains into the meningeal lymphatic system to join with the peripheral lymphatic system [39, 41].
Another proposed pathway for waste clearance within the brain parenchyma is the intramural periarterial drainage (IPAD) pathway [42]. Rather than CSF exchanging and merging with ISF in the paravascular or VRS like in the glymphatic pathway, in the IPAD pathway, CSF and ISF merge in the subarachnoid space along the basement membrane of capillary and arterial walls to then drain into the cervical lymph nodes and blood [42].
Like in the IPAD pathway, the glymphatic system transports neurotoxic extracellular proteins such as Aβ and tau to the cervical lymph nodes [38]. Iliff et al. [38] provided strong evidence of this protein outflow after injecting radiolabeled Aβ1–40 into the mouse striatum. Fluorescent tagged Aβ was rapidly cleared from the brain in wild-type mice along the para-venous efflux route, however, clearance was significantly reduced in mice with an AQP4 deletion [38]. One manifestation of decreased glymphatic function is depolarized AQP4 channels leading to increased formation of senile plaques and neurofibrillary tangles caused by accumulated tau proteins [43]. In other words, depolarized AQP4 channels are defective and non-functional similar to being eliminated in Iliff’s mouse study. Therefore, the depolarization of AQP4 leads to insufficient clearance of soluble Aβ, reinforcing the importance of the glymphatic system for removing Aβ from the CNS [38]. In AD, depolarized and therefore defective AQP4 channels, normally expressed in astrocytes’ end feet (Figure 1) are instead distributed throughout the astrocyte cell body in the cerebral parenchyma leading to dysregulation of glymphatic flow [3]. This mislocalization reduces the glymphatic system’s clearance efficiency, impeding fluid exchange and leaving traces of toxic protein monomers, oligomers, and aggregates within the cerebral parenchyma contributing to AD [3].
Glymphatic dysfunction, such as the disruption of glymphatic flow, significantly precedes the appearance of misfolded Aβ and fibrillary tangles. This indicates the early development of neurode-generative diseases by leading to the accumulation of harmful substances, most notably, Aβ accumulation and tau pathology in AD [13]. As a result, we postulate that impaired glymphatic clearance leads to neurotoxic amyloid plaque buildup and disruption of synaptic function, contributing to the cognitive decline and progression of AD. Thus, it is critical to maintain glymphatic circulation to prevent the development of neurological diseases such as AD.
During sleep, the rate of glymphatic clearance is significantly amplified to remove potentially harmful metabolites. The interstitial space increases and allows greater fluid movement with more efficient clearance of waste, which is essential for maintaining brain health, preventing the accumulation of potentially toxic substances, and preserving overall brain homeostasis [3]. Since the glymphatic system is most active during sleep, both chronic and acute sleep deprivation cause waste accumulation in the brain. One particularly notable study demonstrated only one night of sleep deprivation causes significant Aβ accumulation [2]. Consequently, repeated instances of missed sleep can cause the buildup of toxic metabolites in the brain.
Age is an additional factor affecting glymphatic clearance. It is estimated that renewal of CSF in young adults occurs four to five times a day but decreases to two to three times in the elderly [44]. Pulsatile forces from the vessels in the brain, which aid in driving CSF into the brain parenchyma, weaken over time due to arteriosclerosis, further decreasing filtration [3]. As elderly populations have the highest instances of AD, impairment of the glymphatic system may play an important role in contributing to its development. Previous studies demonstrated that in the glymphatic pathway, para-arterial influx, and AQP4 polarization are impaired in the aging brain [45] further demonstrating aging as a risk factor for impaired glymphatic clearance.
In addition to APOE being the primary genetic risk factor of AD, it is also associated with impaired glymphatics. In the immunohistochemistry of mice brains, APOE proteins were observed to be concentrated around astrocyte end-feet surrounding glymphatic vessels similar to AQP4 [46]. Further experiments evaluating the pathway by which APOE4 was transported to the brain were performed in vivo using fluorescently labeled APOE4 protein and injecting the sample into the cisterna or anesthetized mice and observed with two-photon laser microscopy. They observed APOE4 traveling along the cerebral surface arteries to the penetrating arterioles and into the VRS within various sites of the cerebral parenchyma [46]. The clearance APOE proteins were confirmed to exit the brain through parenchymal veins. Additionally, previous studies highlighted that the APOE ε4 allele reduces Aβ clearance from the ISF resulting in brain parenchymal deposition and CAA further disrupting glymphatic flow [47]. This reinforces the hypothesis that the APOE gene, a critical genetic risk factor of AD, is also strongly associated with the glymphatic system.
Like arteriosclerosis, disease states impacting the brain’s vessels reduce waste clearance and therefore impair glymphatics. Additionally, damage to vessels can result in edema, microhemorrhages, and hypoxia, further affecting cognition. As mentioned previously, TBI leads to compromised cerebral vasculature and disruption of the BBB due to Aβ and p-tau deposition [32]. Specifically, following TBI, Aβ deposition occurs around the paravascular space, while p-tau proteins in neurofibrillary tangles accumulate around small cortical vessels, such as the penetrating arteries, also known as the VRS (Figure 1) [32]. An uncompromised flow between the ISF into the VRS is necessary to filter waste products throughout the glymphatic system. However, protein depositions from TBI disrupt the glymphatic flow, ultimately leading to AD.
Other brain injuries disrupting glymphatic flow include subarachnoid hemorrhages (SAH). A unique case study following an SAH demonstrates the effect of vascular disease on the clearance of Aβ [4]. The 59-year-old patient presented in 2016 with an acute SAH due to a rupture of an aneurysm affecting the right posterior communicating artery (PCoM). The patient recovered well following surgery and functioned independently for several years after, until she returned reporting memory impairment. A PET scan performed in 2021 showed a significant diffuse deposition of amyloid in the brain, more prominent on the right side, as compared to a previous scan performed in 2018. The development of cognitive decline was markedly rapid, as AD typically takes a decade or more to develop following biochemical detection [5], suggesting another phenomenon was responsible for the exponential neurocognitive decline. We postulate this is due to the disruption of the glymphatic circulation.
There have been previous studies that showcased significantly impaired glymphatic flow through disruption of the para-arterial influx in SAH animal models compared to control subjects just after 24 hours [48]. Essentially, the hemorrhagic cerebral injury as a result of SAH leads to fibrin and fibrinogen aggregation surrounding the VRS which occludes para-arterial influx, leading to impaired glymphatic clearance, diminished waste clearance, disruption of AQP4 polarity [49], and early onset dementia [4, 32]. In instances where there is an insult or disruption of the normal physiological barriers such as the BBB in TBI and SAH, the body’s initial response will be inflammation following the deposition of fibrin and fibrinogen to heal that process. Although this is the body’s way of healing cerebral injury, it has deleterious effects on cerebral blood flow and therefore glymphatic circulation. This suggests that impaired glymphatic clearance as a result of SAH contributed to the exponential neurocognitive decline and early signs of AD in our 59-year-old patient [4].
The well-established pathologic hallmark of AD is Aβ accumulation and p-tau in neurofibrillary tangles [6, 9]. In the healthy brain, soluble Aβ and tau proteins travel through the glymphatic system via the CSF, where it is exchanged with the ISF on astrocytes via AQP4 from the VRS into the brain parenchyma to eventually be removed paravenously into the peripheral lymphatic system [31]. Conversely, in AD, Aβ aggregates within the paravascular space disrupt the polarity of the glymphatic system required for proper clearance, ultimately hindering glymphatic flow.
Notable risk factors of AD, such as genetic predispositions, cause increased Aβ production, whereas other AD risk factors affecting the intracranial vessels most likely affect the clearance of Aβ aggregates. Non-modifiable risk factors such as presenilin mutations and Down syndrome lead to increased Aβ production, whereas other risk factors such as APOE ε4 genotype, TBI, SAH, and sleep deprivation lead to decreased Aβ clearance and tau accumulation. Both these categorical risk factors lead to Aβ and neurofibrillary tangle deposition in the cerebral parenchyma and around the cerebral vasculature, eventually inhibiting glymphatic flow and leading to the hallmark cognitive decline seen in AD.
Aβ: amyloid β
AD: Alzheimer’s disease
APOE ε4: apolipoprotein E ε4
APP: amyloid precursor protein
AQP4: aquaporin-4
BBB: blood-brain barrier
CNS: central nervous system
CSF: cerebrospinal fluid
IPAD: intramural periarterial drainage
ISF: interstitial fluid
MCI: mild cognitive impairment
p-tau: hyperphosphorylated tau
SAH: subarachnoid hemorrhages
TBI: traumatic brain injury
VRS: Virchow-Robin space
IH, MB, TL, and CC: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. FL: Conceptualization, Investigation, Validation, Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Rafael Franco, Joan Serrano-Marín
Karolina Armonaite ... Luigi Laura
Jesús Avila ... Félix Hernández
Glòria Salort ... Jesús A. García-Sevilla
Milind Watve, Ashwini Keskar Sardeshmukh
